




 , Wei-Li Zhao 1,*
, Wei-Li Zhao 1,*1 Shanghai Institute of Hematology, State Key Laboratory of Medical Genomics, National Research Center for Translational Medicine at Shanghai, Ruijin Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, 200025 Shanghai, China
Abstract
Lymphoma is a common and aggressive form of hematopoietic malignancies with diverse clinical and pathological features due to its heterogeneity. Although the current immunochemotherapeutic regimens improve clinical outcomes, many patients still display poor prognosis and frequent relapse. Epigenetic alterations contribute to the progression of lymphoma. DNA methylation and histone methylation are the most common epigenetic alterations and regulate the gene expression involved in lymphoma pathogenesis, including silencing of tumor suppressor genes or activation of proto-oncogenes. Dysregulation or mutation of genes related to DNA methylation, including DNMTs, TET2, IDH2, and genes related to histone methylation, including EZH2, KMT2D has been observed. Most of these alterations are associated with inferior outcomes of patients with diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL), peripheral T-cell lymphoma (PTCL), and other subtypes of lymphoma. To overcome the pathogenetic consequence induced by aberrant DNA methylation and histone methylation, novel targeted drugs including azacitidine and decitabine have been gradually applied in practice to enhance the efficacy of current therapy and improve the prognosis of lymphoma patients. Investigating and targeting epigenetic mechanisms in lymphoma could be a key point of future research. Therefore, we mainly summarize the methylation alterations in lymphoma and their respective targeted therapies in this review.
Keywords
- Lymphoma
- DNA methylation
- Histone methylation
- Targeted therapies
Lymphoma is an aggressive and prevalent hematological malignancy which has diverse clinical characteristics and pathological features. According to the evolving knowledge in genetic alterations of lymphoma, classification and therapeutic strategies for lymphoma are also improved [1]. Lymphoma is pathologically classified into Hodgkin’s lymphoma (HL) and non-Hodgkin’s lymphoma (NHL). NHL mainly includes B cell lymphomas such as diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL), mantle cell lymphoma (MCL), as well as T cell lymphomas such as peripheral T-cell lymphoma not otherwise specified (PTCL, NOS), anaplastic large cell lymphoma (ALCL), and angioimmunoblastic T-cell lymphoma (AITL) [2]. The standard therapy for B cell lymphoma, especially DLBCL, FL, is R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone), which has remarkably improved the prognosis of patients [3, 4]. First-line therapy for T cell lymphoma is the CHOP regimen or CHOP-like regimen [4, 5]. Patients have different response to first-line treatment due to the heterogeneity of each subtype. Emerging targeted agents have also been explored in the treatment of lymphoma patients, in which epigenetic alterations are important targets [6].
Epigenetic changes refer to alterations of gene expression through mechanisms that do not involve changes in DNA sequence. These epigenetic mechanisms include DNA methylation, histone methylation, histone acetylation and chromatin remodeling [7]. Methylation alterations are the most common epigenetic modifications that regulate lymphoma genesis and progression, result in silencing of tumor suppressor genes and activation of proto-oncogenes. Of note, DNA methylation is mainly carried out by DNA methyltransferases (DNMTs) and tightly regulated by ten-eleven translocation dioxygenases (TETs) and isocitrate dehydrogenases (IDHs) [8]. Overexpression or mutation in DNMT3A, TET2 and IDH2 result in hypermethylation or hypomethylation in the promoter region of target genes and have functions in several physiological processes, including cell cycle, apoptosis, or immune response [9, 10]. Also, histone methylation plays a vital role in lymphoma progression [11]. The core genes involved in histone methylation include enhancer of zeste homolog 2 (EZH2) and the lysine methyltransferase 2 family genes (KMT2s) [12]. Methylation of histone H3K4 is related to transcriptional activation, while methylation of H3K27 corresponds to gene silencing [9]. Importantly, these aberrant DNA methylation or histone methylation can be reversed by specific inhibitors. Targeted inhibitors of methylation alterations such as DNMTs inhibitors azacitidine and decitabine and EZH2 inhibitor tazemetostat (EPZ6438) have been approved for clinical practices [13]. Thus, this review highlights the recent study of methylation alterations and possible therapeutic targets in lymphoma.
DNA methylation is a crucial epigenetic alteration in normal cells and in the process of tumorigenesis. It can affect gene expression by directly controlling the activities of DNA regulatory elements, including the cytosine-guanine (CpG) islands of the promoter regions. In normal cells, DNA methylation at centromeric sequences and transposable elements can stabilize the genomic structure, but during malignant transformation, abnormal DNA methylation leads to silencing of many tumor suppressor genes. However, the upregulation of DNA methylation promotes the progression of multiple lymphomas and negatively influences clinical outcomes. Moreover, it has been proven that using DNA methylation inhibitor has a significant anti-tumor effect.
DNA methyltransferases (DNMTs) are the predominant factors in the regulation of DNA methylation (Fig. 1). Within the DNMT family, DNMT1, DNMT3A and DNMT3B have been associated with tumorigenesis. DNMTs catalyzes 5-cytosine to generate 5-methylcytosine (5mC), which regulates gene silencing, imprinting, and genome stability [14]. The domain structures of DNMTs can be divided into the conserved catalytic domain on the C-terminus and diverse regulatory domains on the N-terminus. Different functions of DNMT1 and DNMT3A, DNMT3B are determined by the diversity of their N-terminal regions. The regulatory region of DNMT1 consists of several components such as a nuclear localization signal (NLS), a replication foci-targeting sequence (RFTS) for locating in the DNA replication fork, a CXXC domain for recognizing unmethylated CpG, as well as two bromo-adjacent homology (BAH) domains for protein-protein interaction and gene silencing, and a glycine-lysine (GK) domain as the linker of the C-terminal and N-terminal regions. The regulatory regions of DNMT3A and DNMT3B contain a proline-tryptophan-tryptophan-proline (PWWP) domain which is responsible for heterochromatin localization, and an ATRX-DNMT3A/3B-DNMT3L (ADD) domain which interacts with H3 when H3K4 is unmethylated [15, 16] (Fig. 2).
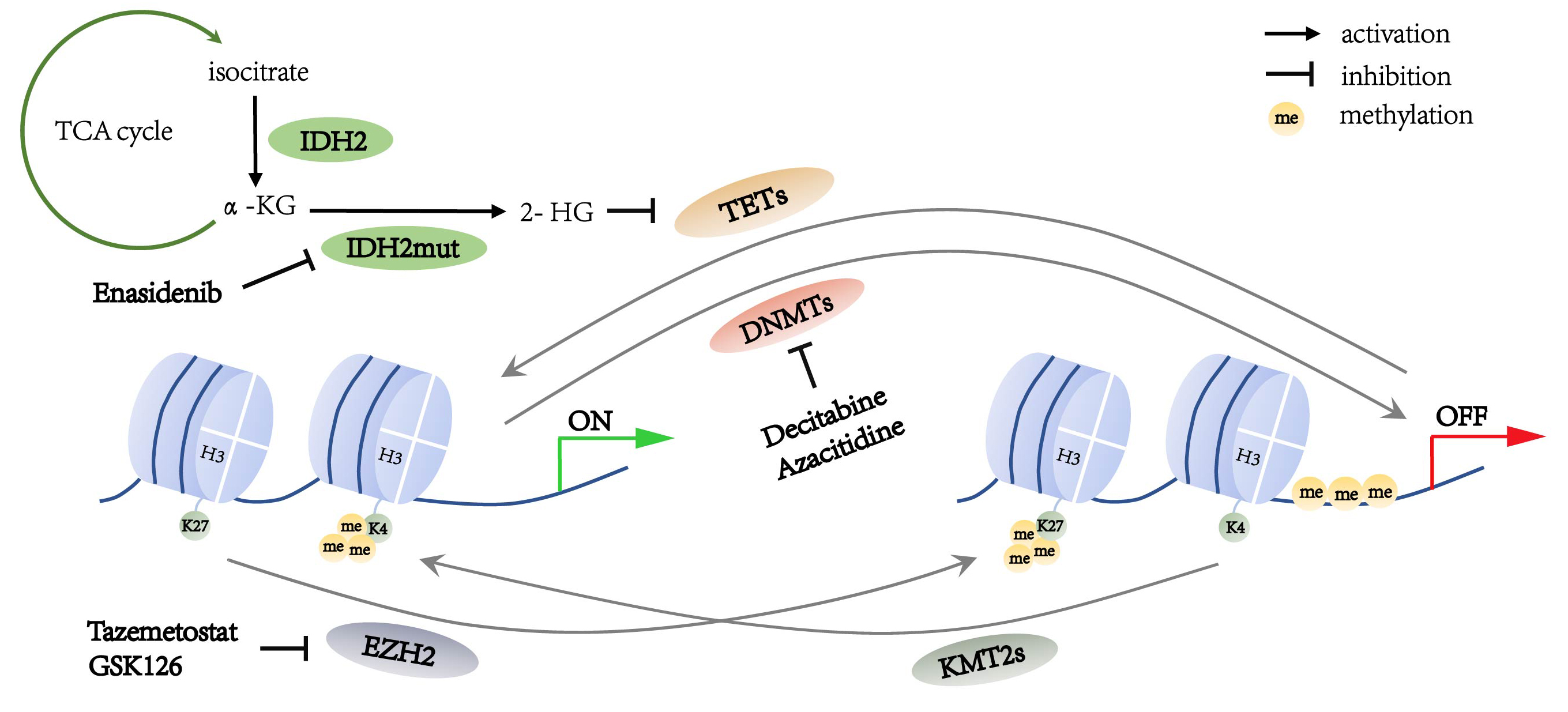 Fig. 1.
Fig. 1.Summary of the methylation alterations in lymphoma cells.
 Fig. 2.
Fig. 2.Domain architectures of the DNA methyltransferases DNMT1, DNMT3A and DNMT3B.
Previous studies have revealed that DNMT1 participates in maintaining DNA methylation in the synthesized DNA strand, while DNMT3A and DNMT3B regulate de novo methylation [17]. Abnormal cytosine methylation in gene promoters results in gene silencing. DNMT mutations mainly occur in DNMT3A but is rarely reported in DNMT1 and DNMT3B. However, altered expression of DNMT1 has been implicated in lymphomagenesis.
DNMT1 overexpression was demonstrated to regulate cell cycle progression in DLBCL, with an increase of the proliferation marker Ki-67. It was observed that knocking down DNMT1 reduces several genes’ expression in regulating cell cycle, such as CDK1, CCNA2 and E2F2 [18]. Among these genes, CDK1 mediates the phosphorylation of DNMT1 and regulates its enzymatic activity [19]. During the S phase of the cell cycle, DNMT1 is recruited to methylate the hemimethylated DNA strand [18]. In MCL, DNMT1 expression is increased, and it can be downregulated by treatment with arsenic trioxide [20]. In Burkitt’s lymphoma, MYC translocation is also related to DNMTs dysregulation. MYC can bind directly to the promoter region of DNMT1 and upregulate DNMT1 expression, which results in lymphoma progression [21]. In T cell lymphoma, DNMT1 is also reported to be involved in the maintenance of MYC-induced T-lymphoma in mice, as knocking out DNMT1 reduces the tumor development [22].
Although DNMT3A overexpression is not as common as DNMT1 and DNMT3B in lymphoma,
it possesses the highest mutation rate [23]. DNMT3A mutation is detected
in 11–20% of T cell lymphoma [24]. Loss-of-function DNMT3A mutations
include mostly missense mutation on R882 or other residues, and less commonly
nonsense or frameshift mutations which induce truncated protein [15, 25]. In PTCL
and AITL, 20% of DNMT3A mutation occurs in the R882 position, other
mutation positions such as D686, F731, G762 and G890 mainly locate in the
methyltransferase domain of DNMT3A (Fig. 3) [24]. In PTCL, DNMT3A
mutation is correlated with TET2 mutation, as 73% of patients with
DNMT3A mutation have TET2 mutation [23, 24]. The coexistence of
TET2 and DNMT3A
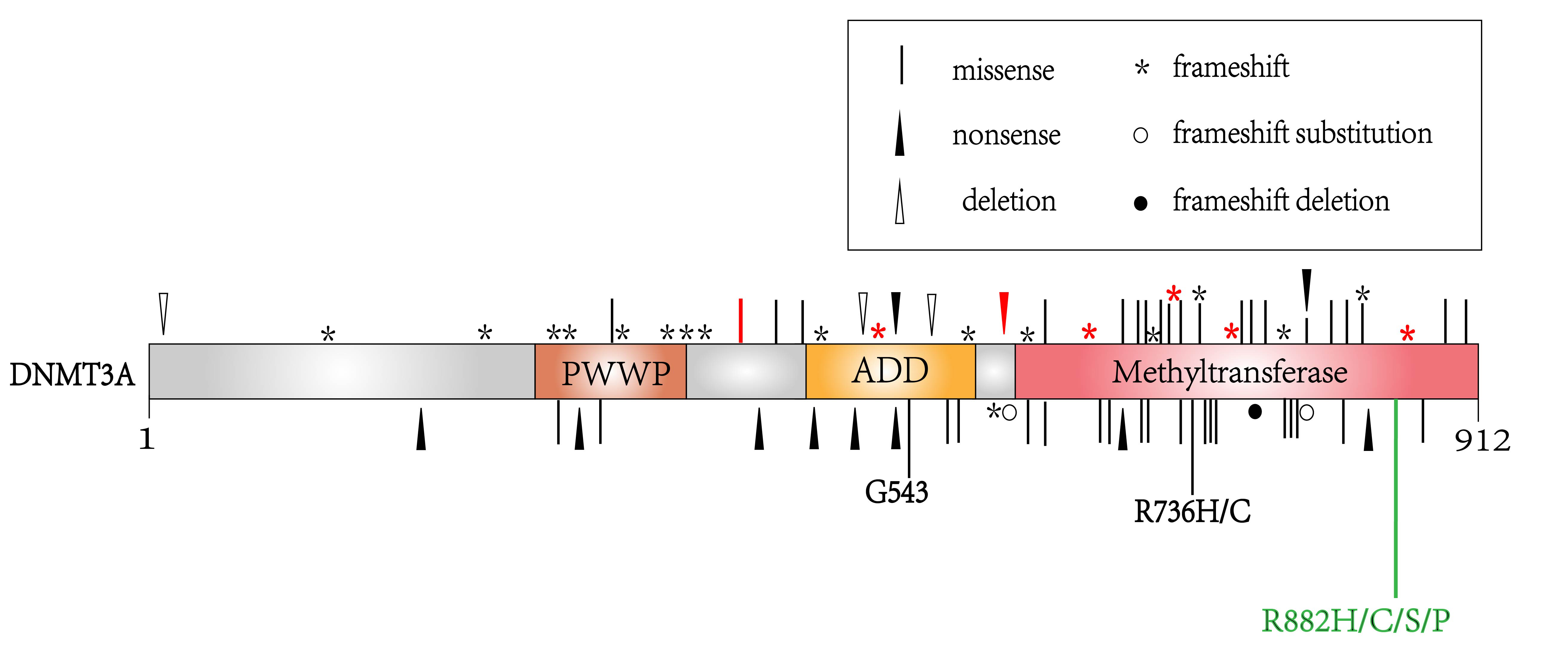 Fig. 3.
Fig. 3.The location of DNMT3A mutations in hematological malignancies. Labels in red represent mutations detected in T cell lymphoma, while labels in black represent mutations in acute myeloid leukemia or myelodysplastic syndromes. The R882 mutation labeled in green is a mutational hotpot detected most frequently in lymphoma and other hematological malignancies.
DNMT3B is overexpressed in patients with Burkitt’s lymphoma [28]. MYC is reported to directly bind to the promotor region of DNMT3B, resulting in DNMT3B overexpression in Burkitt’s lymphoma. Knocking down DNMT3B decreases the proliferation of tumor cell through blocking cell cycle and increasing lymphoma cell apoptosis [21]. In DLBCL, DNMT3B overexpression is significantly related with unfavorable overall survival (OS) and progression-free survival (PFS) [29]. DNMT3B7, a splice variant of DNMT3B, is also elevated in DLBCL and MCL. DNMT3B7 causes hypomethylation of a proto-oncogene described as MEthylated in Normal Thymocytes (MENT), which increases MENT expression in lymphomagenesis [30].
DNMT inhibitors mainly include decitabine, azacitidine, guadecitabine, MG98, RG108, and SGI-1027. The clinical trials related to these DNMT inhibitors are listed in Table 1. Azacitidine is an analog of cytidine which can substitute the nucleoside in DNA and RNA and bind covalently to DNMTs to inhibit DNA methylation [31]. The overall response rate (ORR) is 6.7% when relapsed or refractory (R/R) DLBCL patients received combined therapy of azacitidine plus vorinostat (NCT01120834). Azacitidine plus R-ICE (NCT03450343) or azacitidine plus R-GDP (NCT03719989) are currently ongoing in DLBCL patients. A phase 1/2 trial of azacitidine combined with R-CHOP achieves 91.7% CR in DLBCL (NCT01004991). In T-cell lymphoma, clinical trials of oral azacitidine in AITL (NCT03593018), azacitidine plus CHOP in untreated PTCL (NCT03542266), azacitidine combined with voriostat in NK/T-cell lymphoma (NKTCL, NCT00336063) are ongoing. In the trial of Aza-SAHA-GBM [azacitidine, vorinostat (suberoylanilide hydroxamic acid, SAHA), gemcitabine, busulfan, and melphalan] combined with stem cell transplantation for refractory lymphoma, the event-free-survival (EFS) was reported to be 65.4% in DLBCL, 100% in other B-cell lymphomas, 87.5% in T-cell NHL, and 76.2% in Hodgkin lymphoma (NCT01983969).
Decitabine is a deoxyribonucleoside that can incorporate into DNA and occupy DNMTs to induce DNA hypomethylation [32]. Several clinical trials using decitabine are currently ongoing. Decitabine improved the complete remission rate from 32% to 79% and prolonged the median PFS from 15.5 months to 35.0 months when combined with anti-PD-1 agent in Hodgkin lymphoma patients (NCT02961101) [33]. Furthermore, a phase 2 clinical trial investigated decitabine combined therapy with SHR-1210 in the treatment of R/R Hodgkin lymphoma (NCT03250962). In that study, the combined therapy achieved 95% ORR and 71% CR in R/R Hodgkin lymphoma patients who did not previously receive anti-PD-1 therapy. In patients had previously received anti-PD-1 therapy, the ORR is 52%, and CR rate is 28% [34]. A phase 4 clinical trial is exploring the efficacy of decitabine therapy in relapse and refractory DLBCL (NCT03579082). There is a phase 1 and 2 clinical trial estimating the efficacy of using decitabine plus R-CHOP therapy in newly diagnosed DLBCL (NCT02951728). Combined therapy of GVD (gemcitabine, vinorelbine, and doxorubicine) plus PD-1 antibody (SHR-1210) with or without decitabine on PMBCLs is being investigated in a trial (NCT03346642). Regarding T-cell lymphoma, there is a study on decitabine combined therapy which applies decitabine and pembrolizumab in PTCL and CTCL patients (NCT03240211). Also, studies of decitabine combined with CHOP in newly diagnosed PTCL (NCT03553537), decitabine combined with sintilimab in R/R NKTCL (NCT04279379) and decitabine combined with chidamide, or camrelizumab in NHL relapsed after chimeric antigen receptor T cells infusion (NCT04337606) are ongoing.
| Treatment | Disease | Trial name | Phase | Status | NCT ID | Results |
| Azacitidine + vorinostat | Relapsed or refractory DLBCL | Study of 5-azacitidine in Combination With Vorinostat in Patients With Relapsed or Refractory Diffuse Large B Cell Lymphoma (DLBCL) | Phase1 Phase2 | completed | NCT01120834 | ORR 6.7% |
| Azacytidine + R-ICE | Relapsed or refractory DLBCL | Oral Azacitidine Plus Salvage Chemotherapy in Relapsed/Refractory Diffuse Large B Cell Lymphoma | Phase1 | recruiting | NCT03450343 | - |
| Azacytidine + R-GDP | Relapsed or Refractory DLBCL | Azacitidine and Rituximab-GDP Immunochemotherapy in Patients With Relapsed/Refractory Diffuse Large B-Cell Lymphoma (EPIC) | Phase2 | Not yet recruiting | NCT03719989 | - |
| Azacitidine + R-CHOP | DLBCL | Phase I/II Trial of R-CHOP + Azacytidine in Diffuse Large B Cell Lymphoma | Phase1 Phase2 | completed | NCT01004991 | CR 91.7% |
| Azacitidine | Relapsed or Refractory AITL | Efficacy and Safety of Oral Azacitidine Compared to Investigator’s Choice Therapy in Patients With Relapsed or Refractory AITL | Phase3 | recruiting | NCT03593018 | - |
| Azacitidine + CHOP | Untreated PTCL | CC486-CHOP in Patients With Previously Untreated Peripheral T-cell Lymphoma | Phase2 | recruiting | NCT03542266 | - |
| Azacitidine + vorinostat | NK/T-cell lymphoma | Vorinostat and Azacitidine in Treating Patients With Locally Recurrent or Metastatic Nasopharyngeal Cancer or Nasal Natural Killer T-Cell Lymphoma | Phase1 | Active, not recruiting | NCT00336063 | - |
| Stem cell transplant + | Refractory lymphoma | Aza-SAHA-GBM With AutoSCT for Refractory Lymphoma | Phase1 Phase2 | completed | NCT01983969 | Event-free-survival (EFS): |
| Azacitidine + Vorinostat + | DLBCL 65.4%; | |||||
| Gemcitabine + Busulfan + | Hodgkin lymphoma 76.2%; | |||||
| Melphalan | T-cell NHL 87.5%; | |||||
| Other B-cell lymphoma 100% | ||||||
| Decitabine + Anti-PD 1+chemotherapy | Non-Hodgkin lymphoma | Anti-PD-1 Antibody Alone or in Combination With Decitabine/Chemotherapy in Relapsed or Refractory Malignancies | Phase1 Phase2 | recruiting | NCT02961101 | - |
| Decitabine + SHR-1210 | Hodgkin lymphoma | SHR-1210 Alone or in Combination With Decitabine in Relapsed or Refractory Hodgkin Lymphoma | Phase2 | recruiting | NCT03250962 | - |
| Decitabine | DLBCL | A Clinical Trial of Decitabine in Relapse and Refractory Diffuse Large B Cell Lymphoma | Phase4 | recruiting | NCT03579082 | - |
| Decitabine + R-CHOP | DLBCL | Decitabine Plus R-CHOP in Diffuse Large B-cell Lymphoma (DR-CHOP) | Phase1 Phase2 | Active, not recruiting | NCT02951728 | - |
| GVD and SHR-1210 with or without Decitabine | PMBCL | Two Stage Study of Combination of Chemotherapy, SHR-1210 and/or Decitabine for Relapsed/Refractory PMBCLs | Phase1 Phase2 | recruiting | NCT03346642 | - |
| Decitabine + Pembrolizumab | PTCL, CTCL | Study of Pembrolizumab Combined With Decitabine and Pralatrexate in PTCL and CTCL | Phase1 | recruiting | NCT03240211 | - |
| Decitabine + CHOP | PTCL | Efficacy and Safety of Decitabine Plus CHOP vs CHOP in Patients With Untreated Peripheral T-Cell Lymphoma | Phase3 | recruiting | NCT03553537 | - |
| Decitabine + Sintilimab | NK/T-cell lymphoma | Sintilimab and Decitabine for Patients With Relapsed/Refractory or Advanced NK/T-cell Lymphoma | Phase2 | Not yet recruiting | NCT04279379 | - |
| Decitabine + Chidamide + Camrelizumab | Non-Hodgkin lymphoma | Chidamide in Combination With Decitabine in Non-Hodgkin’s Lymphoma Relapsed After Chimeric Antigen Receptor | Phase1 Phase2 | recruiting | NCT04337606 | - |
The ten-eleven translocation (TET) family enzymes regulate DNA demethylation (Fig. 1) [8]. Mutations in the TET family genes are found to be involved in DNA hypermethylation in both DLBCL and PTCL [35]. The TET family enzymes include TET1, TET2, and TET3, in which TET2 is important in the DNA methylation alterations in lymphoma [36].
TET proteins are highly conserved on the C-terminal region and varied on the
N-terminal region. There is a CXXC domain on the C-terminal region responsible
for recognizing CpG sites, although this domain is absent from TET2. Their
N-terminal regions, which are responsible for the catalytic function, consist of
a double-stranded
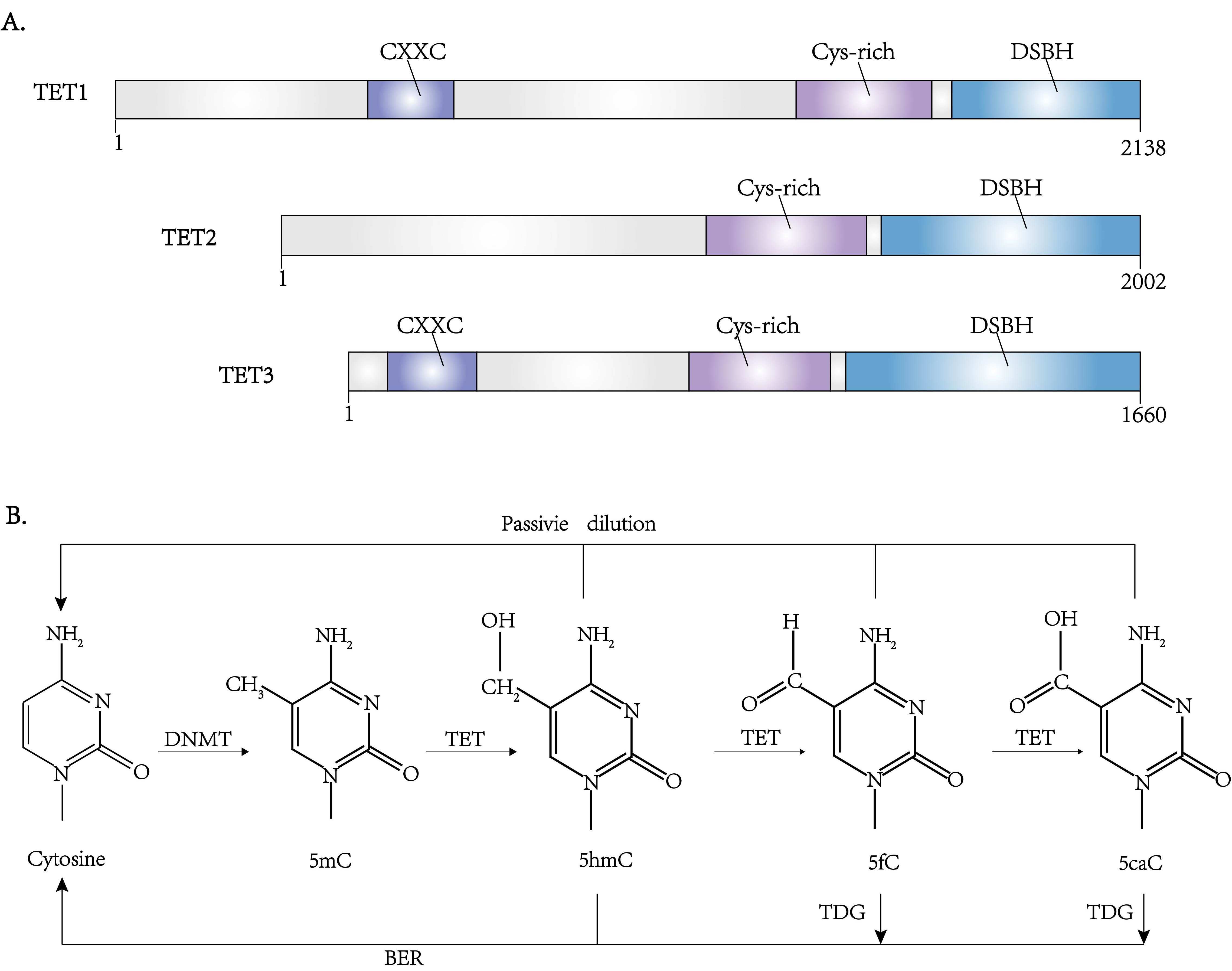 Fig. 4.
Fig. 4.The structure and function of TET family proteins. (A) Domain architectures of the TET family proteins. (B) The pathway of DNA methylation and demethylation. Besides DNMTs and TETs, another enzyme TDG (thymine DNA glycosylase) is also involved in regulating these processes through BER (base excision repair).
TET1 is involved in B cell lymphomagenesis as a tumor suppressor gene. It is reported that TET1 is hypermethylated and silenced in DLBCL and FL in mice [39]. In mouse models, deficiency of TET1 drives the formation of B cell lymphoma, while TET1 deficiency in human hematopoietic stem cells (HSCs) leads to the differentiation towards B cell lineage and might induce secondary mutations or overexpression of BCL2 which attenuates apoptosis [39]. Several gene mutations related to histone modification, such as KMT2D mutations, are frequently found in TET1-knockout lymphoma mouse models [40].
TET2 is a well-known tumor suppressor. TET2 deficiency promotes B cell lymphomagenesis via impairing B cell differentiation in the germinal center (GC) and inducing GC hyperplasia [41]. Mutations in the C-terminal and N-terminal regions of TET2 catalytic domain lead to the disruption of the catalytic function, and most are heterozygous mutations [42]. Loss-of-function mutation of TET2 is displayed in about 10% of DLBCL, 50–80% of AITL, and 40–50% of PTCL [38]. TET2 mutations include deletion, missense, nonsense, and frameshift mutations. TET2 modulates the demethylation in DLBCL and AITL, and the mutations are predominantly observed in germinal center B-cell (GCB) DLBCL [43]. In GCB cells, TET2 mutations restrain gene expression in the GC exit and plasma cell differentiation [44]. Deficiency of TET2 promoted B cell tumor generation in mice models through activation-induced deaminase (AID)-mediated mutation in the B-cell receptor (BCR) signaling pathway [45]. TET2 mutations are primarily detected in T cell lymphoma derived from follicular helper T cells (TFH) including AITL and PTCL, in which TFH function as B-cell stimulator and promote B cells expansion [46]. TET2 mutation causes the altered methylation of BCL6 locus and increases the expression of BCL6, which promotes proliferation of TFH cells [47]. In AITL and PTCL, TET2 and DNMT3A mutations coexist, but whether these TET2/DNMT3A mutations cause hypermethylation or hypomethylation remains controversial in different reports [48]. TET2 mutations are related to the clinical features and prognosis in patients with AITL and PTCL-NOS. In a previous study, patients with TET2 mutations possessed advanced stage, more involved extranodal sites, higher possibility of B symptoms, and higher international prognosis index (IPI) than those with wild-type TET2 [49]. AITL patients with TET2 mutation are reported with decreased overall survival rate [50]. However, AITL patients with TET2 mutation reached an ORR of 75% upon azacitidine treatment [51].
Isocitrate dehydrogenase (IDH) can irreversibly convert isocitrate into
Enasidenib (AG-221), a drug inhibitor of mutant IDH2, is approved in AML [59, 60]. The study of enasidenib in the therapy of AITL is still under clinical investigation. A phase1 and phase2 study on enasidenib in AITL patients with IDH2 mutation has been completed, but the results are not yet published (NCT02273739).
Histone methylation alteration is also detected in lymphoma through mutations of histone methyltransferases, which include Enhancer of Zeste Homolog 2 (EZH2) and mixed-lineage leukemia family proteins (MLLs; also known as lysine methyltransferase 2, KMT2s). EZH2 results in H3K27me3, which is related to the suppression of gene transcription. MLL alters the chromatin state through not only its direct trimethylation of histone H3 lysine 4 (H3K4me3) but also recruitment of demethylases to reduce H3K27me3 (Fig. 1). Therefore, the functions of EZH2 and MLL are opposite. A balance between the states of H3K27me3 and H3K4me3, and the transforming of these states is associated with gene repression and activation [61].
EZH2 is the catalytic component of the polycomb repression complex 2 (PRC2), mediating H3K27me3 and recruiting the DNA methyltransferase to inhibit gene transcription (Fig. 1) [62]. EZH2 acts on cell proliferation and differentiation in tumorigenesis. During the development of B-lymphocyte, EZH2 downregulates genes which have negative effect on cell cycle to promote proliferation of immature B cells [63]. EZH2 represses anti-proliferative gene CDKN1A and inhibits IRF4 and PRDM1 expression in the terminal differentiation [64, 65].
EZH2 overexpression is more common in aggressive B-cell lymphoma than in
indolent B-cell lymphoma [66]. In a study of various types of B-cell lymphoma,
EZH2 was positive in 97% of cases of lymphoma, including DLBCL, Burkitt
lymphoma, and double-hit lymphoma. This study also reveals that EZH2 expression
is positively correlated with phosphorylated ERK1/2 (p-ERK1/2) in DLBCL and MYC
expression in Burkitt lymphoma and double-hit lymphoma [66]. Coexpression of EZH2
and BCL-2 was related to worse OS and PFS and a higher frequency of relapse in
DLBCL [67]. In a study of primary gastrointestinal DLBCL, EZH2 overexpression is
indicated with a more progressive clinical stage and inferior outcomes [68].
EZH2 gain-of-function mutation is mainly detected in GCB-DLBCL and
follicular lymphoma (FL), including predominantly heterozygous mutation on
tyrosine 641 (Y641) [63]. Somatic mutation of EZH2 in Y641 enhances H3K27me3
[69]. EZH2
EZH2 is also found overexpressed in T-NHL and can be targeted by EZH2 inhibitors [74]. In some studies, EZH2 upregulation is mediated by high MYC protein expression. In PTCL, EZH2 is overexpressed in 64.6% (53/82) patients, which is related to advanced clinical stage and lower OS [75]. In NKTCL, EZH2 is overexpressed in most of the patients, which is related to poor prognosis [76]. EZH2 expression can be downregulated by JAK inhibitor, which indicates that EZH2 could be downstream of the JAK/STAT pathway.
Several small-molecule inhibitors of EZH2 have been discovered, and the effects of these EZH2 inhibitors such as tazemetostat (EPZ6438), GSK343, GSK503, GSK126, CPI-1205 are evaluated in some experimental studies and clinical trials [77]. Tazemetostat (EPZ6438) selectively inhibits EZH2 function and decreases H3K27 methylation in B lymphoma cell lines and Y646 mutated cells. Oral administration of EPZ6438 also shows EZH2 inhibition in the mice model [78]. There are multiple clinical trials to investigate the efficacy of tazemetostat in lymphoma. A study (NCT01897571) explores the effect of tazemetostat combined with prednisolone in DLBCL or tazemetostat alone in FL. During the assessment, eight DLBCL or FL patients’ median time of partial response was 3.5 months; three patients with initial partial response achieved complete response at 9, 22, 24 months [79]. In the phase 2 trial (NCT01897571), tazemetostat monotherapy achieved the objective response rate of 69% at EZH2 mutated FL patients; the median PFS was 13.8 months [80].
GSK126 is an EZH2 inhibitor that can inhibit the activity of both wild-type and mutant EZH2 in several DLBCL and FL cell lines. GSK126 treatment leads to decreased H3K27me3 level and tumor regression [81]. It is also observed that GSK126 can reduce H3K27me3 in multi-drug resistant B lymphoma cell lines and increase the sensitivity to etoposide in the combined therapy [82]. A phase 1 clinical trial (NCT02082977) has explored the safety, pharmacokinetics, pharmacodynamics and clinical efficacy in R/R DLBCL, transformed FL, and other NHL with GSK126. At the end of the trial, 1 of the 20 (5%) lymphoma patients achieved partial response, while 6 patients had stable disease (30%) [83].
The lysine methyltransferase 2 (KMT2; also known as MLL) family proteins methylate histone H3K4 and positively regulate gene transcription (Fig. 1). They include KMT2A, KMT2B, KMT2C, KMT2D, KMT2F, and KMT2G; each protein is involved in multiple subunit protein complexes with diverse components [84]. However, only KMT2A, KMT2C, and KMT2D are reported to be mutated in hematological malignancies. KMT2A and KMT2D induce H3K4me3 in the promoter region of the genes involved in hematopoietic cell development and differentiation. KMT2C induces H3K4me1 in the enhancer regions to regulate gene expression [85].
In the KMT2 family, KMT2D and KMT2C are found to be involved in malignant
lymphomas such as DLBCL and FL [84]. KMT2D is supposed to be a tumor suppressor
gene, as 91% of mutations result in silencing the enzymatic function [86].
KMT2D mutations are detected in 24–32% of DLBCL and 72–89% of FL
patients, mostly are nonsense or frameshift mutations resulting in
down-regulating the expression of KMT2D protein [87]. However, KMT2C
mutations are detected in 8.2% of DLBCL [88] and 13% of FL patients [87]. KMT2D
and KMT2C mutations are considered to loss their functions of histone
methylations. KMT2D deficiency results in alteration of several genes, including
TNFAIP3, SOCS3, SGK1, TRAF3,
TNFRSF14 and ARID1A, and subsequently influences the JAK-STAT,
Toll-like receptor and B-cell receptor pathways in B-lymphoma cells [89]. In MCL,
KMT2D mutations indicate dismal prognosis. The 4-year PFS and OS are
lower in KMT2D mutated patients than wild-type patients (PFS 33.2% vs.
63.7%, OS 62.3% vs. 86.8%). MCL patients with KMT2D mutations have
higher
As reported in previous studies, chidamide combined with decitabine could target KMT2D mutation and inactivate the constitutively activated MAPK pathway in T-cell lymphoma [11].
RNA methylation is a cotranscriptional or posttranscriptional modification which
has become an emerging regulatory mechanism of gene expression, termed as
“epitranscriptomics”, commonly occurs in N
Methyltransferase METTL3 expression and m
RNA demethylase ALKBH5 and RNA binding protein YTHDF3, activated by MYC, reduce
the m
RNA m
DNMT2 is an important RNA methyltransferase involved in tRNA m
Improved understanding of epigenetic and epitranscriptomic alterations in lymphoma has raised new insights into lymphoma pathogenesis and progression. DNA and histone methylation are highly variable processes in which the DNA methylation functions mainly by silencing tumor suppressor genes, while the histone methylations on H3K4 and H3K27 play opposite roles in gene transcriptional regulation. RNA methylation is also a novel mechanism involved in regulating RNA metabolism. Overexpression or mutation of the enzymes such as DNMT3A, TET2, EZH2, and KMT2D are frequently detected in lymphoma. Survival data also supported that most of these epigenetic alterations are associated with poor clinical outcomes in lymphoma patients. Current studies have shown that aberrant epigenetic modification could be targeted by multiple agents such as DNMT inhibitors azacitidine and decitabine, and EZH2 inhibitor tazemetostat. Most of the inhibitors are under investigation through clinical trials. However, the clinical efficacy of these epigenetic drugs and whether they could significantly improve lymphoma patient prognosis have yet to be verified. New target agents for epigenetic alterations are still worth investigating.
WLZ and LW developed the original idea of the article; MKL, XJS, XDG and YQ collected literature and data; and WLZ and LW wrote the paper. All authors approved the final manuscript.
Not applicable.
We thank Peter Liao for English editing.
This study was supported, in part, by research funding from the National Natural Science Foundation of China (81830007, 81520108003, 81670176, and 82070204), Chang Jiang Scholars Program, Shanghai Municipal Education Commission Gaofeng Clinical Medicine Grant Support (20152206, and 20152208), Clinical Research Plan of Shanghai Hospital Development Center (SHDC2020CR1032B), Multicenter Clinical Research Project by Shanghai Jiao Tong University School of Medicine (DLY201601), Collaborative Innovation Center of Systems Biomedicine, and the Samuel Waxman Cancer Research Foundation.
The authors declare no conflict of interest.