




1 School of Biomedical Engineering, University of British Columbia, Vancouver, BC V6T 1Z3, Canada
2 Department of Cellular and Physiological Sciences, University of British Columbia, Vancouver, BC V6T 1Z3, Canada
Academic Editors: Brian A. Mendelsohn, Yutaka Matsuda and Josef Jampílek
Abstract
Finding the ideal epitope to target is a key element for the development of an antibody-drug conjugate (ADC). To maximize drug delivery to tumor cells and reduce side effects, this epitope should be specific to cancer cells and spare all normal tissue. During cancer progression, glycosylation pathways are frequently altered leading to the generation of new glycosylation patterns selective to cancer cells. Mucins are highly glycosylated proteins frequently expressed on tumors and, thus, ideal presenters of altered glycoepitopes. In this review, we describe three different types of glycoepitopes that are recognized by monoclonal antibodies (mAb) and, therefore, serve as ideal scaffolds for ADC; glycan-only, glycopeptide and shielded-peptide glycoepitopes. We review pre-clinical and clinical results obtained with ADCs targeting glycoepitopes expressed on MUC1 or podocalyxin (Podxl) and two mAbs targeting glycoepitopes expressed on MUC16 or MUC5AC as potential candidates for ADC development. Finally, we discuss current limits in using glycoepitope-targeting ADCs to treat cancer and propose methods to improve their efficacy and specificity.
Keywords
- glycoepitope
- mucin
- antibody-drug conjugate
- cancer treatment
With an aging global population, cancer incidence is on the rise, and, in most developed countries, cancer is the leading cause of death [1]. Conventional cancer therapies include surgery, radiotherapy, and chemotherapy [2]. While for many patients, these therapeutic interventions can effectively control tumor growth and there have been great strides in tailoring these to the molecular subtypes of neoplasia, there remains a large proportion of cancers that are refractory to treatment. Metastatic disease remains as a leading cause of cancer deaths with very few therapeutics that target this systemic phase of the disease [2]. Even when successful, chemotherapy-associated toxicity can often have a major impact on patient quality of life and drug-resistant relapse is common [3, 4]. Antibody-based targeting of tumors offers more efficacious treatments with fewer adverse side effects. One of the ways to do this is using specific antibodies to tumor-restricted antigens to direct toxic payloads to the tumor while sparing normal tissue. In this review we will define such “tumor antigens” as antibody targets (rather than T cell receptor peptide antigens).
Antibody-drug conjugates (ADC) represent a relatively new class of cancer treatments that seek to avoid the off-target toxicity associated with chemotherapy by linking a cytotoxic drug (“payload”) to a tumor reactive monoclonal antibody (mAb) [5, 6]. The goal of this site-specific delivery method is to minimize systemic chemotherapy-associated side effects and maximize delivery of the cytotoxic agent to tumor cells [5, 6]. Ideal payloads are ones with limited toxicity in the circulation while covalently linked to an antibody but that are highly toxic when internalized and released intracellularly. Several factors such as linker properties, drug-antibody-ratio, stability and biodistribution, and drug dosing, dictate clinical success of an ADC [7]. Of course, the selection of the target antigen and the mAb tissue/tumor specificity are crucial elements for the efficacy of this approach.
Ideal ADCs target an epitope highly expressed on cancer cells but absent or weakly expressed on normal cells [7]. Furthermore, an effective ADC epitope should be expressed on the surface of the cell (extracellular) and become internalized upon mAb binding [8]. Most ADC targets currently approved or in development are “tumor-associated” instead of “tumor-specific”, as they are also weakly expressed on normal tissue [9]. One interesting example of a true tumor-specific target is the epidermal growth factor receptor (EGFR) variant III (EGFRvIII) with deletions in EGFR exons 2–7. EGFRvIII is a tumor neoantigen that has attracted many efforts to generate mAbs, ADCs, vaccines, and chimeric antigen receptor T cell (CAR-T) candidate therapies [10, 11]. Due to the important list of requirements for the generation of an optimal ADC, and primarily the scarcity of true tumor specific epitopes, optimal targets are extremely rare.
Intriguingly, the dysregulation of glycosylation pathways is a frequent feature of tumor progression as the resulting altered glycan structures participate in an array of biological processes involved in cancer development [12]. One important consequence of this dysregulated glycosylation is the generation of tumor-associated carbohydrate antigens (TACA) that comprise an array of potential tumor-specific neo-epitopes which, in theory, represent ideal ADC targets [13]. Mucins are a family of highly glycosylated extracellular proteins of particular interest as several members of this protein family are abundantly overexpressed on the extracellular surface of cancer cells, likely reflecting their role in altering adhesion and facilitating migration [14, 15]. In this review, we provide an overview of the mechanisms that lead to TACA expression on mucins, the type of glycoepitope suitable for ADC-targeting and summarize results of select pre-clinical and clinical mucin-targeted ADC candidate therapies.
Mucins are typically large glycoproteins (200 kDa–200 MDa) expressed by
epithelial cell membranes [16]. This protein family is characterized by the
presence of one or more modular mucin proline (Pro), threonine (Thr), serine
(Ser) (PTS)-rich domains with a high frequency of Thr or Ser amino acid residues
covalently conjugated with an
Mucins are frequently expressed, shed (proteolytic cleavage) or secreted on the apical surface of barrier structures of tissues, especially epithelial barriers contacting the extra-tissue environment (mucosal surface epithelium) and endothelial layers contacting the circulation [18]. Their presence maintains the integrity of these barriers while also limiting contact with pathogens, toxins and antigens and inflammatory agents that may cause damage and trigger inflammatory responses. Secreted/shed mucins can help neutralize pathogens and transmembrane mucins play a role in sensing the environment and maintaining tissue architecture. Tumorigenesis exploits the function of mucins (stochastically) to promote their own growth and survival; enhance motility; limit adhesion; and evade immune surveillance [18]. In addition, dysregulated expression of mucins during tumor progression may be accompanied by changes to glycosylation and metabolic machinery that result in aberrant mucin glycoforms with altered physical and functional properties including immune evasion [18, 21]. In this review, we will selectively focus on four mucins that have been explored extensively for their role in cancer: transmembrane mucins Mucin 1 (MUC1), podocalyxin (Podxl), and MUC16 and secreted mucin MUC5AC (Table 1, Ref. [22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43]).
| Mucin | Normal tissue expression | Tumor expression pattern |
| MUC1 | Glandular or luminal epithelial cells of the mammary gland, esophagus, stomach, duodenum, pancreas, uterus, prostate, and lungs, and to a lesser extent hematopoietic cells [22, 23, 24] | Breast, lung, endometrium, endocervix, ovary, bladder, kidney, esophagus, stomach, pancreas, colon and bile duct carcinomas [25, 26] |
| PODXL | Kidney glomeruli, surface of vascular endothelial cells, megakaryocytes and platelets, mesothelial cells, hematopoietic progenitors, and a subset of neurons [27, 28, 29, 30, 31, 32] | Embryonal, oral squamous, esophageal, lung, gastric, colorectal, pancreatic, prostate, bladder, thyroid, uterine and renal cell carcinoma as well as astrocytoma and glioblastoma [33, 34] |
| MUC16 | Epithelial cell surface lining the upper respiratory tract, cornea and conjunctiva, female reproductive organs, the pleura, the peritoneum, and the pericardium, the abdominal cavity, and the cervical mucus [35, 36] | Pancreatic, esophageal, gastric, colorectal, breast, ovarian and non-small-cell lung cancers [35, 37, 38, 39] |
| MUC5AC | Goblet cells in the lung, eyes, stomach and endocervix [40, 41] | Pancreatic, colorectal, esophageal, gastric, mucinous ovarian and bronchoalveolar cancers [41, 42, 43] |
MUC1 is the founding member of large family of mucins normally expressed on glandular or luminal epithelial cells in a variety of tissues, where its extended negatively charged sugar branches create a selective biophysical barrier with anti-adhesive properties, limiting pathogen accessibility and preventing colonization of mucosal surfaces [23]. MUC1 has proved to be an interesting target for ADC development as aberrantly glycosylated MUC1 is overexpressed in most human epithelial cancers [25]. Podxl is a highly glycosylated cell surface sialomucin of the CD34 family of stem cell antigens and plays important roles in cell adhesion and transendothelial migration in normal and cancer tissues [44, 45]. Its expression is upregulated by a wide variety of cancer types and Podxl overexpression is consistently linked to poor prognosis, more aggressive tumor progression, unfavorable treatment outcomes, and possibly chemoresistance [32, 33, 34]. In keeping with its normal distribution on hematopoietic and early embryonic tissue stem cell populations, Podxl appears to be expressed by a highly mobile subset of tumor initiating cells. Furthermore, inactivation of the PODXL gene or dampening of its expression in cell lines cripples their ability to form tumors in xenografts and dampens their “tumorsphere” forming potential in vitro [46, 47]. MUC16, a MUC1 relative, is the largest of all known mucins and is the carrier of the Cancer Antigen 125 (CA125) epitope, which is widely used as a serum marker for the detection of ovarian cancer [35, 48, 49]. MUC5AC is a secreted, gel-forming, mucin that normally composes the airway mucus layer [50, 51]. In cancer, its expression is upregulated by an array of tumor types and its expression is predominantly cytoplasmic or at the apical pole of tumor cells [41].
O-linked glycosylation of mucins is initiated with the monosaccharide
GalNAc
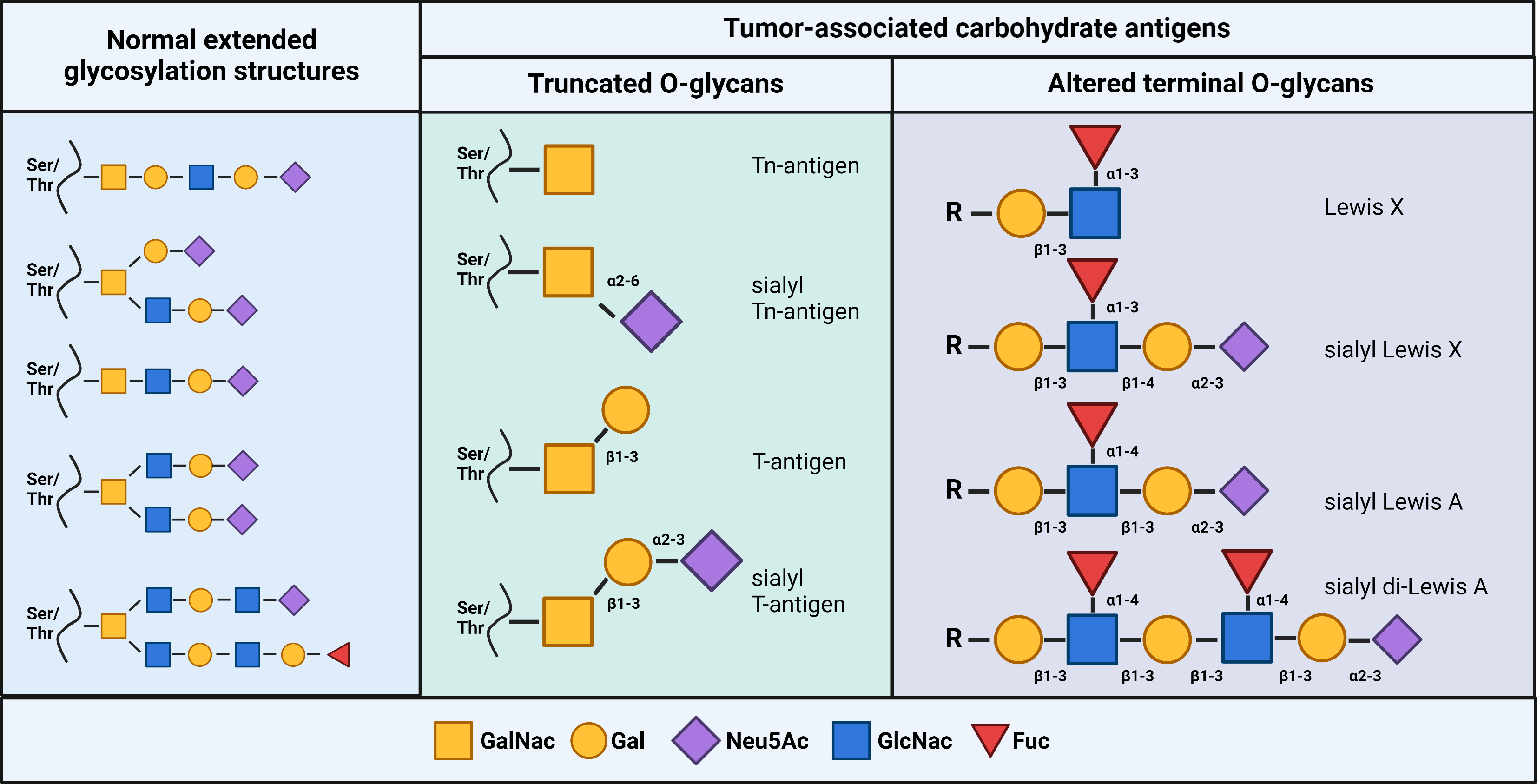 Fig. 1.
Fig. 1.Glycan structures of tumor-associated carbohydrate antigens. (Left) Examples of extended glycans expressed on normal cells. (Middle) Examples of common truncated O-linked glycans. (Right) Examples of altered terminal O-linked glycans. Created with BioRender.com.
By definition, a glycoepitope refers to a carbohydrate moiety that is recognized
by a mAb or other glycan-binding protein [60]. While mAbs that only bind
monosaccharide units (glycan-binding mAbs) exist, their affinity is usually much
lower than protein-specific mAbs with equilibrium dissociation constant
(K
Another class of TACA-binding mAbs that, in general, tend to exhibit improved binding affinities and specificity are those mAbs that recognize a glycoepitope formed by combinations of a glycan and a defined peptide epitope (i.e., glycopeptide glycoepitopes) [67, 68]. These tend to show specificity and affinities more typically associated with protein antigens. A third type of glycoepitope that we have named “shielded-peptide glycoepitopes” are not true glycoepitopes per se as the mAb does not directly bind the glycan, but instead recognize polypeptide epitopes whose accessibility to the peptide sequence is restricted by the altered glycosylation status of the glycoprotein expressed on normal cells. A schematic representation of these three classes of epitopes is presented in Fig. 2.
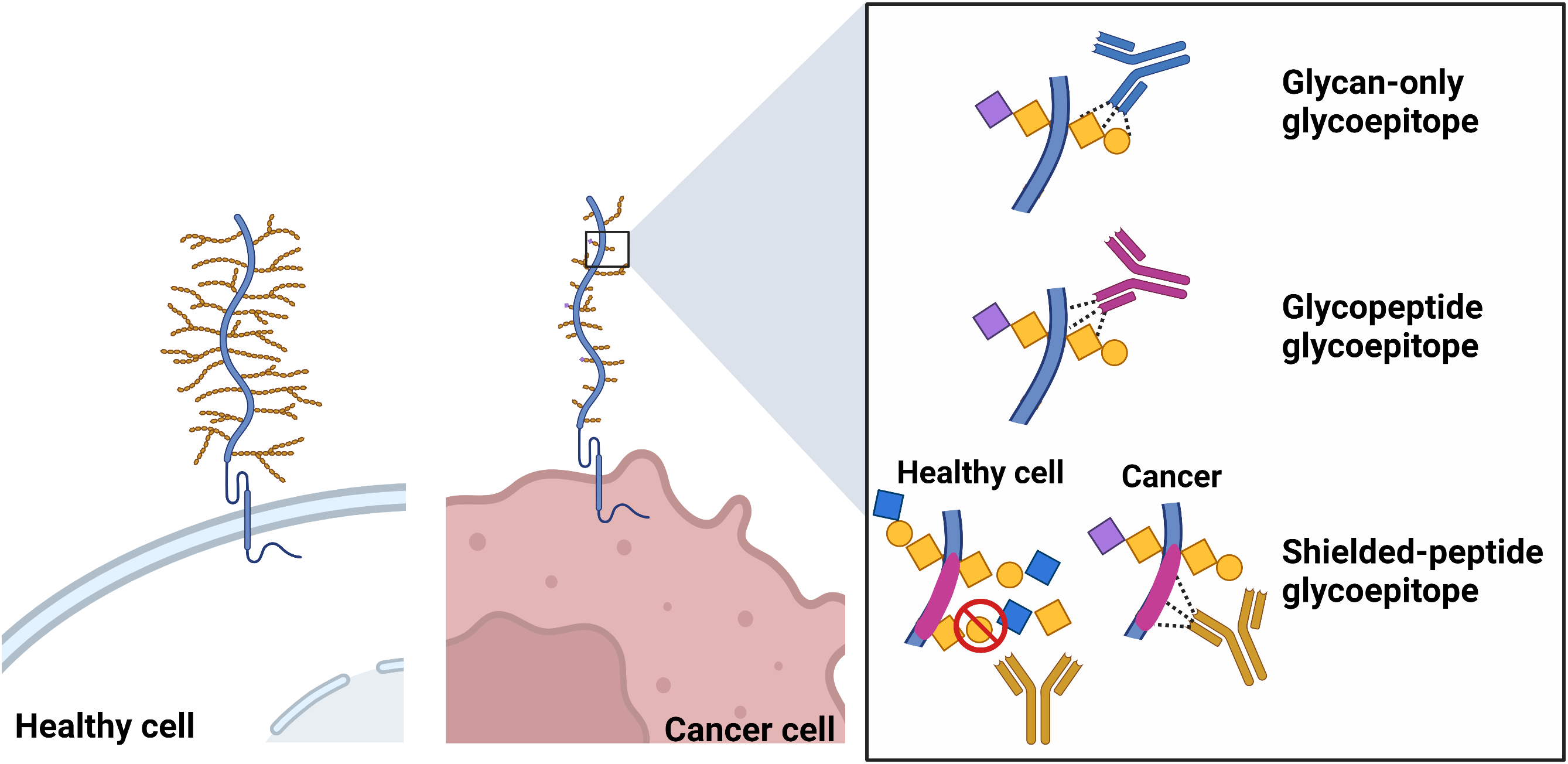 Fig. 2.
Fig. 2.Schematic representation of glycan-only, glycopeptide and shielded-peptide glycoepitopes. Created with BioRender.com.
While the identification of the amino acid sequence of a regular peptide epitope is now relatively easy, the characterization of a glycoepitope structures remains complex and challenging due to their complex and branched chain structure. For this reason, the exact glycoepitope structure of most mAb/ADC targets discussed in this review is still unknown. Nevertheless, we have included mAbs/ADCs with sufficient epitope mapping data to suggest that their epitope falls within one of three categories described in Fig. 2.
Therapeutic mAbs and associated ADCs included in this review (summarized in Table 2, Ref. [69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106]) were selected to provide illustrative examples of tumor-specific mucin glycoepitopes that may be ADC targets. As the glycoepitope nature of the target is the central focus of this review, several interesting anti-mucin ADCs were excluded due to the peptide nature of their epitope or the lack of sufficient epitope mapping information to support the contention that their epitope falls within one of the three types of glycoepitopes described in Fig. 2.
| I. Antibodies to unknown glycan epitopes | |||
| Antibody/Target | ADC | Epitope expression | Pre-clinical/clinical development stage |
| mJAA-F11 | huJAA-F11 H2aL2a-DM1 | Targets glycans of an O-linked glycosylated protein expressed in breast, lung, prostate, colon, bladder, and ovarian tumors [69, 70] | In vitro, in vivo mouse models [71] |
| huJAA-F11 H2aL2a | |||
| FG129 (m) | CH129-DM1 | Putative mucin expressed glycans in pancreatic, gastric, colorectal, ovarian, and NSCLC tumors | In vitro and in vivo mouse models |
| CH129 (ch IgG |
CH129-DM4 | ||
| CH129-MMAE | |||
| II. Antibodies binding glycoepitope-dependent epitopes of MUC1 | |||
| Antibody | ADC | Epitope expression | Pre-clinical/clinical development stage |
| mAb 16A | 16A-MMAE | Breast tumor, NSCLC and gastric tumors [73, 74] | In vivo mouse models [74] |
| PankoMab (m) | Cervical, ovarian, lung, breast, gastric, colorectal, liver, kidney, and thyroid tumors [75, 76, 77, 78] | ||
| Gatipotuzumab/PankoMab-GEX (hu) | gatipotuzumab /PankoMab-GEX | ||
| humanize unconjugated form | |||
| Phase I (NCT0122624) [80, 81] | |||
| Phase II (NCT01899599) [82, 83] | |||
| (m)DS6 | huDS6-SPDB-DM4 (SAR566658) | Breast, ovary, lung, bladder, and pancreas tumors [84, 85, 86] | In vivo mouse models [86] |
| (hu)DS6 | Phase I (NCT01156870) [87] | ||
| Phase II (NCT02984683) (discontinued) [88] | |||
| C242 (m) | huC242-DM1 (cantuzumab mertansine) | Pancreas and colorectal tumors [89, 90, 91] | In vivo mouse models (DM1) [92, 93] (DM4) [94] |
| huC242 or cantuzumab (hu) | huC242-DM4 (cantuzumab ravtansine/IMGN242) | huC242-DM1 | |
| Phase 1 [95, 96, 97] | |||
| huC242-DM4 | |||
| Phase I [98] | |||
| Phase II [99] | |||
| III. Antibodies to non-MUC1 glycoepitopes | |||
| Antibody | ADC | Epitope expression | Pre-clinical/clinical development stage |
| PODO447 | huPODO447-Vedotin (MMAE) | Tumor specific glycoform of PODXL expressed in ovarian tumors [47] | In vitro and in vivo [47, 100] |
| AR9.6 | ADC not yet developed | MUC16 expressed in pancreatic tumors [101] | unconjugated mAb, in vivo mouse models [101, 102] |
| NPC-1 | ADC not yet developed | MUC5AC expressed in colon and pancreatic tumors [103] | unconjugated mAb, in vivo mouse model [103] |
| NPC-1C | Phase I [104] | ||
| ensituximab | Phase II [105] | ||
| (NEO-102) | Phase I/II (NCT01040000) [106] | ||
| m, mouse; hu, humanized; rb/hu, rabbit/human chimera; NSCLC, non-small cell lung carcinoma; ch, chimeric human; MMAE, monomethyl auristatin E; DM1, maytansinoid DM1/mertansine; DM4, maytansinoid DM4. | |||
It is difficult to compare mAb affinities across examples especially when
disparate methods of assessment are used between different research groups. While
still imperfect, here we report antibody-antigen affinity values
(K
A prime example of a glycan-binding mAb is the murine JAA-F11 IgG
Another interesting glycan-binding mAb is the mouse IgG
Several MUC1-directed mAbs and single-chain variable fragments (scFv) have been generated as potential cancer therapeutics for CAR-T, radioimmunotherapy (RIT) and mAb therapy that may have applications as ADCs (reviewed in ref [109]). These mAbs, as a collection, target glycan or peptide epitopes as well as glycoepitopes [109]. Below we highlight examples of established MUC1-directed ADC candidates that target glycoepitopes.
16A is a murine IgG
Gatipotuzumab, formerly known as PankoMab-GEX, is the humanized version of the
mouse IgG
The murine MuDS6 IgG
The C242 mouse IgG
While MUC1 glycoepitopes are interesting targets for the development of ADCs, the few targeting mAbs that reached the clinical trial stage have, to date, failed to be approved for cancer treatment. Possible explanations for these disappointing results will be discussed at the end of this review.
Abnormal mucin-glycosylation patterns in cancer are not restricted to MUC1. We
recently developed a highly tumor specific rabbit/human chimeric IgG
AR9.6 is a murine mAb that binds a conformational epitope of the SEA domain 5 of
MUC16 that is influenced by O-linked glycosylation [101]. Interestingly,
binding of unconjugated AR9.6 with MUC16 leads to a significant reduction of
tumor growth in a pancreatic tumor model by blocking MUC16-dependent activation
of ErbB (EGF) receptors on the cancer cell surface and downstream attenuation of
oncogenic AKT and GSK3
Finally, NEO-102 (ensituximab) is a chimeric mouse/human IgG
While most mucin glycoepitope-targeting ADCs described in this review were highly effective at eliminating tumor cells in vitro and in animal models, the few that reached clinical trials have so far failed to provide convincing results and some have revealed unexpected side effects. These pitfalls are not unique to glycoepitope-specific antibodies and there are several factors that might explain these unfortunate results (summarized in Fig. 3). First, the shedding or secretion of the extracellular mucin domain containing the ADC glycoepitope can reduce the specific binding of the ADC to its target. This can simultaneously lead to off-target antigen-ADC interaction with cells that bind the shed antigen leading to toxicity while, at the same time, lessening the portion of the administered drug reaching the tumor microenvironment, reducing efficacy, and potentially leading to an unnecessary dose escalation to deliver drug on target [124, 125, 126, 127, 128]. With that in mind, careful measurement of the target glycoepitope entering the circulation during the clinical trials to assess potential of shedding to undermine efficacy could prove to be an important further criterion and evaluating potential of these types of targets in evaluating the potential of these types of targets. This approach was used in the IMGN242 trial to adjust dosing in patients with low CanAg circulating in plasma to mitigate off-target toxicity [99].
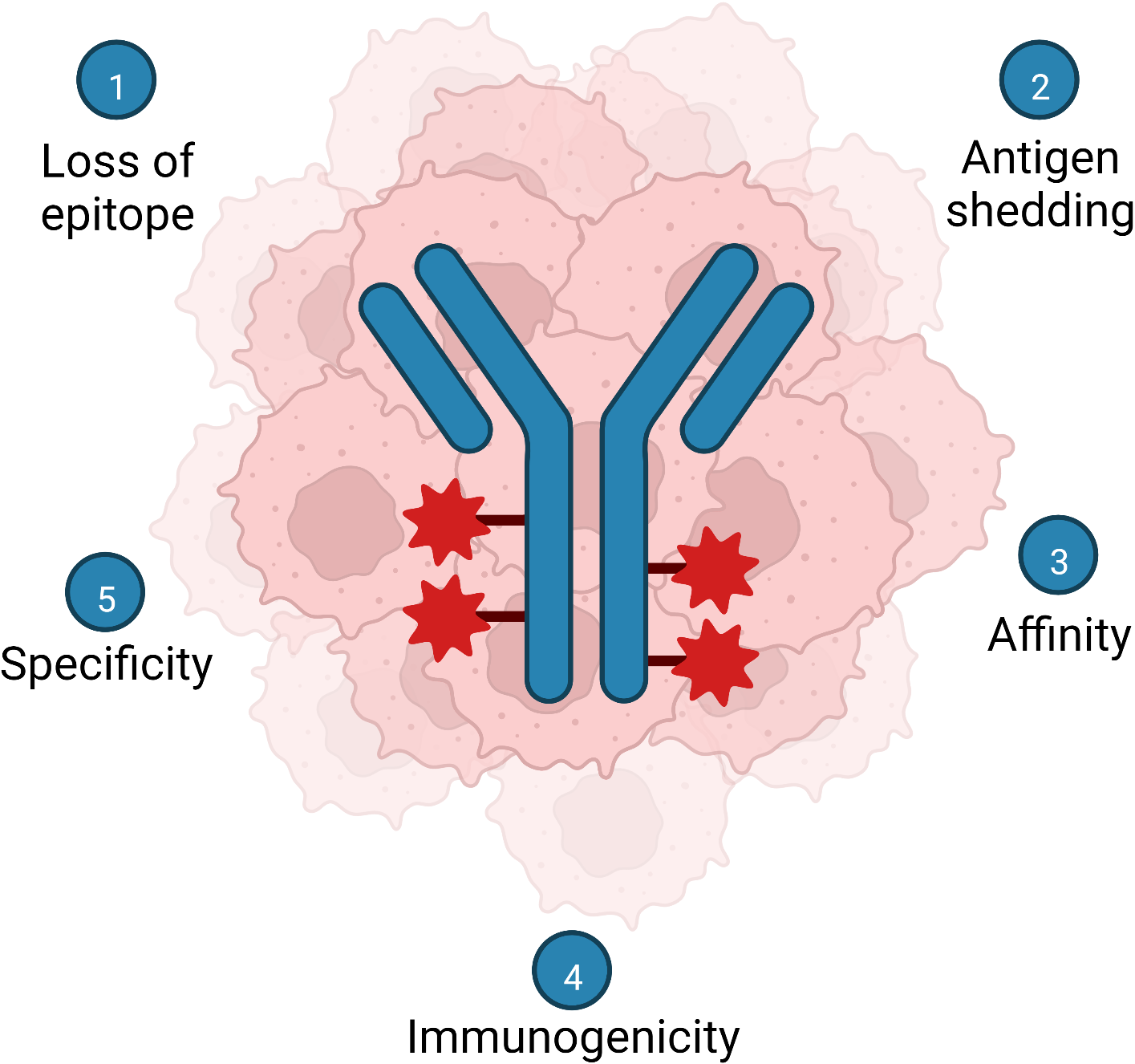 Fig. 3.
Fig. 3.Summary of notable challenges facing development of mucin glycoepitope therapeutic mAb and ADC. (1) Loss of the target epitope by the tumor and tumor heterogeneity. (2) Shedding or secretion of the epitope. (3) Carbohydrate mAbs tend to bind epitopes with lower affinities compared to peptide epitopes. (4) Carbohydrate epitopes are less immunogenic than peptide epitopes. (5) Identifying truly tumor-restricted mucin glycoforms (tumor antigens) Created with BioRender.com.
Another consideration should be the tumor specificity of the glycoepitope. While the expression of all the glycoepitopes targeted by the ADCs described in this review are predominantly present on cancer cells, many are also expressed at lower levels on subsets of normal tissue cells [72, 74, 84]. Depending on the type of tissue and the level of non-tumor expression, this could lead to significant normal tissue toxicity. The cBR96-doxorubicin (BR96-DOX/SGN-15), a “first generation” ADC directed against a putative “tumor-specific” glycoepitope (Lewis Y), is a good example of how the expression of the ADC target on normal tissue can negatively influence its efficacy. This ADC failed clinical trials due, in part, to the expression of Lewis Y in gastrointestinal (GI) mucosa that caused both toxicity (adverse GI events) and poor therapeutic performance (an “antigen sink” that sequesters the drug) [129].
Glycan immunogenicity is generally inferior to peptide antigens and, therefore, the affinity of mAbs is stronger for peptide sequences compared to glycans [61]. Since much of the tumor specificity of the epitope is provided by the glycan structure, the optimal glycoepitope target is likely a glycopeptide epitope rather than one comprised purely from glycans. Shielded-peptide glycoepitopes are also good alternatives, but as several altered glycosylation profiles could provide mAbs with access to an otherwise, shielded-peptide epitope, the tumor-specificity might be compromised in some normal contexts. Currently, the characterization of the exact glycan structure of the epitope and the identification of the interactions between the mAb and the glycoepitope remain challenging to obtain. New technologies such as tandem mass spectrometry (MS/MS) used in combination with new computational tools will help predict the exact structure of glycoepitopes and might help select candidate mAbs that are more likely to be tumor-specific [130]. Likewise, the screening of cellular arrays bearing lesions in known GTFs (as was done for the PODO447 mAb [100]) could prove to be a highly effective method of fine mapping the glycoepitopes on target antigens.
Another factor that can limit the efficacy of mucin glycoepitope targeting ADCs is the possibility that, at some point during the treatment, tumors undergo selective pressure to alter their expression of GTFs or that existing variants lacking such enzymes expand when reactive subsets are eliminated. Either of these mechanisms would be expected to allow tumors to escape therapeutic targeting. To better characterize this phenomenon in mAb and target specific scenarios, the expression of the glycoepitope should be carefully monitored following the treatment in pre-clinical models and during clinical trials whenever possible. Furthermore, a potential therapeutic approach to circumvent this problem could be to combine the ADC treatment with other therapies, such as immunotherapies, to help eradicate cells negative for the ADC-glycoepitope [131]. In this regard, it is intriguing that preclinical studies with the PODO447-ADC targeting Podxl have shown that residual tumors remaining after ADC treatment fail to express the Podxl polypeptide rather than maintaining its expression and simply altering its glycosylation [100]. At face value, this may suggest that the pattern of GTF expression by aggressive tumors plays a more critical role to their biology than expression of the individual polypeptide itself. More strikingly, the cells that lose expression of the Podxl core protein are known to grow more slowly and are less invasive. Finally, these residual Podxl-negative tumors appear to be more prone to clearance by the endogenous immune response (i.e., the ADCs are much more efficacious in Nude mice than in the more severely immunocompromised NSG mice) suggesting that killing of Podxl+ subset tumor cells may have an outsized effect on tumor clearance. These observations bode well not only for the PODO447-ADC but also for development of any other tumor-specific glycoepitope antibodies that detect a similar glycan-modification.
In conclusion, ADCs targeting mucin’s glycoepitopes represent a novel opportunity for highly selective tumor-targeting. While the ability to generate antibodies to identify, and fine map these target epitopes have all been challenging, once identified, the data suggest that these may prove to be highly specific at targeting the most relevant subsets of tumor cells that drive disease. The few emerging mucin targets described here may indeed be the vanguard of a much larger constellation of potential glycoepitopes. With that in mind, a well-developed, stepwise pathway for systematic evaluation of the tumor glycoepitope space could prove highly revealing of new actionable targets.
JB realized the original draft preparation. JB and MRH designed the figures. MRH, CDR and KMM reviewed and edited the manuscript. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
This work was supported by the Canadian Institutes of Health Research (PJT-166180) and supported by a Canadian Cancer Society Challenge Grant (#707343). JB was supported by a Research Trainee Award from the Michael Smith Foundation for Health Research.
MRH, CDR and KMM are co-inventors on PODO447 patent application(s) assigned to the Centre for Drug Research and Development University of British Columbia, the University of British Columbia and, the University of Copenhagen.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.