



 , Wanghua Gong 2, Jiaqiang Huang 1,3, Teizo Yoshimura 4, Ji Ming Wang 1,*
, Wanghua Gong 2, Jiaqiang Huang 1,3, Teizo Yoshimura 4, Ji Ming Wang 1,*1 Laboratory of Cancer ImmunoMetabolism, Center for Cancer Research, National Cancer Institute at Frederick, Frederick, MD 21702, USA
2 Basic Research Program, Leidos Biomedical Research, Inc., Frederick, MD 21702, USA
3 College of Life Sciences, Beijing Jiaotong University, 100044 Beijing, China
4 Department of Pathology and Experimental Medicine, Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama University, 700-8558 Okayama, Japan
Abstract
Human cathelicidin antimicrobial peptide LL-37 (LL-37) is an antimicrobial peptide derived from its precursor protein hCAP18, which is an only cathelicidin in human. LL-37 not only serves as a mediator of innate immune defense against invading microorganisms, but it also plays an essential role in tissue homeostasis, regenerative processes, regulation of proinflammatory responses, and inhibition of cancer progression. Therefore, LL-37 has been considered as a drug lead for diseases. However, high levels of LL-37 may reduce cell viability and promote apoptosis of osteoblasts, vascular smooth muscle cells, periodontal ligament cells, neutrophils, airway epithelial cells and T cells. Recent evidence reveals that LL-37-derived short peptides possess similar biological activities as the whole LL-37 with reduced cytotoxicity. Thus, such small molecules constitute a pool of potential therapeutic agents for diseases.
Keywords
- LL-37
- Short peptides
- Immunoregulation
- Infection
- Cancer
LL-37 is constitutively expressed or produced in almost all tissues and organs [1, 2]. These cells expressing LL-37 are either in direct contact with the exterior environment or operating at sites of infection and immune responses, indicating the importance of LL-37 to host defense. LL-37 plays multifunctional roles in host defense. LL-37 has a broad spectrum of microbicidal activities and is effective against Gram-positive and negative bacteria, fungi, and some viruses [3, 4, 5, 6]. LL-37 also acts as a potent chemoattractant to guide immune cells. Upon infection, LL-37 acts as a danger signal and bridges the innate and adaptive immune system by guiding immune cells such as monocytes, neutrophils, T cells to the site of infection. Moreover, LL-37 modulates the levels of inflammatory cytokines, serving to control the delicate balance between pro- and anti-inflammatory responses [7, 8, 9]. In addition, LL-37 is involved in many key biological processes implicating non-immune cells such as apoptosis, angiogenesis, re-epithelialization, wound closure and the maintenance of the intestinal epithelial barrier integrity [1, 9, 10, 11, 12, 13].
Accumulating knowledge indicates the possibility of LL-37 as a potential therapeutic agent for infection and cancer. Synthetic LL-37 was used for the topical treatment of hard-to-heal venous leg ulcers (VLUs) with safety and marked therapeutic effects [14]. A preliminary evaluation of orally administered recombinant L. lactis containing LL-37 peptide was performed in SARS-CoV-2-infected patients with mild symptoms. The recombinant L. lactis containing LL-37 peptide improved symptoms of CoV-2-infected patients in fever, and fatigue, myalgia, as well as gastrointestinal (abdominal discomfort, nausea, poor appetite, and diarrhea) and respiratory symptoms (pharyngalgia, cough, chest distress, shortness of breath) without adverse reactions [15]. In experimental colon cancer metastasis, mice injected intravenously with LL-37-overexpressing adeno-associated viruses (CAMP-HA-AAVs) reduced human-specific cytokeratin 18 positive tumor colonies in the lung, indicating that LL-37 produced in vivo inhibited colon cancer metastasis. An in vitro study showed that LL-37 inhibited the migration of human colon carcinoma cells by disrupting the tubulin structure inside the cells [16].
However, the therapeutic application of LL-37 is limited due to its low cell
selectivity and high production cost due to its large size [17]. Cytotoxic
effects of LL-37 were also detected in vitro on erythrocytes,
lymphocytes, and fibroblasts. LL-37 damages the plasma membrane of human cells at
concentrations similar to those required for its antimicrobial activity [2, 18],
constituting a potentially detrimental side effect. High levels of LL-37 (
The precursor protein hCAP18 of LL-37 is composed of three parts: A N-terminal signal peptide, a highly conserved cathelin like domain, and an antimicrobial peptide domain at the C-terminus [21, 22, 23]. The full peptide was first isolated from the bone marrow and subsequently from the secretions of neutrophils [24, 25]. The full-length hCAP-18 (Inactive LL-37 precursor) is cleaved by proteinase 3 [26], elastase [24], or skin-derived kallikreins [27] to release a 37-amino acid residue to form of the mature LL-37. However, hCAP-18 in seminal plasma is cleaved by prostate-derived gastricsin to release a 38-amino acid antimicrobial peptide, ALL-38. The antimicrobial spectrum and potency of ALL-38 are similar to those of the prototype LL-37 [28]. LL-37 may be further processed to release shorter fragments with biological activities. In human sweat, the mature LL-37 has been shown to be degraded to shorter peptic fragments, including RK-31, KS-30 and KR-20 [29] by two distinct kallikreins: kallikrein 5 and kallikrein 7 [27, 30].
These natural and recently developed synthetic LL-37-derived short peptides have contributed to a better understanding of the full LL-37 peptide and the replacement for LL-37 to apply in treatment of diseases [31]. For example, KS-30 (LL-8-37), KR-20 (LL-18-37), KR-12 (LL-18-29), and FK-16 (LL-17-32) released from LL-37 all exhibit microbiocidal capacity [32, 33]. Moreover, replacement of amino acid residues can enhance the activity as compared with the prototype LL-37 [33]. Engineered LL-37-derived peptides inhibited Ebola [34] and Zika virus [35] infection. Interestingly, a synthetic peptide, called LL37-analogous peptide (LLAP) and with the sequence of GRKSAKKIGKRAKRI, possesses more potent antibacterial activity by targeting ATPases in bacterial cell membrane [36]. The sequences of some natural LL-37-derived short peptides and the engineered short peptides are listed in Table 1.
| AMP | Sequence | AA numbers |
| LL-37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | 37 |
| LL-27 | FRKSKEKIGKEFKRIVQRIKDFLRNLV | 27 |
| RK-31 | RKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | 31 |
| KS-30 | KSKEKIGKEFKRIVQRIKDFLRNLVPRTES | 30 |
| KR20 | KRIVQRIKDFLRNLVPRTES | 20 |
| FK16 | FKRIVQRIKDFLRNLV | 16 |
| KR12 | KRIVQRIKDFLR | 12 |
| GF17 | GFKRIVQRIKDFLRNLV | 17 |
| 17BI | GXKRLVQRLKDXLRNLV | 17 |
| GF18 | GEFKRIVQRIKDFLRNLV | 18 |
| GI-20 | GIKEFKRIVQRIKDFLRNLV | 20 |
| GI-20d | GIKEFKRIVQRIKDFLRNLV | 20 |
| Red italic: Engineered short peptides or amino acid residues. | ||
Aghazadeh and colleagues [37] investigated the effects of LL-37-derived short peptides; P38 (TSVRQRWRWRQRVRTS─NH2), P22 (KRSKRKRRIHQRVRIS─NH2) and P7 (TLSKEKERIVQRVRTS─NH2), against MDR clinical isolates of bacteria including five E. coli and five S. aureus strains. They found that the antibacterial activity of P38 against MDR clinical isolates of E. coli was higher than that of P7 and P22. The P38 as well as P22 and P7 have no antimicrobial activity against all S. aureus isolates. Mechanistic studies revealed that P38 had a much higher affinity for the outer membrane of Gram-negative bacteria compared with both P22 and P7, and P38 killed E. coli by disrupting the bacterial membrane. Thus, P38 has a better potential for clinical applications against infectious diseases, especially MDR clinical isolates of E. coli.
KR-12 (KRIVQRIKDFLR), corresponding to the residues 18–29 of LL-37, is the smallest peptide of LL-37 known to possess antimicrobial activities. KR-12 displayed a selective toxic effect on bacteria but not human cells [38, 39]. KE-18 (KEFKRIVQRIKDFLRNLV) is another peptide corresponding to the residues 15–32 of LL-37. Luo and colleagues [40] compared the effects of KR-12 and KE-18 against Candida albicans, Staphylococcus aureus, and E. coli. All three microorganisms have been demonstrated to be involved in ventilator-acquired pneumonia (VAP) [41]. KE-18 showed a significant inhibitory activity against C. albicans and S. aureus for the biofilm-development. In contrast, KR-12 did not display any antibiofilm properties against C. albicans, S. aureus, or E. coli. Thus, KE-18 appears a good therapeutic agent for prevention of multi-species biofilm-related infections such as VAP.
Kim and colleagues [42] designed and synthesized three analogs of KR-12-a5 (KRIVKLILKWLR-NH2); KR-12-a5(5-DK) (KRIVKLILKWLR-NH2) and KR-12-a5(7-DL) (KRIVKLLLKWLR-NH2) and KR-12-a5(6-DL) (KRIVKLILKWLR-NH2). D-Lys (K) and D-Leu (L) substituted the Lys5 and Ile7 in the central position of the polar and non-polar face of KR-12-a5, respectively. KR-12-a5(6-DL) was designed by substituting D-Leu for Leu6 in the polar-nonpolar interface of KR-12-a5 [42]. KR-12-a5 and its analogs show higher antimicrobial activities against all tested antibiotic-resistant bacterial strains than LL-37, including methicillin-resistant Staphylococcus aureus strains (MRSA; CCARM 3089, CCARM 3090, and CCARM 3095), multidrug-resistant Pseudomonas aeruginosa strains (MDRPA; CCARM 2095, and CCARM 2109) and vancomycin-resistant Enterococcus faecium (VREF; ATCC 51559). Antibiotics and chemicals have difficulty penetrating and killing the bacteria which have formed biofilm. Biofilm is formed by bacteria with bacteria-secreted slimy, glue-like substances, adhering to the surface of some object in a moist environment [43]. KR-12-a5 and its analogs KR-12-a5(5-DK), KR-12-a5(6-DL), and KR-12-a5(7-DL) inhibited biofilm development of MDRPA (CCARM 2095). By contrast, the parental peptide, LL-37, did not show a significant anti-biofilm activity. KR-12-a5 and its analogs also displayed a remarkable synergy with antibiotics, including chloramphenicol, ciprofloxacin, and oxacillin against MDRPA. Thus, KR-12-a5 and its analogs can be developed further as novel antimicrobial/anti-inflammatory agents to treat antibiotic-resistant infections.
Rajasekaran and colleagues [17] synthesized a series of FK13 (FKRIVQRIKDFLR-NH2)
analogs based on the sequence of the 13-meric short FK13 peptide (residues 17–29
of LL-37), including FK-13-a1 (WKRIVRRIKRWLR-NH2), FK13-a2 (WKRIVRWIKRWLR-NH2),
FK13-a3 (WKRIVRRIWRWLR-NH2), FK13-a4 (FKRWVQRWKRFLR-NH2), FK13-a5
(WKRWVQRWKRFLR-NH2), FK13-a6 (WKRWVQRWKRWLR-NH2) and FK-13-a7
(WKRWVRRWKRWLR-NH2). FK13-a1 was designed by replacing Phe1 and Phe11 of FK13
with Trp, FK13-a2 and FK13-a3 were designed by Arg7 to Trp7 and Lys9 to Trp9
substitution in FK13-a1, respectively. FK13-a4 was obtained from FK13 by Trp
substitution of the two consecutive Ile residues at positions 4 and 8 (Ile4,8
FK-16 (FKRIVQRIKDFLRNLV) is a potently active peptide corresponding to the residues 17–32 of human LL-37 [44]. FK-16-coated titanium surface also demonstrated a significant anti-biofilm activity against methicillin-resistant S. aureus and E. coli [44].
Biofilm-related infections by implanted medical devices cause implant failures, increase treatment costs, and lead to higher patient morbidity and mortality [45]. In the past years, both metals (e.g., silver, zinc, copper, and zirconium) and non-metals (e.g., selenium and antibiotics) have been used for coating biomaterial surface to prevent such infections [46]. The effective use of metals, however, is complicated by leaching and cytotoxicity, whereas a prolonged use of antibiotics results in reduced efficacy due to the emergence of multi-drug resistant-pathogens [47]. Titanium surface coated with LL-37-derived FK16 showed a broad-spectrum of activity against ESKAPE pathogens, including Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter cloacae. FK-16-coated titanium surface also demonstrated a significant anti-biofilm activity against methicillin-resistant S. aureus and E. coli[44]. Antimicrobial effects of LL-37-derived peptides are achievable at the concentrations not toxic to human cells. Thus, the titanium surfaces covalently immobilized with LL-37-derived peptides with a potent antimicrobial activity may be harnessed to prevent biofilm-related infection on medical devices [44]. On the other hand, the compounds conjugated with both metal and a LL-37-derived peptide exerted more potent antimicrobial activities with improved water solubility and air stability of LL-37-derived short peptides through structural changes of peptides upon metal conjugation and redox chemistry [48].
The LL-37-derived synthetic peptide OP-145 (another name is P60.4Ac)
(acetyl-IGKEFKRIVERIKRFLRELVRPLR-amide) was more effective than LL-37 in
eradicating S. aureus in wound infection models in vitro and proved to
be safe and successful for treatment for patients with chronic suppurative otitis
media [49, 50]. De Breij and colleagues incorporated OP-145 into a Polymer-Lipid
Encapsulation MatriX (PLEX)-coating to obtain high peptide levels for prolonged
periods at the implant-tissue interphase [50]. They found that
OP-145 incorporated PLEX coating on Tricalcium
phosphate (TCP) granules showed a burst release of approximately 55% of the
peptide by the coating during the first 48 h followed by a daily release of about
1% for 30 days, and completely killed 1
Nowadays, most patients with upper airway infections are treated with antibiotics because bacterial products, like lipopolysaccharide (LPS) from Gram-negative bacteria and lipoteichoic acid (LTA) from Gram-positive bacteria are involved in a large part of symptoms of these infections [51, 52]. Nell and colleagues found that P60.4, a 24 amino acid peptide they designed and synthesized, had a similar efficacy as LL-37 in terms of LPS and LTA neutralization and lower pro-inflammatory activity. Moreover, the acetylated and amidated version of this peptide showed no toxicity and displayed higher or equal antimicrobial activity compared to LL-37 [52]. FK13 nearly lost its ability to bind LPS, but all FK13 analogs (FK-13-a1, FK-13-a2, FK-13-a3, FK-13-a4, FK-13-a5, FK-13-a6 and FK-13-a7) showed much higher LPS-binding ability than FK13. Among the FK13 analogs, FK13-a4 and FK13-a5 showed the lowest LPS-binding ability [17]. Other LL-37 derived short peptides KE-18 and KR-12 retained the LPS-binding activity of LL-37. However, KE-8 showed significantly enhanced LTA-binding activity, while KR-12 retained the weak LTA-binding activity of the parent LL-37 peptide [40].
KR-12-a5 and its analogs KR-12-a5(5-DK), KR-12-a5(6-DL), and KR-12-a5(7-DL)
inhibited the expression and production of iNOS, TNF-
Several mimetics derived from LL-37 or other Amtimicrobial peptides (AMPs) have been used as antiviral agents. For example, GI-20, GI-20d, GF-17, 17BI, RI-10, BMAP-18, and DASamP2 were used for Zika virus (ZIKV) infection [35]; GF-17, 17BI, GI-20, GI-20d and RI-10 were for Ebola virus (EV) infection [34] and PG-1, SMAP-29 were for Human rhinoviruses (HRVs) infection [53]. Anti-virus effects of some of LL-37 and LL-37-derived short peptides were listed in Table 2.
| Virus | Peptides | Proposed mechanism |
| SARS-CoV-2 | LL-37 | Binding virus and inhibiting viral replication. |
| Dengue virus (DENV) | LL-37 | Binding E-protein dimmers of the virus to prevent cell–virus interaction. |
| Human rhinoviruses (HRVs) | LL-37, PG-1, SMAP-29 | Direct antiviral effects on HRVs and reducing the metabolic activity of infected cells. |
| Respiratory syncytial viruses (RSV) | LL-37 | Binding RSV to reduce the level of colocalization of the F- and N-proteins. |
| Vaccinia virus (VV) | LL-37 | Inhibiting the replication of VV. |
| Venezuelan equine encephalitis virus (VEEV) | LL-37 | Inhibiting the VEEV replication. |
| Ebola virus (EV) | LL-37, GF-17, 17BI, GI-20, GI-20d and RI-10 | Acting as cathepsin B (Cat-B) inhibitors to block the endosomal processing of EBOV glycoprotein (GP). |
| Zika virus (ZIKV) | LL-37, GI-20, GI-20d, GF-17, 17BI, RI-10, BMAP-18, and DASamP2 | Interfering with ZiKV entry host cells by increasing IFN- |
| Neurotropic herpes simplex virus 1 (HSV-1) | LL-37 | Suppressing HSV-1 replication. |
| Human immunodeficiency virus (HIV) | LL-37 | Binding HIV-1 reverse transcriptase to block its activity. |
| Notes: PG-1, Porcine Cathelicidin Protegrin-1 (RGGRLCYCRRRFCVCVGR); SMAP-29,
Sheep Myeloid Antimicrobial Peptide 29
(RGLRRLGRKIAHGVKKYGPTVLRIIRIAG); BMAP-18, Bovine Myeloid
Antimicrobial Peptide-18 (GRFKRFRKKFKKLFKKLS). DASamP2: DASamP2 was discovered by screening a library of representative peptides selected from the Antimicrobial Peptide Database (IKWKKLLRAAKRIL-NH | ||
17BI was engineered based on the sequence of GF-17, a fragment of LL-37 with three D-form amino acids and two biphenylalanines, whereas GI-20d is a novel peptide engineered with all D-form amino acids with identical sequence to GI-20, another short peptide derived from LL-37. Ebola virus (EV) is a single-stranded, negative-sense RNA virus, which causes Ebola virus disease (EVD) characterized by fever, gastrointestinal signs, and multiple organ dysfunction syndrome [54]. HeLa cells and human macrophages, including primary and blood CD4+ monocyte-derived macrophages, were infected with VSV-eGP (a system with a recombinant vesicular stomatitis virus (rVSV) expressing EBOV-GP and green fluorescence protein) together with AMPs (LL-37, GF-17, 17BI, GI-20, GI-20d and RI-10). 17BI and GI-20d showed the highest level of inhibition of VSV-eGP infection. 17BI and GI-20d also blocked the infection of wild-type (WT) EBOV at a much higher level than the peptides containing only L-amino acids, such as GF-17 and GI-20. Mechanistic studies revealed that 17BI and GI-20d act as cathepsin B (Cat-B) inhibitors to block the endosomal processing of EBOV glycoprotein (GP); thus, preventing virus entry because EBOV requires the cleavage of EBOV-GP by cathepsins within host cell endosomes [34] (Fig. 1).
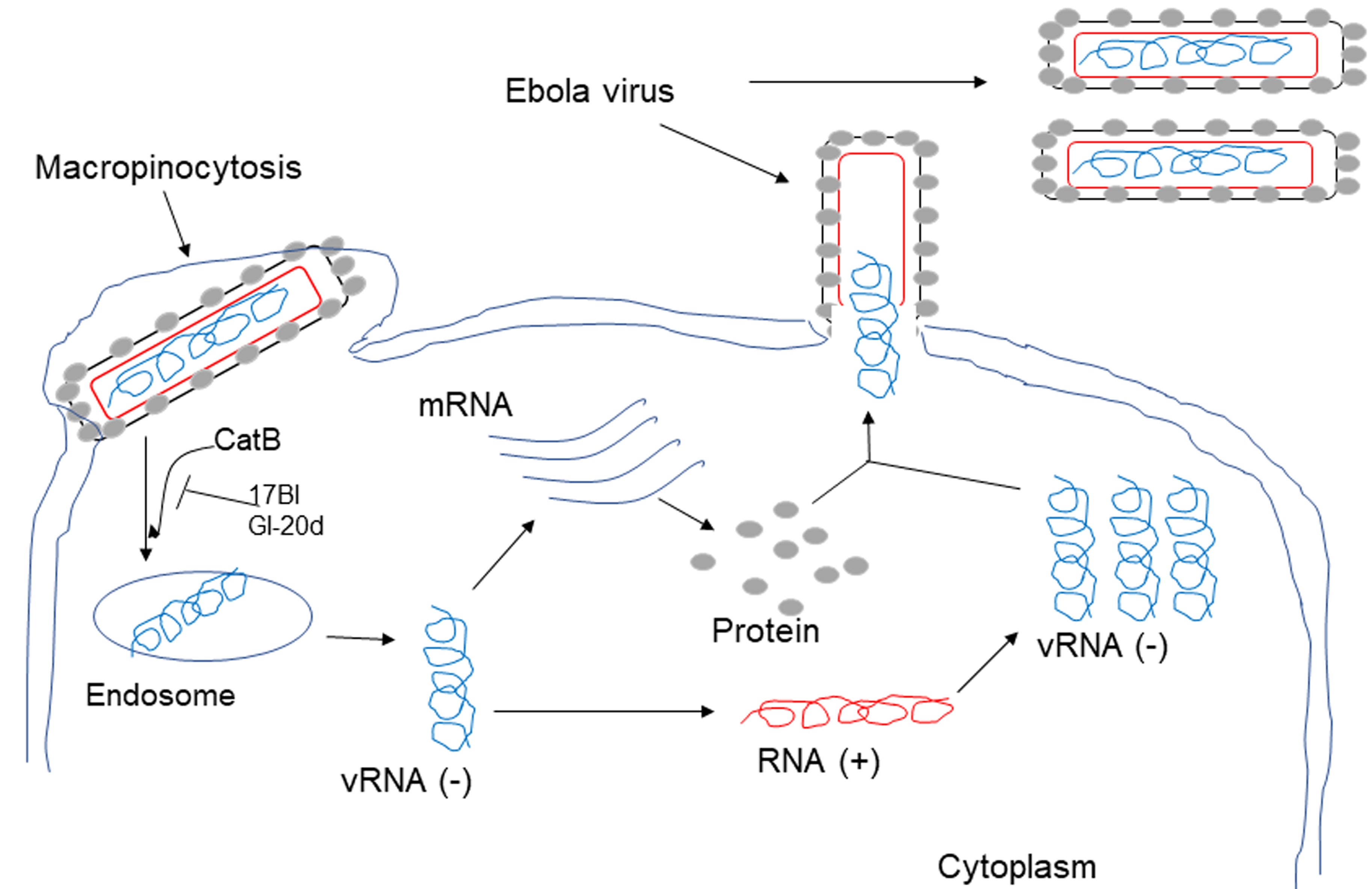 Fig. 1.
Fig. 1.17BI and GI-20d inhibit Ebola virus infection of host cells. 17BI and GI-20d act as cathepsin B (Cat-B) inhibitors to block the endosomal processing of EBOV glycoprotein (GP), thus preventing virus entry because EBOV requires the cleavage of EBOV-GP by cathepsins within host cell endosomes.
FF/CAP18, also known as LL-27, is a 27mer peptide that lacks the first and the
last 5 amino acids of LL-37. FF/CAP18 was designed by replacing a glutamic acid
and a lysine residue with phenylalanine of hCAP18 to enhance its antimicrobial
and anticancer activity [55, 56]. Human colorectal carcinoma HCT116 cells
cultured in the presence or absence of FF/CAP18 secreted exosomes in the
supernatant. The exosomes indicated by CD63 and CD81 expression were 40–100 nm
in size with 3.8
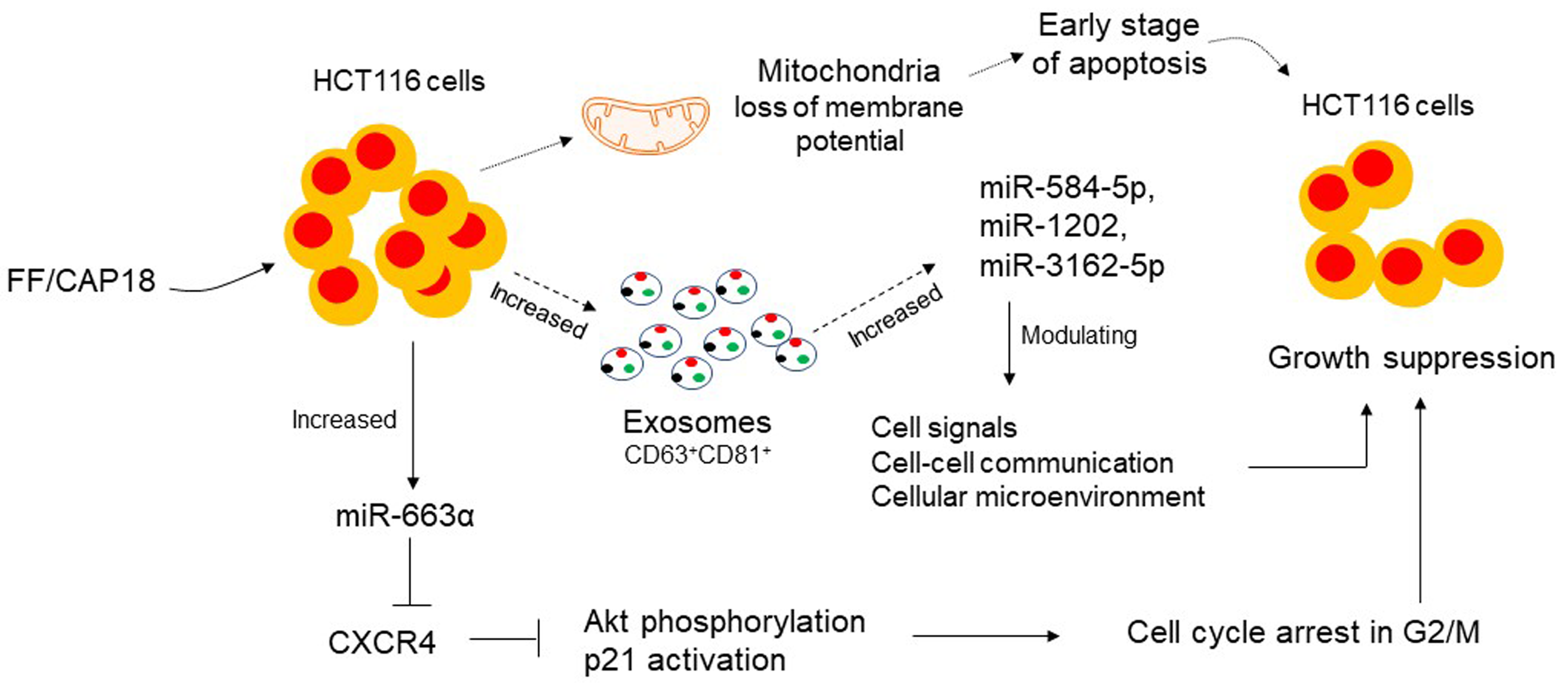 Fig. 2.
Fig. 2.FF/CAP18 inhibits the growth of human colon cancer cells. Human
colorectal carcinoma HCT116 cells cultured in the presence or absence of FF/CAP18
secreted exosomes in the supernatant. FF/CAP18 treated HCT116 and exosomes
expressed miR-584-5p, miR-1202 and miR-3162-5p, which reduced the growth of
HCT116. FF/CAP18 treated HCT116 cells increased miR-663
Both autophagy and apoptosis are programmed cell death processes and may occur within the same cell. Normally autophagy is followed by apoptosis [61]. DNA fragmentation, ER stress, hypoxia, and metabolic stress initiate intrinsic death pathways through activation of the proteins BAX and BAK, the release of cytochrome c from mitochondria and activation of caspase-9 which in turn cleaves Caspase-3 into the effector Caspase-3. Once caspases-3 is activated, cell apoptosis becomes irreversible [61]. Autophagy in colon cancer cells is activated by the miRNA (or LncRNA)-induced PI3K/Akt/mTOR signaling pathway or the Reactive oxygen species (ROS)-induced AMPK/mTOR signaling pathway [61]. For example, LncRNA is upregulated in clinical CRC tissues. Blockage of LncRNA in HCT116 cells induces apoptosis and inhibits autophagy by regulating the mir-100/ATG5 axis and the PI3K/AKT/mTOR pathway [62]. Oridonin, a compound isolated from medicinal herb Rabdosia rubescens, initiates colon cancer cell apoptosis through upregulation of caspase-3 cleavage and activation of autophagy by increasing the expression of Becin1 and LC3-II via AMPK/mTOR/ULK1 [63].
FK-16 is a short peptide corresponding to the residues 17–32 of human LL-37. Both FK-16 and LL-37 significantly reduced the viability of the human colon cancer LoVo and HCT116 cells [32]. Notably, FK-16 displayed a more potent activity against colon cancer cells than LL-37. Moreover, FK-16 had a minimal effect on the viability of normal human colon mucosal epithelial NCM460 cells. FK-16 upregulated nuclear p53 expression, thereby upregulating expression of Bax and downregulating Bcl-2 expression. The Bax signal was increased followed by increased nuclear levels of AIF and EndoG, which originally localized in mitochondria and translocated into the nuclei upon activation to trigger chromatin condensation and DNA fragmentation in cancer cells to induce programmed cell death. As FK-16 induced downregulated Bcl-2 expression, HCT116 cells increased autophagy-related proteins LC3-I and LC3-II as well as Atg5 and Atg7, which is required for the formation of autophagosomes at an early stage in cancer cells, to trigger cancer cell death. This fact was supported by the evidence that increased formation of LC3+ autophagic vacuoles and knockdown of Atg5 or Atg7 in HCT116 cells significantly reduced the cytotoxic effect of FK-16 for cancer cells. However, the full-length LL-37 had a minimal effect on LC3-I/II expression, indicating the importance of the short peptide FK-16 to induce autophagic death of colon cancer [32] (Fig. 3).
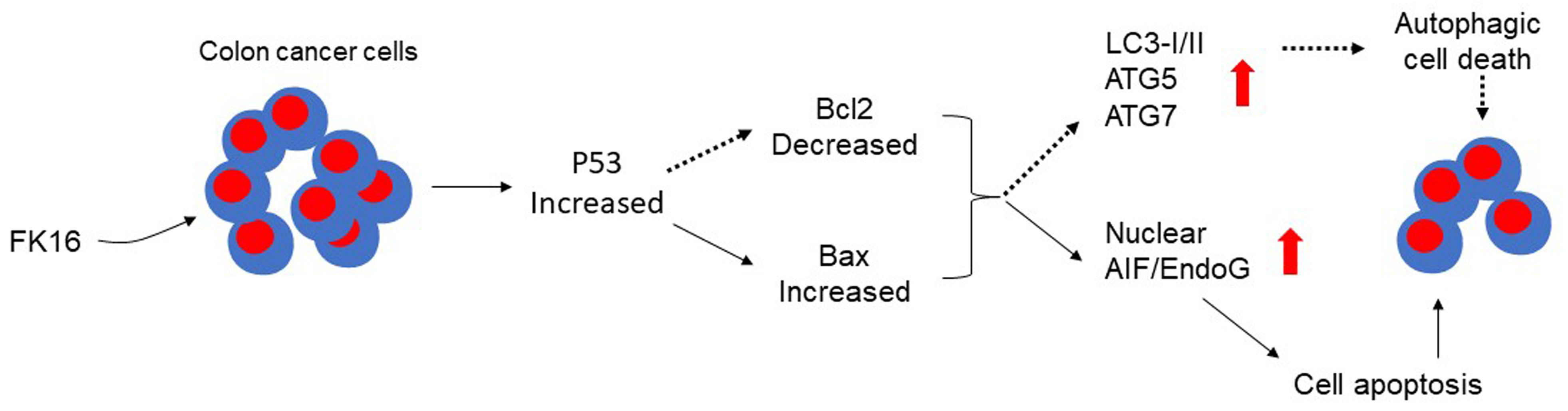 Fig. 3.
Fig. 3.FK16 induces the apoptosis and autophagic cell death in human colon cancer cells. FK16 treatment induced up-regulation of p53 expression, then increased the expression of BAX, AIF/EndoG to induce HCT116 cell apoptosis. FK16 induced Bcl2 down-regulation, resulting in up-regulation of LC3-I/II, ATG5, ATG7 expression, then activating the autophagic death of HCT116 cells. AIF, Apoptosis-inducing factor; EndoG, Endonuclease G.
LL-37-drived short peptides have several potential advantages over the
full-length LL-37 as future therapeutics. They have the capacity to neutralize
endotoxins released by pathogens and enhance host immune responses to infection
as well as their broad spectrum of antimicrobial activities. There are numerous
AMPs in clinical development to treat infections associated with pathogenic
microbes, but most of them are intended for topical applications. OP-145
(P60.4Ac) is a synthetic antimicrobial short peptide derived from LL-37. OP-145
has the capacity to bind LPS and LTA [52] and was formulated in eardrops
to treat chronic bacterial middle ear infections [64]. Chronic suppurative otitis
media (CSOM) is a chronic infectious disease with worldwide prevalence that
causes hearing loss and decreased quality of life. Application of
P60.4Ac-containing ototopical drops in the ear canal of patients suffering from
CSOM was found to be safe and well-tolerated as well as treatment success in 47%
of cases versus 6% in the placebo group [65]. The optimal dose of P60.4Ac was
selected for the subsequent phase II
As discussed earlier, a high number of LL-37-drived short peptides have been discovered with promising and potent activities to combat infections and colon cancer, but their translation into the clinic has been difficult. One of the greatest challenges restricting the development of LL-37-drived short peptides into therapeutics is their low metabolic stability. There is a low oral bioavailability for oral administration of peptide drugs because of degradations by proteolytic enzymes of the digestive system and their poor ability for penetration across the intestinal mucosa [66]. In the same way, intravenous administration is also limited due to rapid cleavage by proteolytic enzymes in the blood plasma and rapid removal from the circulation by excretory organs (liver and kidneys) before distribution to the surrounding tissues [66].
LL-37-derived short peptides can be obtained by purification from natural sources, chemical synthesis and expression using biological systems. Isolation from natural sources is not viable to produce in bulk quantities for clinical trials. Expression systems can also be used to produce short peptides in bacteria, yeast, insect, plants and mammalian cells but with lower yields and limitations to introduce chemical modifications into the peptide sequence [67]. Chemical synthesis is generally considered as the most mature technology available to produce linear peptides up to 50 residues with sufficient yields [68]. Moreover, the short peptides can be engineered to improve the bioavailability [17, 34]. Currently, head-to-tail cyclisation and incorporation of D-amino acids are good strategies to improve stability and antimicrobial activity and to reduce salts sensitivity of various LL-37-derived short peptides. However, to overcome some of the hurdles like high biodegradability and rapid removal from circulation, new strategies are needed to facilitate short peptide antibiotic development.
In addition, the mechanisms of action of LL-37-derived short peptides need
further studies. First, synergistic effects of LL-37-derived short peptides with
other factors are worth further investigations. Several antimicrobial agents,
such as human
For the challenges posed by the nonspecific distribution and short half-lives of LL-37 and its short peptides, an important consideration is to develop degradable, controlled-release polymers and polymeric nanoparticles as carriers. Polymeric drug delivery system is represented by a formulation or a medical device that is able to deliver drugs to target cells or organs, while the degradation and release of therapeutic molecules are controlled [81, 82]. Since the first generation of polymeric drug delivery system passively carrying PEGylated molecules to the second-generation system which was able to selectively bind specific targets, the third-generation system has emerged [81], which has evolved into the utilization of local biochemical changes in aberrant disease states to trigger activation-driven drug release [81]. It is also important to better understand the role of LL-37 in the pathogenesis of inflammation and tumorigenesis because LL-37 may display opposing activities in different tumors [33]. In addition, it should be kept in mind that LL-37 has been reported to interact with several receptors expressed by different cells or tissues to elicit diverse biological activities [8, 74, 75, 76, 77, 78, 79, 80]. Therefore, use of LL-37 and its short peptides as future therapeutic agents must be based on the development of more efficient delivery system and understanding of the precise mechanisms of human diseases.
KC, WG, JH—writing original draft; TY, JMW—editing the manuscript. All authors have read and agreed to the published version of the manuscript.
Not applicable.
This project was funded in part by federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E and was supported in part by the Intramural Research Program of the NCI, NIH, and by the fund from LCIM, CCR, NCI-Frederick. The authors thank Ms. C. A. Rhoderick for secretarial assistance.
This research received no external funding.
The authors declare no conflict of interest.