


1 Cellular and Molecular Signaling, New York, NY 10022, USA
Abstract
Introduction: Dementia and cognitive loss impact a significant proportion of the global population and present almost insurmountable challenges for treatment since they stem from multifactorial etiologies. Innovative avenues for treatment are highly warranted. Methods and results: Novel work with biological clock genes that oversee circadian rhythm may meet this critical need by focusing upon the pathways of the mechanistic target of rapamycin (mTOR), the silent mating type information regulation 2 homolog 1 (Saccharomyces cerevisiae) (SIRT1), mammalian forkhead transcription factors (FoxOs), the growth factor erythropoietin (EPO), and the wingless Wnt pathway. These pathways are complex in nature, intimately associated with autophagy that can maintain circadian rhythm, and have an intricate relationship that can lead to beneficial outcomes that may offer neuroprotection, metabolic homeostasis, and prevention of cognitive loss. However, biological clocks and alterations in circadian rhythm also have the potential to lead to devastating effects involving tumorigenesis in conjunction with pathways involving Wnt that oversee angiogenesis and stem cell proliferation. Conclusions: Current work with biological clocks and circadian rhythm pathways provide exciting possibilities for the treating dementia and cognitive loss, but also provide powerful arguments to further comprehend the intimate and complex relationship among these pathways to fully potentiate desired clinical outcomes.
Keywords
- Alzheimer’s disease
- Autophagy
- Circadian rhythm
- Dementia
- Erythropoietin
- Forkhead
- FoxO
- Mechanistic target of rapamycin (mTOR)
- Parkinson’s disease
- Silent mating type information regulation 2 homolog 1
- wingless
- Wnt
Neurodegenerative disorders pose a significant challenge for diagnosis,
preventing disease progression, and providing treatment. Cognitive loss in
relation to Alzheimer’s disease (AD) is an excellent example since diseases that
include AD are the result of multiple underlying mechanisms [1, 2, 3, 4, 5, 6] (Table 1). For
example, many pathways may lead to memory loss and involve neuronal and vascular
cell injury related to metabotropic receptors, lipid dysfunction, cellular
metabolic dysfunction with diabetes mellitus (DM), astrocytic cell injury,
In addition, cognitive disorders raise significant financial concerns [1, 31, 32, 33, 34]. Greater than 800 billion United States dollars (USD) per year are required to treat dementia equaling approximately 2 percent of the global Gross Domestic Product. Social and medical services by the year 2030 may possibly equal 2 trillion USD per year in the United States. Currently, greater than 5 million patients have AD and it is estimated that 4 million receive care at a yearly cost of 3.8 billion USD. Furthermore, the market revenue to provide treatments for AD may not be fully appreciated, but at minimum it may be greater than 11 billion USD. Many new social and medical services will be necessary to meet this challenge such that 60 million additional care workers will be needed [35, 36, 37]. These projections do not consider that all cases of dementia may not have been identified and diagnosed at this time [38, 39].
| Neurodegeneration and dementia: circadian rhythm biological clock gene pathways |
Cognitive loss impacts a large spectrum of the population. Dementia in the
United States affects greater than 5 million people [4]. Many of these cases, 60
percent, are diagnosed as AD [4, 6, 17, 40, 41, 42, 43]. Case of AD that are familial in
origin comprise under 2% of all cases [4]. In familial AD that affects 200
families worldwide, mutations in the presenilin 1 or 2 genes occurs and an
autosomal dominant mutated amyloid precursor protein (APP) gene exists. In these
familial AD patients, illness can present prior to 55 years of age [44, 45, 46].
Familial AD can be the result of mutations in chromosome 21 leading to changes in
APP, mutations in chromosome 14 causing changes in presenilin 1, and mutations in
chromosomes 1, 14, and 21 such that mutations in chromosome 1 lead to changes in
presenilin 2. However, it is the sporadic version of AD that leads to illness in
patients over age 65 and represents the cases of AD in ten percent of the
population in the world. The
Current attempts to treat dementia such as with cholinesterase inhibitors may lead to a decrease in the presenting symptoms but ultimately do not block the progression of the disease, such as in AD [27, 45, 47, 48]. Other treatments for cognitive loss can focus on metabolic disorders, such as diabetes mellitus (DM) [1, 20, 27, 41, 49, 50], and on vascular disease [19, 45, 51, 52, 53]. Yet, there exist other risks for developing vascular cognitive loss that can affect the efficacy of treatments such as tobacco use, alcohol consumption, hypertension, and a low level of education [20, 39, 54, 55, 56, 57]. With reference to metabolic disease, tight glucose control in the serum in combination with early diagnosis of DM may assist to limit the progression of the disease, but complications from DM can still ensue [6, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69]. Given the need for novel strategies directed against memory loss and dementia, exciting new avenues of development are now focusing upon biological clock mechanisms and include the pathways of the mechanistic target of rapamycin (mTOR), its associated pathways of mTOR Complex 1 (mTORC1), mTOR Complex 2 (mTORC2), the silent mating type information regulation 2 homolog 1 (Saccharomyces cerevisiae) (SIRT1), mammalian forkhead transcription factors (FoxOs), the growth factor erythropoietin (EPO), and the wingless pathway of Wnt pathway (Fig. 1).
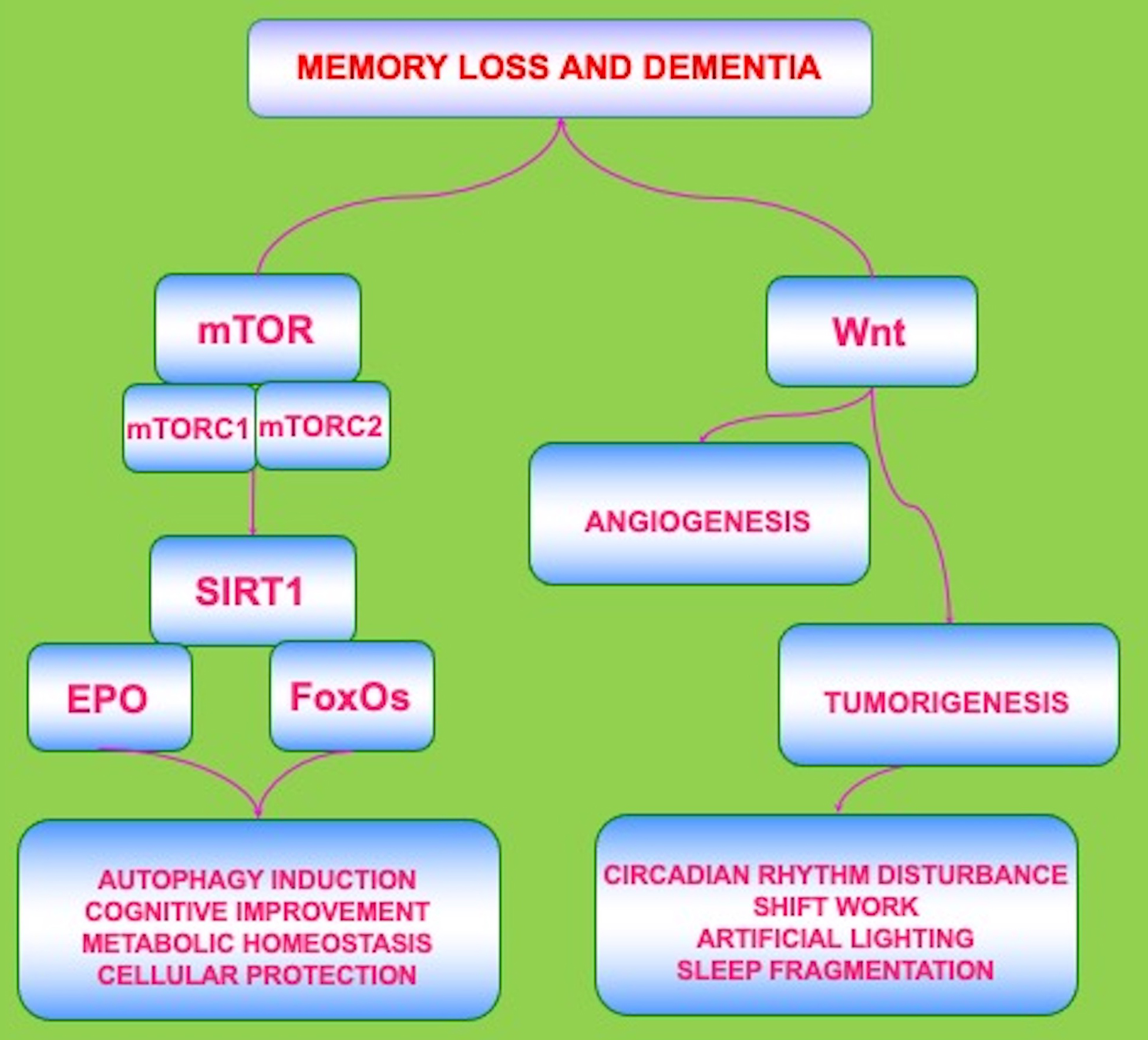 Fig. 1.
Fig. 1.Biological Clock Pathways Are Complex and May Yield Variable
Outcomes. The circadian biological clock gene pathways are intricately related
but complex in nature. The pathways of the mechanistic target of rapamycin
(mTOR), the silent mating type information regulation 2 homolog 1(Saccharomyces cerevisiae) (SIRT1), mammalian forkhead transcription
factors (FoxOs), the growth factor erythropoietin (EPO), and the
wingless Wnt/
Biological clocks and circadian rhythm pathways are vital components in the
onset of nervous system disorders, memory loss, and dementia [6, 34, 39, 70, 71, 72, 73, 74, 75, 76]
(Table 1). Changes in the function of biological clock pathways can impact
cellular metabolic homeostasis [6, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85], cancer [6, 80, 81, 84, 86, 87, 88, 89], energy
metabolism and aging [70, 74, 77, 84, 90], mitochondrial energy maintenance [76, 81, 91, 92], renal disease [78, 86], and viral diseases [72, 93, 94, 95, 96, 97, 98, 99, 100, 101]. Circadian
rhythm in mammals is controlled in a region over the optic chiasm that detects
light with retinal photosensitive ganglion cells in the suprachiasmatic nucleus
(SCN) [6, 84, 98]. With the exposure to external light, biological clock genes
oversee biochemical cell transmissions, physiological process in the body, and
changes in behavior. The SCN controls the temperature of the body, cortisol and
melatonin release, and oxidative stress responses through a connected system
among the hypothalamic nuclei, pineal gland, and vasoactive intestinal peptide
[88, 102, 103]. As part of the biological clock gene group, members of the basic
helix-loop-helix-PAS (Period-Arnt-Single-minded) transcription factor family,
that include CLOCK and BMAL1 [104], control gene expression of
Cryptochrome (Cry1 and Cry2) and Period (Per1,
Per2, and Per3) [6, 78, 84, 86, 105, 106, 107]. Modulation of these
pathways and auto-feedback interactions are controlled by PER:CRY heterodimers
that block transcription during nuclear translocation promoted by CLOCK:BMAL1
complexes. Other regulatory pathways that can be activated by CLOCK:BMAL1
heterodimers include ROR
With neurodegeneration and aging studies, experimental studies with Parkinson’s
disease (PD) using 6-hydroxydopamine (6-OHDA) during chronic treatment with
levodopa show depressed levels of BMAL1 and ROR
Lifespan can be affected by biological clock genes. Lifespan in
Drosophila melanogaster is decreased through three arrhythmic mutants
involving ClkAR, cyc0 and tim0. In addition, mutations in ClkAR with increasing
age can result in dysfunction with ambulation. Through the promotion of Clk
function, the locomotor deficits in Drosophila were reversed. This loss
of function appears linked to the absence of dopaminergic neurons instead of
insults from oxidative stress [75]. Other studies in Drosophila also
suggest negative effects with alterations in circadian rhythm [6, 80, 84] (Table 1). For example, TIMELESS, a mammalian homolog of Drosophila circadian
rhythm gene, can lead to cell death and has increased expression in
nasopharyngeal carcinoma. During increased TIMELESS expression, cell growth
pathways are fostered that involve the wingless pathway of
Wnt/
Circadian clock genes rely upon pathways of both autophagy and the mechanistic target of rapamycin (mTOR) [6, 84, 124, 125, 126] (Table 1). Circadian rhythm dysfunction can lead to changes in the induction of autophagy especially during cognitive loss [72, 81, 84, 92, 127, 128, 129]. Autophagy plays a vital role in multiple diseases of the nervous system and can sequester and remove intracellular deposits during AD [19, 41, 130, 131], amyotrophic lateral sclerosis [48, 132, 133], Huntington’s disease (HD) [19, 134], traumatic brain injury [135, 136, 137], and PD [83, 130, 135, 138, 139, 140]. This removal of toxic intracellular substances may be important to maintain memory and cognition. As part of a programmed cell death pathway, autophagy is tied to oxidative stress [2, 29, 66, 67, 71, 141, 142, 143, 144, 145]. Autophagy pathways can recycle cytoplasmic organelles and components for tissue remodeling [19, 146] and can eliminate non-functional organelles [6, 71, 142, 147]. Macroautophagy reuses organelles in cells and packages cytoplasmic proteins into cellular components termed autophagosomes. Once associated with lysosomes, the autophagosomes are degraded to begin another process for the recycling of organelles [19]. Microautophagy promotes invagination of lysosomal membranes to allow for the digestion of cell cytoplasm components. Chaperone-mediated autophagy employs cytosolic chaperones to transport cytoplasmic cell components across lysosomal membranes.
Previous studies also suggest in experimental studies with AD that a baseline
cyclic circadian rhythm that controls autophagy is necessary to reduce A
In regard to the mTOR pathway, mTOR is a 289-kDa serine/threonine protein kinase
and is vital during nervous system disease and memory loss [2, 19, 20, 25, 49, 155, 156, 157]. mTOR is also known as the mammalian target of rapamycin and the
FK506-binding protein 12-rapamycin complex-associated protein 1 [19, 85, 158, 159]. mTOR is the main component of the protein complexes mTOR Complex 1 (mTORC1)
and mTOR Complex 2 (mTORC2) [160, 161, 162]. mTORC1 and mTORC2 are then divided into
additional components [2, 107, 163, 164, 165]. mTORC1 is composed of Raptor, Deptor
(DEP domain-containing mTOR interacting protein), the proline rich Akt substrate
40 kDa (PRAS40), and mammalian lethal with Sec13 protein 8, termed mLST8 (mLST8)
[20, 40, 166]. mTORC1 activity is controlled through a number of pathways that
includes PRAS40 by blocking the association of p70 ribosomal S6 kinase (p70S6K)
and the eukaryotic initiation factor 4E (eIF4E)-binding protein 1 (4EBP1) with
Raptor [167, 168]. Rapamycin is an agent that can inhibit mTOR activity [164, 169, 170, 171, 172]. Rapamycin blocks the activity of mTORC1 through its association with
immunophilin FK-506-binding protein 12 (FKBP12) that attaches to the FKBP12
-rapamycin-binding domain (FRB) at the carboxy (C) -terminal of mTOR to impede
the FRB domain of mTORC1 [4]. mTORC2 is composed of Rictor, Deptor, mLST8, the
mammalian stress-activated protein kinase interacting protein (mSIN1), and the
protein observed with Rictor-1 (Protor-1) [167, 173, 174]. mTORC2 oversees
remodeling of the cytoskeleton through PKC
mTOR also maintains a relationship with the silent mating type information regulation 2 homolog 1 (Saccharomyces cerevisiae) (SIRT1). SIRT1 maintains an inverse relationship with mTOR [19, 176, 177, 178, 179, 180]. SIRT1 can also affect pathways of autophagy [49, 65, 163, 178, 181, 182, 183, 184, 185, 186]. SIRT1 activity can lead to the expansion of neurites and promote the survival of neurons during conditions that limit nutrients that involves mTOR inhibition [187]. SIRT1 can foster growth of tumors during autophagy induction that requires the blockade of mTOR, indicating that autophagy and SIRT1 can be targeted to control tumorigenesis [183]. SIRT1 is necessary to foster autophagy and mTOR inhibition during oxidative stress to preserve mitochondrial function in embryonic stem cells [188]. During periods of elevated serum glucose, SIRT1 can block mTOR to offer vascular cell protection [189]. SIRT1 with the blockade of mTOR activity can increase photoreceptor cell survival [177] and limit cell senescence [190]. It is also important to note that some pathways that lead to nerve cell injury require a relationship between mTOR and SIRT1 that is symbiotic. During the loss of dopaminergic neuronal cells, it has been observed that a balance in activities of SIRT1, mTOR, and forkhead transcription factors are required to promote neuronal cell survival [191]. It also has been demonstrated that SIRT1 and mTOR absence during obesity can suppress core circadian components CLOCK and BMAL1 and lead to loss of metabolic cellular homeostasis. The agent metformin, an inhibitor of mTOR activity [4, 65, 72], can prevent such processes during obesity in experimental mouse models and can reverse the loss of SIRT1 function during inhibition of the circadian components CLOCK and BMAL1 [192].
Biological clock pathways closely rely upon SIRT1 [6, 84, 85, 91, 193, 194]
(Table 1). SIRT1 is a histone deacetylase that can transfer acetyl groups from
Through SIRT1 pathways, the coenzyme ß-nicotinamide adenine dinucleotide
(NAD
SIRT1 regulation of biological clock genes also can affect cognitive function
though growth factors, such as EPO [161, 197, 212, 213, 214]. The EPO gene is
present on chromosome 7 and represents a single copy in a 5.4 kb region of the
genomic DNA [215, 216]. The gene encodes for a polypeptide chain protein that has
193 amino acids [64, 217]. EPO later undergoes the removal of a carboxy-terminal
arginine
In relation to SIRT1, EPO prevents metabolic dysfunction by modulating adipose
energy homeostasis in adipocytes through the combined activation of peroxisome
proliferator-activated receptor-
EPO also relies upon mTOR to affect cellular survival. EPO employs mTOR to
foster neuronal regeneration through autophagy and apoptotic pathways [20, 203, 236, 237, 238, 239]. EPO prevents apoptosis during A
Mammalian FOXO proteins of the O class are transcription factors and play a
significant role in the nervous system. FoxO family members include FOXO1, FOXO3,
FOXO4, and FOXO6 [67, 164, 245, 246, 247] (Table 1). FoxO proteins bind to
deoxyribonucleic acid (DNA) through the FoxO-recognized element in the
C-terminal basic region of the forkhead DNA binding domain. With the
binding to DNA by FoxOs, target gene expression is blocked or promoted through
fourteen protein-DNA contacts with the primary recognition site located at
In regard to SIRT1, blockade of the activity of FoxOs by SIRT1 can promote cell
survival [19, 67, 249, 250, 251]. However, FoxOs can attach to the SIRT1 promoter
region to further change forkhead transcription [181]. This mechanism permits
FoxOs to use auto-feedback mechanisms to regulate the activity of SIRT1. FoxO
proteins, including FoxO1, can oversee SIRT1 transcription and increase the
expression of SIRT1 [261]. These studies suggest an intimate relationship between
SIRT1 and FoxOs. Interestingly, SIRT1 and FoxOs can synergistically increase cell
survival. SIRT1 and FoxO3a can work in unison to block memory loss and A
Neurodegenerative disorders that involve cognitive loss and dementia impact a
significant proportion of the world’s population and lead to a large financial
burden for all nations. Adding to these concerns is the knowledge that cognitive
disorders present almost insurmountable challenges for treatment since they are
multifactorial in origin and can result from multiple pathways that involve
A
The pathways that impact circadian rhythm have an intricate relationship that
can lead to both beneficial as well as detrimental clinical effects. For example,
blockade of mTOR activity can change circadian rhythm, affect memory function,
and increase neuronal cell injury such as during stroke. SIRT1 can oversee the
production of NAD
These observations serve to form a strong foundation for the further investigation of biological clock genes and circadian rhythm in regards to their significant role in neurodegenerative disorders such as dementia. The circadian pathways involving mTOR, SIRT1, FoxOs, EPO, and the Wnt can offer considerable potential for the understanding and treatment of memory loss and neurodegnerative disorders. Yet, it is the intimate and complex relationship among these pathways that is most intriguing and potentially offers the greatest insight to harness this knowledge for the innovative treatment of dementia.
KM conceptualized and produced this work.
Not applicable.
We appreciate the reviewers for their opinions and suggestions.
This research was supported by the following grants to Kenneth Maiese: American Diabetes Association, American Heart Association, NIH NIEHS, NIH NIA, NIH NINDS, NS053956, and NIH ARRA.
The author declares no conflict of interest.
AD, Alzheimer’s disease; DM, diabetes mellitus; EPO, erythropoietin; FoxOs, mammalian forkhead transcription factors; HD, Huntington’s disease; NCDs, non-communicable diseases; mTOR, the mechanistic target of rapamycin; mTORC1, mTOR Complex 1; mTORC2, mTOR Complex 2; PER2, period2; PRAS40, proline rich Akt substrate 40 kDa; SIRT1, the silent mating type information regulation 2 homolog 1 (Saccharomyces cerevisiae); US, United States; USD, United States Dollars; wingless, Wnt