

Frontiers in Bioscience-Landmark (FBL) is published by IMR Press from Volume 26 Issue 5 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.

1 School of Life Sciences, B.S. Abdur Rahman Crescent Institute of Science and Technology, Chennai, India
Abstract
This review is a concise summary of studies involving the design, synthesis and characterization of potential inhibitors against thymidylate kinase of Mycobacterium tuberculosis. Tuberculosis inspite of being an ancient disease still continues to be a leading cause of death in the world. The increasing emergence of drug resistant Mycobacterium tuberculosis is one of the challenges in the complete elimination of tuberculosis. Thus, there is an undeniable need to develop novel treatment strategies to combat this deadly pathogen. Several protein targets are being investigated in Mycobacterium tuberculosis with an aim to develop the most potent and selective antituberculosis agents. It is not surprising that protein kinases, the key regulators of metabolism in almost all organisms are one of the major targets explored in antituberculosis drug discovery. Thymidylate kinases, a well established antiviral and anticancer target has garnered significant attention in the past twenty years in the development of prospective antituberculosis agents. A comprehensive analysis of such studies will provide a better understanding on the druggability of thymidylate kinase and also, will enable to refine the future drug designing studies on this attractive drug target of Mycobacterium tuberculosis.
Keywords
- Thymidylate kinase
- review
- Mycobacterium tuberculosis
- antituberculosis
- Kinase inhibitors
- Review
Mycobacterium tuberculosis (Mtb) is the etiological agent of Tuberculosis (TB), which caused an estimated 1.3 million deaths worldwide in the year 2017 (1). Existence of large gaps in diagnosis, treatment and prevention strategies has led to 6.4 million new cases of TB and 160,684 cases of multi drug resistant (MDR) TB in 2017. All the more alarming is the existence of 1.7 billion people with latent TB infection (LTBI). Thus, timely action is needed to manage drug resistant TB and to offer early treatment to latently infected people, thereby preventing them from acquiring active TB.
There are approximately 30 drugs falling into different lines of defence that are used in the current TB regimen for drug susceptible and resistant TB (2). But still such an armamentarium is inefficient in the case of extensively drug resistant (XDR) and total drug resistant (TDR) TB. In this scenario, the recent conditional approval of the two new antituberculosis drugs Delamanid and Bedaquiline is considered to be historical as it is almost four decades since the discovery of an anti-TB drug. Further, there are 20 more drugs in different phases of clinical trial in the TB drug discovery pipeline. However, on a broader perspective this is indeed a slow progress considering the intensity of TB pandemic. The major hurdle in designing new antituberculosis agents can be attributed to the complexity and uniquness of Mtb’s physiology (3). Firstly, the lipid rich, complex cell envelope protects Mtb from the hostile environment of human macrophages and lethal drugs. Secondly, the massive array of genes dedicated for metabolism is critical for the survival and persistent state of Mtb. Given these attributes of Mtb, most of the current TB drugs including isoniazid the most potent and safe first line drug are targeted against cell wall biosynthesis (4). Apart from rifampicin that inhibits RNA synthesis (5), there are very few TB drugs that have targets beyond cell wall biosynthesis. With ever increasing reports of drug resistant TB, there is immense need to develop candidate drugs with novel action mechanisms.
Protein kinases (PKs) are the key regulators of cellular processes spanning all life forms. Hence, PKs are the most sought after drug targets in the pharmaceutical industry after G-protein coupled receptors for several human and animal diseases. With the advent of genome sequencing projects of many infectious pathogens, PKs have gained popularity and reaped benefits as successful therapeutic targets. The sequencing of Mtb genome in 1998 (6) accelerated the drug discovery investigations on several novel protein targets in Mtb (7). Significant studies have also been carried out with the PKs family of Mtb such as serine/threonine PKs, shikimate kinases, pantothenate kinases and thymidylate kinases. There are around eleven eukaryotic-like serine/threonine PKs in Mtb, which are thought to play pivotal roles in cell growth, signal transduction and pathogenesis. Shikimate kinase is a key enzyme involved in the biosynthesis of aromatic amino acids through the shikimate pathway and is proven to be essential for the survival of the microbe. Pantothenate kinase, an essential enzyme in bacteria and eukaryotes, is involved in catalysing the first step of conversion of pantothenate to coenzyme A. It exists in three isoforms (type I, II and III) which can be differentiated from each other on the basis of their biochemical and structural characteristics. Thymidylate kinases of Mtb (TMPKmt) have been shown in vitro to be an essential enzyme in DNA synthesis. TMPKmt is an essential thymidylate synthase that is mechanistically and structurally unrelated to the human enzyme and hence best among the other kinases to be targeted as inhibitor.
The purpose of this review is to comprehensively analyse the different methodologies used to design inhibitors against the thymidylate kinases of Mtb (TMPKmt) and also, to shed light on the possible future developments to improvise the drug designing strategies.
Signal transduction systems in bacteria are equally essential as in higher organisms. Bacteria are expected to demonstrate a wide variety of regulatory responses to the changing environments (8). Phosphorylation/dephosphoylation activities modulated by a myriad of protein kinases are key players in bacterial signal transduction systems (9). Thus, it is natural that successful pathogens like Mtb have 60 different protein kinases to support their host evasion and survival mechanisms (Table 1). Two major PKs family in Mtb are the two component regulatory systems (2CRS) and serine/threonine protein kinases (STPKs), each consisting of 11 members. The 2CRS primarily comprises of a histidine kinase and a response regulator. Most of the Mtb 2CRS play a crucial role in virulence, whereas the first identified 2CRS, MtrA-MtrB system is essential in regulating cell growth (10). The eleven STPks are important for cell wall biosynthesis, cell division and most importantly in the regulation of metabolism in Mtb (11). Most of the STPKs share only 20% to 30% similarity with human homologues and it is strongly believed that the perturbation of the signalling cascade driven by these STPKs could significantly affect Mtb survival (12). Hence, there are numerous studies carried out to identify potent inhibitors of these STPKs in Mtb. Apart from these two major classes of PKs Mtb has other kinases that are involved in essential metabolic activities such as energetics, amino acid synthesis, purine/pyrimidine biosynthesis and synthesis of cofactors and other prosthetic groups. It is interesting to note that several of the kinases involved in these essential pathways share low similarity with the human counterparts and hence explored as anti-TB targets. Pantothenate kinase, which catalyses the key rate limiting step in the coenzyme A biosynthesis has been studied considerably for the discovery of novel antituberculosis compounds (13, 14). Shikimate kinase involved in aromatic amino acid biosynthesis has also been studied effectively as an important drug target. Finally, several of the kinase enzymes in the purine/pyrimidine biosynthesis have also garnered significant attention in Mtb owing to their success in other pathogens. Adenosien kinase and thymidylate kinase have derived significant attention and success in TB drug discovery studies owing to their exclusive features. This review is focused solely on the analysis of the drug discovery investigations of TMPKmt inorder to assess the current status of this kinase, which has earlier been proved to be a fruitful target in many other pathogens. The readers are requested to refer to many excellent reviews on the drug discovery status of other PKs in Mtb (12) (15).
| Class | Locus | Name | Product |
|---|---|---|---|
| 1.Purine/pyrimidine Biosynthesis | Rv2883c | pyrH | Probable uridylate kinase PyrH (UK) (uridine monophosphate kinase) (UMP kinase) |
| Rv2202c | adoK | Adenosine kinase | |
| Rv3247c | tmk | Thymidylate kinase Tmk (dTMP kinase) (thymidylic acid kinase) (TMPK) | |
| Rv1712 | cmk | Cytidylate kinase Cmk (CMP kinase) (cytidine monophosphate kinase) (ck) | |
| Rv1389 | gmk | Probable guanylate kinase Gmk | |
| Rv2445c | ndkA | Probable nucleoside diphosphate kinase NdkA (43) (NDP kinase) (nucleoside-2-P kinase) | |
| Rv0733 | adk | Adenylate kinase Adk (ATP-AMP transphosphorylase) | |
| Rv1017c | prsA | ribose-phosphate pyrophosphokinase | |
| Rv1286 | cysN | Probable bifunctional enzyme CysN/CysC: sulfate adenyltransferase (subunit 1) + adenylylsulfate kinase | |
| 2. Amino Acid Biosynthesis | Rv1296 | thrB | Probable homoserine kinase ThrB |
| Rv2539c | aroK | Shikimate kinase AroK (SK) | |
| Rv2439c | proB | Probable glutamate 5-kinase protein ProB (gamma-glutamyl kinase) (GK) | |
| Rv3709c | ask | Aspartokinase Ask (aspartate kinase) |
|
| Rv1654 | argB | Probable acetylglutamate kinase ArgB | |
| 3. Energy Metabolism | Rv2252 | Rv2252 | Diacylglycerol kinase |
| Rv1437 | pgk | Probable phosphoglycerate kinase Pgk | |
| Rv1617 | pykA | Probable pyruvate kinase PykA | |
| Rv0729 | xylB | Possible D-xylulose kinase XylB (xylulokinase) (xylulose kinase) | |
| Rv3232c | ppk2 | Polyphosphate kinase Ppk2 (polyphosphoric acid kinase) | |
| Rv1617 | pykA | Probable pyruvate kinase PykA | |
| Rv2984 | ppk1 | Polyphosphate kinase PPK (polyphosphoric acid kinase) (ATP-polyphosphate phosphotransferase) | |
| Rv0409 | ackA | Probable acetate kinase AckA (acetokinase) | |
| Rv0620 | galK | Probable galactokinase GalK (galactose kinase) | |
| Rv0115 | hddA | Possible D-alpha-D-heptose-7-phosphate kinase HddA | |
| Rv3696c | glpK | Probable glycerol kinase GlpK (ATP:glycerol 3-phosphotransferase) (glycerokinase) (GK) | |
| Rv1011 | ispE | Probable 4-diphosphocytidyl-2-C-methyl-D-erythritol kinase IspE |
|
| Rv0114 | gmhB | Possible D-alpha,beta-D-heptose-1,7-biphosphate phosphatase GmhB (D-glycero-D-manno-heptose 7-phosphate kinase) | |
| Rv1496 | Rv1496 | Possible transport system kinase | |
| 4. Biosynthesis of Cofactors, prosthetic groups and others | Rv1631 | coaE | Probable dephospho-CoA kinase CoaE (dephosphocoenzyme a kinase) |
| Rv2977c | thiL | Probable thiamine-monophosphate kinase ThiL (thiamine-phosphate kinase) | |
| Rv1695 | ppnK | Inorganic polyphosphate/ATP-NAD kinase PpnK (poly(P)/ATP NAD kinase) | |
| Rv0254c | cobU | Probable bifunctional cobalamin biosynthesis protein |
|
| Rv2786c | ribF | Probable bifunctional FAD synthetase/riboflavin biosynthesis protein |
|
| Rv1092c | coaA | Probable pantothenate kinase CoaA (pantothenic acid kinase) | |
| Rv3606c | folK | 7,8-dihydro-6-hydroxymethylpterin pyrophosphokinase | |
| Rv0422c | thiD | Probable phosphomethylpyrimidine kinase ThiD (HMP-phosphate kinase) (HMP-P kinase) | |
| Rv0650 | Rv0650 | Possible sugar kinase | |
| Rv2232 | ptkA | Protein tyrosine kinase transcriptional regulatory protein PtkA | |
| 5. Two Component Systems | Rv0982 | mprB | Two component sensor kinase MprB |
| Rv0845 | Rv0845 | Possible two component sensor kinase | |
| Rv3220c | Rv3220c | Probable two component sensor kinase | |
| Rv0758 | phoR | Possible two component system response sensor kinase membrane associated PhoR | |
| Rv0490 | senX3 | Putative two component sensor histidine kinase SenX3 | |
| Rv0600c | Rv0600c | Two component sensor kinase [second part] | |
| Rv0601c | Rv0601c | Two component sensor kinase [first part] | |
| Rv0902c | prrB | Two component sensor histidine kinase PrrB | |
| Rv3132c | devS | Two component sensor histidine kinase DevS | |
| Rv3245c | mtrB | Two component sensory transduction histidine kinase MtrB | |
| Rv2027c | dosT | Two component sensor histidine kinase DosT | |
| Rv1032c | trcS | Two component sensor histidine kinase TrcS | |
| Rv3764c | tcrY | Possible two component sensor kinase TcrY | |
| Rv1028c | kdpD | Probable sensor protein KdpD | |
| 6. Serine/threonine protein kinases | Rv0410c | pknG | Serine/threonine-protein kinase PknG (protein kinase G) (STPK G) |
| Rv0931c | pknD | Transmembrane serine/threonine-protein kinase D PknD (protein kinase D) (STPK D) | |
| Rv0014c | pknB | Transmembrane serine/threonine-protein kinase B PknB (protein kinase B) (STPK B) | |
| Rv0015c | pknA | Transmembrane serine/threonine-protein kinase A PknA (protein kinase A) (STPK A) | |
| Rv3080c | pknK | Serine/threonine-protein kinase transcriptional regulatory protein PknK (protein kinase K) (STPK K) | |
| Rv1743 | pknE | Probable transmembrane serine/threonine-protein kinase E PknE (protein kinase E) (STPK E) | |
| Rv1746 | pknF | Anchored-membrane serine/threonine-protein kinase PknF (protein kinase F) (STPK F) | |
| Rv2088 | pknJ | Transmembrane serine/threonine-protein kinase J PknJ (protein kinase J) (STPK J) | |
| Rv2176 | pknL | Probable transmembrane serine/threonine-protein kinase L PknL (protein kinase L) (STPK L) | |
| Rv1266c | pknH | Probable transmembrane serine/threonine-protein kinase H PknH (protein kinase H) (STPK H) | |
| Rv2914c | pknI | Probable transmembrane serine/threonine-protein kinase I PknI (protein kinase I) (STPK I) |
Thymidylate kinase belongs to the nucleoside mono phosphate (NMP) kinase family and is also referred to as thymidylic acid kinase or thymidine monophosphate kinase or ATP:dTMP phosphotransferase (EC: 2.7.4.9.). It catalyzes the reversible phosphorylation of thymidine 5’-mono phosphate (dTMP) to thymidine 5’-diphosphate (dTDP) in the presence of Magnesium and ATP. The dTDP is then converted to thymidine 5’- triphosphate (dTTP) by nucleoside diphosphate kinase. The dTMP derived from deoxy uridine monophosphate (dUMP) via thymidylate synthase in the denovo pathway and the dTMP derived from thymidine via thymidine kinase in the salvage pathway are both phophorylated by TMPKmt (Figure 1). Therefore, making TMPkmt as the last specific junction enzyme of both the denovo and salvage pathways of pyrimidine biosynthesis (16). The function of thymidylate kinase was first demonstrated in Saccharomyces cerevisiae, encoded by the cdc8 gene (17, 18). Inhibition of TMPK activity affects cell viability and hence its undomitable success as targets of antiviral and anticancer drugs (19, 20).
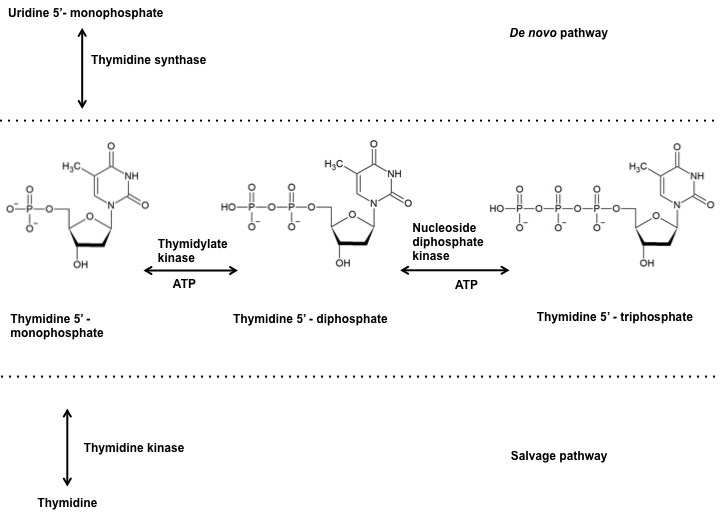 Figure 1
Figure 1Reaction catalyzed by thymidylate kinase at the junction of denovo and salvage pathway.
TMPKmt (Rv3247c) is an essential gene for the in vitro survival (21) of Mtb. Targeting TMPKmt is crucial, as the bacterium will be prevented from synthesizing new NMP through the recycling of DNA/RNA degradation products in its dormant state.
The elucidation of the X-ray crystal structure of TMPKmt was the major initiation factor for the numerous strcucture based drug design investigations (22). The structure of TMPKmt bound with its natural substrate TMP determined at a resolution of 1.95 Å was the first of its kind NMPK structure to be reported in a pathogen. TMPK is a homodimer in solution (monomer molecular mass=24 kDa; 214 residues per monomer), each monomer being composed of nine α-helices surrounding a five-stranded β-sheet core. The TMPKmt structure has three functionally essential regions for its catalytic activity.The P-loop region acts as the controlling centre for the phosphoryl group of the phosphate donor and also features the second essential region, a conserved arginine residue. The third important region is the LID region which functions in a flexible manner to close on the phosphoryl donor upon binding. The presence of arginine residues in both the P-loop and the LID region of TMPKmt is something unlike the other earlier reported TMPK structures. Further, the presence of a Magnesium ion in the active site and the characteristic helical conformation of the LID region are the other features that make the TMPKmt strcuture unique.
Also, noteworthy is that the crystal structure revealed the point of contacts of the natural substrate TMP and the important amino acid residues involved in catalytic activity. Even before the structure determination, these unique characteristics of TMPKmt was predicted by Labesse G and colleagues through their proposed structural model (23). Further, TMPKmt was found to be a stable protein with a melting temperature of 68 °C and similarly resistant to thermal denaturation like other Mtb proteins. The high thermal resistance of TMPKmt is due to its conformational stability as evidenced by its high total standard Gibb’s free energy for dissociation upon urea induced denaturation.
Chemical synthesis of TMPKmt inhibitors were mainly oriented towards structure guided generation of lead compounds due to the availability of its good quality three dimensional structure. Synthesis of nucleotide/nucleoside analogues were the most sought after inhibitors for TMPKmt owing to the simple methods of synthesis and also due to the confidence offered by the success of azidothymidine as an antiviral agent. On the other hand only very few studies explored non-nucleotide/nucleoside based inhibitors against TMPKmt.
Substrate analogues are conventionally the first choice of inhibitors for inhibiting enzymes and also due to their ease of synthesis. Crystallisation of TMPKmt was followed by several investigations involving the synthesis of TMPKmt inhibitors based on its characteristic features (24). Structural analysis of these inhibitors especially the well known TMPK inihibitor 3’-Azido-3’-deoxythymidine 5’-O-monophosphate (AZT-MP) in complex with TMPKmt revealed a competitive inhibition mechanism (Ki =10 µM, (21)) where in the crucial magnesium ion was eliminated (25). Similar modifications were attempted at 2’ and 3’ positions of the ribose moiety in the dTMP scaffold. A 2’-chloro substitution (Table 2-C1) exhibited a Ki of 19 µM, but still less than that of AZT-MP. Neverthless, this work also explored many other dTMP analogues and in fact provided the first structure activity relationship (SAR) insight for the synthesis of better TMPKmt inhibitors (26).
| Compound | Ki | Reference |
| FBS-25-4871-g002.png C1 | 19 µM | 26 |
| FBS-25-4871-g003.png C2 | 10 µM | 29 |
| FBS-25-4871-g004.png C3 | 5 µM | 29 |
| FBS-25-4871-g005.png C4 | 7 µM | 27 |
| FBS-25-4871-g006.png C5 | 12 µM | 27 |
| FBS-25-4871-g007.png C6 | 3.5 µM | 31 |
| FBS-25-4871-g008.png C7 | 13.5 µM | 31 |
| FBS-25-4871-g009.png C8 | 2.3 µM | 32 |
| FBS-25-4871-g010.png C9 | 0.6µM | 33 |
| FBS-25-4871-g011.png C10 | 0.17 µM | 34 |
| FBS-25-4871-g012.png C11 | 12±3 µM | 35 |
| FBS-25-4871-g013.png C12 | 0.42 µM | 41 |
| FBS-25-4871-g014.png C13 | 0.27 µM | 41 |
| FBS-25-4871-g015.png C14 | 0.8 µM | 44 |
Interestingly, when a series of sugar and base modified dTMP analogues were assayed for their affinity towards TMPKmt, it was noticed that the affinities of phosphorylated and non-phosphorylated analogues remained the same (27). This was observed by examining the affinity of a series of 2’, 3’ and 5’ modified analogues, among which 5’-azido (Ki =7 µM) and 5’-amino (Ki =12 µM) (Table 2- C4, C5) substitutions proved to be good candidates. This finding in turn encouraged the synthesis of several nucleoside analogues, in order to circumvent the issues associated with the entry of charged phosphorylated analogues across the highly lipophilic cell wall of Mtb. A series of 3’-C-branched chain substituted dTMP analogues in ribo and 2’-deoxy ribo series were explored for their TMPKmt affinity. Several 2’-deoxy ribo series compounds such as 3’-CH2NH2 (Ki=10.5 µM), 3’-CH2N3 (Ki=12 µM), and 3’-CH2F (Ki=15 µM) were more potent compared to the ribo analogues, proving that 2’-deoxy series are tolerant to large substitutions at the 3’ position (28). In another study, variations of the thymine moiety of thymidine were synthesized. 5-Halogenated 2’-deoxy uridine (Table 2-C2, C3) showed better potency and selectivity towards TMPKmt than to human TMPK (29). But years later, in a study that explored 5,5’-bis-substituted 2’-deoxyuridine analogues, only two compounds with 5’-OH modified as acetonitrile and tetrazolylmethyl moiety respectively showed substantial affinity towards TMPKmt and was rather classified as weak inhibitors (30). A novel class of bicyclic nucleosides were the next set of inhibitors to be explored as potent as well as selective inhibitors of TMPKmt (Table 2-C6, C7) (31). This study further increased the possibility of exploring a diverse set of nucleosides that can be targeted against TMPKmt. Van Calenbergh and colleagues evaluated the structural, functional and conformational attributes of a series of bicyclic 6-membered ring derivatives and another series of 5-membered ring bicyclic nucleosides, in order to achieve better inhibitory activity. 1-(3-aminomethyl-3,5-dideoxy-2-O,6-N-(thiocarbonyl)-b-d ribofuranosyl)thymine was the best of the series synthesized with a TMPKmt inhibitory potency of Ki 2.3 µM (Table 2- C8) and also capable of significantly inhibiting M.bovis growth (IC99=100µg/mL) (32). In 2007, the same group took inspiration form the dinucleoside as well as from thiocarlide, another TB drug to synthesize 3’- branched thiourea-substituted beta-thymidine derivatives. Among those derivatives, 5-aryl thiourea-alpha-thymidine analogue exhibited significant potency and M.bovis growth inhibition (IC99=20µg/mL). Further, this thiourea derivative also achieved 39 % inhibition of Mtb growth at a concentration of 6.25 µg/mL (Table 2-C9) (33).
Given the moderate success achieved with sugar and base modified dTMP, there still was a need to design and identify more potent inhibitors capable of inhibiting Mycobacterium growth. In 2011, a series of alpha and beta derivatives of 3’ and 5’ modified thymidine analogue were synthesized, which specifically had their 4-O replaced by a more lipophilic sulphur atom. The compound 5’-arylthiourea 4-thio-alpha-thymidine analogue exhibited significant inhibition (Ki = 0.17 µM) of TMPKmt and also inhibited the growth of Mycobacterium bovis (MIC-25 µg/ml) (Table 2-C10) (34). The lipophilic attribute of sulphur atoms were further exploited in a later study, where they synthesized sulfamide analogues with resulting Ki values in the micromolar range. Specifically aryl sulfamides with halogen substitutions were capable of inhibiting the growth of Mycobacterium smegmatis (MIC 250 µg/ml).(Table 2-C11) (35).
While almost all the studies reported on thymine derivatives, a couple of studies examined the TMPKmt inhibitory potency of uridine derivatives. In the first study, 5-substituted analogues of 6-aza-2’-deoxyuridine-5’monophosphate were found to interact at the substrate binding site of TMPKmt but with no clear TMPKmt inhibitory activity (36). In another study three series of 5-arylaminouracil derivatives were synthesized and most of those compounds demonstrated 100% inhibition of Mtb H37Rv growth in the concentration range of 5-40 µg/ml. One compound namely, 1-(4’-hydroxy-2’-cyclopenten-1’-yl)-3-(4′′′-hydroxy-2′′′-cyclopenten-1′′′-yl)-5- (4′′-butyloxyphenylamino)uracil was most effective even on the MDR strain of Mtb MS-115 with a MIC90 of 5µg/mL (37).
Success of boron based drugs like the anticancer drug Bortezomib and several other drugs in clinical trails have increased investigations of bioactive molecules contaning boron clusters against varied biological targets (38). In a recent study, a sereis of thymine derivatives bearing novel boron clusters such as 1,2-dicarba-closo-dodecaborane, 1,12-dicarba-closo-dodecaborane or 7,8-dicarba-nido-undecaborate anions were explored for their TMPKmt inhibitory activity. Of the sereies, two compounds were able to completely inhibit the growth of M.smegmatis at a concentration of 100 µg/mL (39).
Design of non-nucleoside inhibitors captured the interest of quite a few scientists owing to the several drawbacks encountered with nucleotide analogues of TMPKmt inhibitors. Based on a denovo fragment based drug designing platform, called LEA3D, Douget and colleagues identified a potential non-nucleoside inhibitor of TMPKmt (40). The molecule, 3-(4-(thymin- 1-ylmethyl)phenyl) propionamide inspired the synthesis of several series of non-nucleoside inhibitors of TMPKmt. First in the list were a series of N’- (4-substituted-benzyl)-pyrimidine derivatives, which exhibited Ki against recombinant TMPKmt in the micromolar range and also inhibited Mycobacterium bovis growth (MIC50-50 µg/mL). Later, Olga Familiar and his colleagues synthesized the first series of acylic nucleosides. Though addressed as nucleosides they were not exact substrate analogues of dTMP but instead retained the thymine ring as (Z)-butenylthymines carrying a naptholactam or napthosultam moiety at position 4 (Table 2-C12, C13) (41). Both these set of compounds showed significant inhibition of TMPKmt with a Ki in the range of 0.27-0.42 µM, but were not as potent in inhibiting mycobacterial growth. The insoluble nature of these compounds was one of the major reason attributed to their failure. Hence, in the next set of compounds the planar naptholactam and napthosultam groups were replaced by a distal imidazoquinolone resuting in two series of compounds such as 5,6-dihydro-1H-imidazo(4,5,1-ij)quinoli- nones and a 5,6-dihydro-1H,4H-1,2,5-thiadiazolo(4,3,2-ij)quinoline-2,2-dioxide (42). However, these compounds also did not exhibit significant inhibitory activity against Mycobacterium. The search for an ideal non-nucleoside inhibitor led by a high throughput screening using fragment based lead generation resulted in two novel classes of TMPKmt inhibitors. 3-cyanopyridones and 1,6-naphthyridin-2-ones were the first set of actual non-nucleoside inhibitors without a thymidine core unlike the earlier set of compounds discussed (43). Both the compounds exhibited a noteworthy nanomolar potency of inhibition in vitro, but however the mycobacterial inhibitory activity is yet to be convincingly established. Later, in two of their investigations Lijun Song and colleagues synthesized a number of non-nucleosde inhibitors of TMPKmt. In the first study the synthesis and optimization of a series of piperidinyl thymine analogues resulted in a compound with a low Ki, 0.8 µM (Table 2-C14) (44). In the second study, three series of compounds were synthesized namely, phenoxybenzoyl analogues, truncated series of quinolin-2-yl analogue and phenoxylquinolin-2yl derivatives. Out of the three sets of compunds, the phenoxylquinolin-2yl showed potent TMPKmt inhibitory activty and also better antimycobacterial activity (45). Collectively these studies bring to light the potential of non-nucleoside inhibitors and more studies are needed to explore novel scaffolds without relying on the conventional substrate analogues.
Computer aided drug design (CADD) has revolutionized drug discovery pipelines in recent times by playing a key role in accelerating the processes of hit identification and lead optimization. It holds true for antituberculsis drug discovery as well. The two main methods of CADD are structure based drug design (SBDD) and ligand based drug design (LBDD). SBDD also referred to as direct drug designing approach relies on the 3-dimensional structural information of the protein target, in order to identify target specific active compounds. On the other hand LBDD, also referred to as indirect method of drug designing is very useful for protein targets with unknown structural knowledge. In LBDD, new potential hits are designed based on the existing set of ligands known to bind with the target. In the case of TMPKmt, the availability of good quality 3D structure and the knowledge of target specific active ligand sets have led to several studies that successfully integrated SBDD and LBDD approaches.
Manallack and colleagues performed the first CADD study on TMPKmt in 2002, shortly after its structure elucidation (22). In this study compounds showing potential binding affinity towards TMPKmt crystal structure were screened from The Available Chemicals Directory database. Though this study did not result in significant findings, they were able to confirm the hydrogen bond forming ability of Asp100 and Tyr39 of TMPKmt. As years progressed, the advent of chemical synthesis projects of TMPKmt inhibitors and availability of different TMPKmt ligand complex crystal structures provided opportunities for several CADD investigations of TMPKmt inhibitors.
Quantitative structure activity relationship (QSAR) is the major LBDD method that describes a mathematical relatonship between the structural attributes of ligands and their respective biological responses on the target. Several studies have been using this method to discover novel drug candidates against well established Mtb targets (46). Gopalakrishnan and colleagues carried two CADD investigation studies on TMPKmt. In the first series of work published in 2005, the group of Gopalakrishnan and Desiraju derived a pharmacophore model from a list of 47 compounds that were earlier reported in literature as thymidine monophosphate kinase inhibitors (47). The pharmacophore model generated with HypoGen was used as a 3D structural query to virtually screen a combined database of 500,000 molecules, which were derived from several publicly available databases like IBS, MDPI etc., Later using three subsequent filtering schemes, a total of 186 compounds were obtained as hits spanning five different chemical clusters. In their second study in 2006, they employed molecular field analysis (MFA), a type of 3D-QSAR method, which involves the alignment of molecular interaction fields of ligands inorder to extract the aligned features that might be related to biological activity (48). Least square alignment, pharmacophore based alignment and receptor based alignment were the three alignment protocols utilized to develop a 3D-QSAR model using a combined training/test set of 47 compounds as defined in the earlier study. The structures of all compounds were built and energy minimized using the coordinates of the PDB structure, 1G3U. Of the three alignment protocls, the 3D-QSAR model obtained from receptor based alignment was found to be superior in predicting as well as mapping the active site residues involved in ligand binding. These two studies provided early information on the structural and chemical features of TMPKmt inhibitors which were further explored using a variety of CADD methods by different research groups.
Kumar A and colleagues screened compounds available in the Maybridge small molecule database for 3D pharmacophore query in a virtual screening protocol (49). The pharmacophore model was derived from a dataset of 110 compounds collected from literature and defined using the various PDB structures of TMPKmt such as 1N5L (50), 1W2G (25), 1W2H (25) and 1MRS (24). The screening protocol also included molecular docking and receptor based weighting using Structure Interaction Fingerprints to yield 8 hit compounds, which were further evaluated for their in vitro antitubercular activity in Mtb H37Rv. This was the first CADD study on TMPKmt to successfully identify 5 compounds that exhibited antitubercular activity with MIC in the range of 3.12 µg/mL to 12.5 µg/mL.
Andrade.C.H and her colleagues adopted a receptor-independent (RI) 4D-QSAR method to derive a 3D-pharmacophore for a set of 34 5-thiourea-substituted α-thymidine inhibitors (51). The most potent inhibitors obtained from the statistically significant RI-4D QSAR models were further validated by docking to the active site of TMPKmt crystal structure (1G3U). This study led to the identification of new regions of TMPKmt inhibitors such as the sugar-pyrimidine ring structure and the 5’-arylthiourea moiety with good pharmacophore sites. In 2010, they performed a similar RI 4D QSAR investigation and derived a 3D pharmacophore for a larger set of 81 thymidine analogues (52). Thus, using 4D-QSAR methods, which is an extension of 3D-QSAR, these two studies explored an ensemble of different sampling features of the chosen TMPKmt inhibitors. The main drawback of 4D-QSAR analyses is the absence of large and chemically diverse datasets. Keeping this in mind, in their next study Andrade.C.H’s group explored a larger dataset of 97 thymidine based analogues utilizing a fragment based QSAR to generate 2D-QSAR models (53). Specifically they employed hologram QSAR (HQSAR) to identify unique molecular finger print patterns to further support the Quantum Mechanics/Molecular Modeling (QM/MM) docking experiment with the crystal structure of TMPKmt (22). Through this study, the authors were able to modify the existing inhibitors with better drug like properties.
In 2010, a combinatorial screening technique was employed to identify more potent analogues of bicyclic thymidine derivatives as inhibitors of TMPKmt (54). The execution of TMPKmt structure-1MRS (24) based focused screening and with the development of a QSAR model, resulted in a subset of bicyclic thymidine analogues with ADME properties and inhibitory potential that were predicted to be favorable.
Trupti and group developed 2D and 3D QSAR models from the molecular modeling studies on 28 substituted benzyl pyrimidine derivatives that were earlier reported by Gasse et al (55). Later pharmacophore optimization was achieved using the information derived from statistically significant SAR models, which further led to the design of new chemical entities (NCE) using the Combi Lib tool. Further molecular docking studies were performed on the most potent NCEs using the GLIDE tool, which revealed their superiority in docking TMPKmt over AZTMP.
Amidst several studies focusing on SAR studies Zaheer Ul Haq and his team optimized a structure based virtual screening protocol for TMPkmt inhibitors (56). To test the protocol they used a collection of 10,000 randomly selected molecules from the National Cancer Institute database along with 105 known substrates and inhibitors of TMPKmt. All the molecules were subjected to rigid docking algorithm using Fast Rigid Exhaustive Docking (FRED) program and scored using various methods such as enrichment curves, ROC curves and consensus scoring.
In 2013, TMPKmt (1GSI) bound conformations of 15 thymidine inhibitors with known activity and as part of a training set were built and later used to develop 3D QSAR pharmacophore model (57). This model was further validated with a PH4 3D QSAR to screen for TMPKmt inhibitors with potencies in the sub-nanomolar range among a library consisting of literature reported thymidine analogue inhibitors of TMPKmt.
In a recent study, Koseki.Y and colleagues performed an in silico pharmacophore screening of 461,883 moelcules in the ChemBridge database (58). After the three step pharmacophore screening and validation with molecular docking of TMPKmt structure, one compound (KTP3) was identified with significant growth inhibitory effect on Mycobacterium smegmatis, a surrogate model organism of Mtb. A further screening was carried out to identify KTP3 analogues and it resulted in two compounds namely KTPS1 and KTPS2 with half maximal inhibitory concentration values of 8.04 μM and 17.1 μM, respectively. However, since these two molecules exhibited only 18 % and 36 % inhibition of TMPKmt activity, they were suspected to have off target effects in M.smegmatis.
While all studies discussed so far designed small molecule inhibitors against TMPKmt, Kumar M and his colleagues have adopted a rational designing approach to identify small peptide inhibitors for TMPKmt (59). Using in silico structure based approach such as molecular docking and molecular dynamic simulations they had designed a tripeptide Trp-Pro-Asp that had high selectivity towards TMPKmt and also with favourable ADMET properties.
Intense research efforts have been expedited globally in search of new antituberculosis drugs in order to combat MDR and XDR TB. The complete genome sequencing of Mtb and other omics studies which unraveled the physiology and pathoegenesis of Mtb have resulted in many novel drug targets. But the search for highly selective and potent antitubeculosis drugs is never ending in the scenario of increasing drug resistant cases even against the latest TB drugs such as Delamanid. At this juncture a thorough investigation of valuable drug targets of Mtb is indispensable. TMPKmt is one such attractive and promising antituberculosis drug target. Early studies were focused on the synthesis of dTMP analogues and nucleotide inhibitors. As pointed out by Van Celnbergh and colleagues in their review, most of these early thymidine based inhibitors were able to achieve a Ki of upto 10 μM or even higher (60). But in the last decade several new chemical synthesis efforts, such as the thiourea substituted thymidine analogues exhibited low Ki of 0.17 μM. These compounds also inhibited the proliferation of Mtb in vitro. Apart from these promising thiourea based inhibitors, several non-nucleotide inhibitors also showed significant Ki in the submicromolar range. Further experiments are required to explore their complete inhibitory potential on the various Mycobacterium tuberculosis strains followed by in vivo studies. These chemically synthesized inhibitors of TMPKmt paved way parallely for several CADD studies. Most of the CADD studies were structure driven with the availability of the 3D structure of TMPKmt and its complexes with varied inhibitors. Inputs from QSAR models resulted in TMPKmt inhibitors with promising ADMET properties but however most of the studies only yielded compounds with Ki in the micromolar range or even higher. One possible reason is that the QSAR models are derived from datasets comprising of inhibitors with only moderate inhibitory activity. Fragment based drug designing studies that take advantage of the unique structural features of TMPKmt could be a fruitful alternative strategy. One such promising study by Panda and colleague resulted in compounds with nanomolar potency. In future such combined investigations incorporating structure based tailored high throughput screening methods could possibly reap benefits. On the other hand several inhibitors with significant TMPKmt inhibitory potential however suffered setbacks in permeating the Mtb cell wall. To address this issue, other non-nucelotide inhibitors proven to be active against Mtb and also with low toxicity profile can be tested against TMPKmt. In this regard, the compounds tested and made publicly available by Glaxosmithkline could be a very valuable resource (61). In the near future, hopefully TMPKmt could also emerge as the successful target of Mtb following its glory as antiviral and anticancer drug target with complementary efforts.