












Frontiers in Bioscience-Landmark (FBL) is published by IMR Press from Volume 26 Issue 5 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.

1 Discipline of Chemistry, Indian Institute of Technology Gandhinagar, Gujarat, India, 382355, Indian Institute of Technology Gandhinagar, Gujarat, India 382355
Abstract
Phosphatidylinositol-3 kinase-related kinases (PIKKs) belong to a family of atypical serine/threonine kinases in humans. They actively participate in a diverse set of cellular functions such as meiotic, V(D)J recombination, chromosome maintenance, DNA damage sensing and repair, cell cycle progression and arrest. ATR, ATM, DNA-PKcs, mTOR and hSMG are the members of the PIKK family that play an important role in in cancer cell proliferation, autophagy, and cell survival to radio and chemotherapy. Thereby targeting these PIKK kinases in cancer along with chemo/radiotherapy agents, can help in differential cytotoxicity towards cancer cell over the normal cell. In this review, we compile the various small molecule kinase inhibitors with respect to structural and strategic targeting of PIKK family members. Rapalogs, AZD8055, AZD2014, OSI-027, INK-128, MLN0128, VX970, NVP-BEZ235, Torin2, AZ20, and AZ31 are the diverse scaffolds which have successfully made into the pre-clinical trials either as mono or combinatorial therapy for the treatment of various human cancers. Their synthesis and pre-clinical trial highlight the challenges associated in the development process.
Keywords
- Carcinoma
- cancer
- inhibitors
- protein kinases
- kinase domain
- catalytic activity
- Phosphatidylinositol-3 kinase-related kinases
- Phosphatidylinositol 3-kinase
- Ataxia telangiectasia mutated kinase
- ATM- and Rad3-related kinase
- DNA dependent protein kinase catalytic subunit
- mammalian target of rapamycin (mTOR)
- SMG: suppressor with morphological effect on genitalia family member (SMG)
- Transformation/transcription-associated protein (TRAAP)
- quinolines
- clinical trials
- pharmacodynamics
- pharmacokinetics
- IC50
- solubility
- bioavailability
- thiaanthrenpyran-4-one
- Rapalogs
- pyrazine derivatives
- pyrimidine derivatives
- schisandrin
- caffeine
- wortmannin
- Auto phosphorylation
- DFG motif
In the 1990s, a series of high molecular weight (280 and 470 kDa in size) serine/threonine kinases have emerged, cloned and classified as Phosphatidylinositol 3-kinase (PI3K)-related protein kinase (PIKK) family (PIKKs). These proteins are closely related to the kinase domain of Phosphatidylinositol 3-kinase (PI3K) family of phospholipid kinases (1-2). Studies on PIKK family members displayed that they are present in all eukaryotes, and none of them were found in prokaryotes to date. Till date, six human PIKK family kinases have been identified, which includes Ataxia telangiectasia mutated kinase (ATM), ATM- and Rad3-related kinase (ATR), DNA dependent protein kinase catalytic subunit (DNA-PKcs), mammalian target of rapamycin (mTOR), suppressor with morphological effect on genitalia family member (SMG1) and transformation/transcription-associated protein (TRAAP) (3-10). All the family members except TRAAP have protein kinase activity, and none of them exhibited lipid kinase activity. In the last twenty years, there have been broad studies on PIKK family protein, several features of PIKK family members have been discovered which lead to consequential insights into their biological function. Members of PIKK family participate in a diverse set of cellular functions such as meiotic and V(D)J recombination, chromosome maintenance, DNA damage sensor and repair, cell cycle progression and cell cycle arrest, and their dysfunction results in a variety of diseases, including cancer and immunodeficiency neurological disorder (11). These features have led to great interest in academic and pharmaceutical industries on inhibiting PIKK family proteins and their signaling pathways for effective cancer therapy. In this review, we provide a general idea of the structure of reported PIKK family inhibitors and a strategic perspective for the revelation and development of PIKK kinase inhibitors for cancer therapy.
Many of the PIKKs are larger proteins (2549 and 4128 amino acids), have high sequence similarity in their catalytic PIKK kinase domain and share common domain architecture (12-13). The essential kinase domain is located close to the C-terminal end and surrounded by the FAT domain (37- 47 amino acids) on N-terminal (named after the kinases FRAP, ATM, and TRRAP) and a unique FATC domain (~35 amino acid) on its C-terminal side. The FAT and FATC domains are observed to be present in combination and suggest that they may interact with each other or participate in intermolecular protein-protein interactions. Also, it is speculated that they fold together in a conformation that ensures the proper functioning of catalytic kinase domain (14). In mTOR, FKBP- rapamycin binding (FRB) domain (100 residues) is assumed to be located in-between the FAT and catalytic domains, and the RAPTOR and RICTOR binding domain are observed on the N-terminal region of the FAT domain. The FATC domain is more conserved when compared to the FAT domain, it could be an essential domain for regulating kinase activity. Nevertheless, the exact function of the FAT and FATC domain are not firmly ascertained and, still need to be revealed experimentally. In ATR, there is an ATR-interacting protein (ATRIP) binding domain on N-terminus (15). ATRIP is an 85 kDa protein, it is required for binding of ATR to replicative protein A (RPA) on single-strand DNA breaks under replication stress. Also, ATRIP has topoisomerase binding protein I (TopBP1) region, which helps in the interaction of ATR with TopBP1. It is essential for activating cell cycle checkpoints for cell survival with respect to DNA damage (16). On the other hand, an FKBP12-rapamycin binding (FRB) domain located between the FAT and kinase domain of mTOR and SMG, has an inhibitory effect on mTOR function (17) (Figure 1).
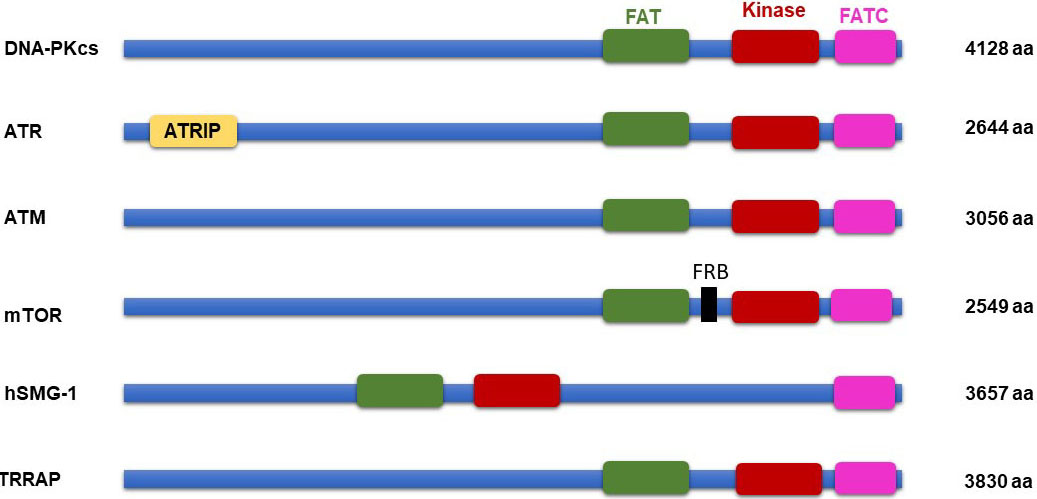 Figure 1
Figure 1PIKK family protein domain architecture.
mTOR (the mammalian or mechanistic target of rapamycin, also known as FRAP, RAFT1, or RAPT) (18-20) is a serine/threonine protein kinase, which takes part in both intracellular and extracellular signals and functions as a master regulator of cell proliferation, survival, metabolism, and growth. mTOR is known to form two distinct protein complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). mTOR Complex 1 (mTORC1) is defined by its RAPTOR (21) (150 kDa) subunit, which is replaced by RICTOR in mTORC2 (22). mTORC1 comprises of mTOR, RAPTOR, and mLST8 as fundamental components and regulates cell growth, stimulating translation, and autophagy. Its activation is controlled by amino-acid levels, growth factors, hypoxia, and cellular nutrients, which results in the co-localization of RAPTOR – mTOR to endomembrane along with its activator GTPase RHEB. mTORC1 substrate includes ribosomal S6 kinases (S6K1), eIF4E-binding protein 1 (4EBP1), and ATG13 (23-24). mTORC2 responds to growth factor and consists of mTOR, RICTOR, mLST8, PRR5, and SIN1 as core subunits. mTORC2 regulates apoptosis, growth, cell cycle control, actin cytoskeleton polarization and the regulation of cell shape and mobility (22). RICTOR -mTOR complex modulates the phosphorylation of other kinases, such as S6 kinase (S6K), Protein Kinase C α (PKCα), and Akt. These kinases share a common hydrophobic motif (HM) phosphorylation site with S6K1 (23, 25).
DNA dependent protein kinase (DNA-PK) is one of the largest serine/threonine protein kinase (465 kDa) discovered in 1985, activated only in response to DSBs (Figure 2) (26). The DNA-PK recruitment and kinase activity was associated with a regulatory Ku heterodimer subunits (Ku70 and Ku80 subunits) (27) and large polypeptide catalytic subunit ( > 460 kDa) named as DNA-PK catalytic subunit (DNA-PKcs) (28). The Ku heterodimer is composed of the Ku70 (73 kDa) and Ku80 (86kDa) subunits, which together form a central cavity to preferentially bind dsDNA end (29). It is hypothesized that the Ku70/80 is the first NHEJ factor that binds to the DNA DSBs and then leads to subsequent recruitment of DNA-PKcs. Thereby, DNA-PKcs acts as a sensor for DNA DSBs and plays a major role in promoting non-homologous end joining (NHEJ) (30-31). NHEJ is a process where DNA ends are ligated together without a concrete request for sequence homology and are error-prone and mutagenic, in contrast with homologous recombination (HR). Further, DNA-PK has been implicated in phosphorylation of various substrates such as histone variant H2AX, autophosphorylation, replication-associated protein (RPA) (32). DNA-PK plays a governing role in the phosphorylation of H2AX on S139 (γH2AX) in response to DNA damage and leads to the activation of the Akt signal pathway. On the other hand GSK3β, a negative regulator of DNA-PK is phosphorylated on Ser9 residue, which prevents GSK3β from dephosphorylating γH2AX. Consequently, in the absence of ATM, DNA-PK directly phosphorylates H2AX and modulates the phosphorylation level of γH2AX through Akt/GSK3 beta signaling pathway (33). Furthermore, DNA-PK plays an important role in the transcriptional activation of the metabolic gene in response to feeding/insulin stimulation (34). Studies on the auto phosphorylation of DNA-PK demonstrated that there are 40 phosphorylation sites on DNA-PKcs identified to date. Auto phosphorylation of DNA-PKcs is very important for the efficient repair of DSBs in DNA. Mechanistic studies uncovered that unphosphorylated DNA-PKcs block DNA ends, and inhibits DNA ligation (35-36). DNA-PKcs auto phosphorylation brings a conformational change in the DNA–protein complex that releases the aligned DNA ends (37-38).
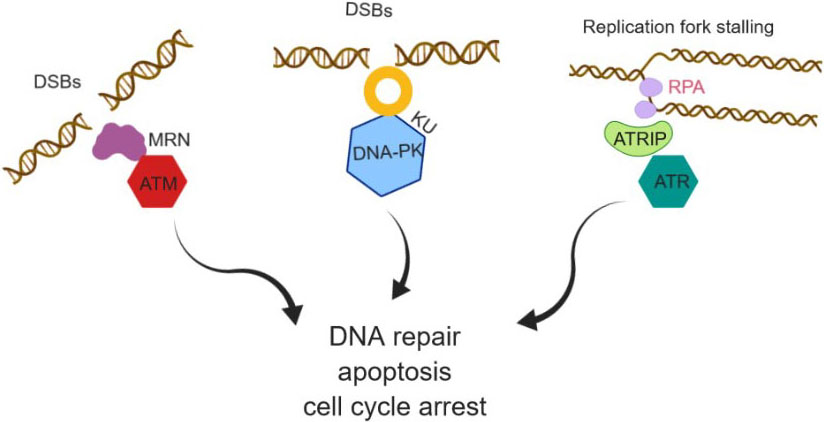 Figure 2
Figure 2DNA damage and response pathway activation, localization and partner proteins.
Replicating mammalian cell is constantly under exogenous and endogenous stress, which subsequently affect the integrity of the genome. To overcome this adverse effect and maintain the genomic stability, eukaryotic cells developed a highly organized and coordinated network of cellular events called a DNA damage response (DDR). ATM and ATR kinase are the first members of the PIKK family serine/threonine kinase senses the DNA damage and activate DDR pathway (Figure 2). DDR signaling pathway is initiated by the recognition of lesions in DNA structures by ATR and ATM kinases. ATR and ATM kinases interact with many other checkpoint proteins that co-localizes at the site of damage. For example, checkpoint kinase 1 (Chk1) and checkpoint kinase 2 (Chk2) activity are mainly modulated by ATR and ATM kinase. This results in the S phase and G2 phase arrest during cell cycle (39-41). ATM and ATR to respond to different types of DNA damage (Figure 2). DSBs (ionizing radiation) activate ATM, which can phosphorylate BRCA1, and Chk2 kinase leading to activation of p53 followed by G1/S arrest or apoptosis. The localization of ATM kinase to DSBs is chiefly regulated by the Mre11-Rad50-Nbs1 (MRN) complex (42). Studies have shown that MRN is one of the first sensors to be recruited to DSBs (43). The unwinding of DNA ends by MRN complex stimulates the activity of dimeric/oligomeric ATM kinase (42). ATM kinase was discovered in ataxia-telangiectasia (AT) patients, characterized by dilated blood vessels and progressive neurological decline. AT results in gait abnormality and a lack of involuntary movement. AT patients are oversensitive to IR radiation and are defective in DSB repair just as the G1/S, intra-S, and G2/M checkpoints. The defective checkpoints in AT cells are mainly due to the lack of ATM kinase (44). ATM was orthologous to Saccharomyces cerevisiae Tel1 (45-46), a protein involved in controlling telomere length, DNA repair, and cell-cycle checkpoint control. ATM additionally phosphorylates the Ser139 in the C-terminal tail of histone variant H2AX in response to DSBs, bringing about discrete γ-H2AX foci at the DNA damage sites (47). H2AX phosphorylation is a general cellular response to processes involving DSB intermediates. p53 is another substrate of ATM which gets phosphorylated on Ser15 in response to DSBs. Phosphorylation of p53 leads to the degradation of Mdm2 and subsequent G2/M cell cycle arrest (48).
ATR activation is associated with UV light irradiation and replication fork stalling. When the DNA damaging agents cause DNA polymerase to stall during replication and helicases continues to unwind the DNA, then it leads to the generation of a stretch of single-strand DNA. The ssDNA is recognized by Replication protein A (RPA) and recruits the ATRIP/ATR complex (49). An 85 kDa, ATR-interacting protein (ATRIP) acts together with ATR on its N-terminus in the recognition and binding to RPA. Further, the ssDNA-RPA complex stimulates the loading of RAD9–HUS1–RAD1 (9–1–1) heterotrimer complex onto the DNA ends (50). Sequentially, the 9-1-1 complex assembles TopBP1 to activate ATR (51). Activated ATR phosphorylates the Ser317 and Ser345 on Chk1 kinase leading to S and G2 arrest (39-40). Consequently, ATR also phosphorylates BRCA1 (breast cancer suppressor protein 1), the MCM2-7, replication factor C complex, and RPA (52-53). Phosphorylation of these substrates is important for inhibition of replication, recovery of replication and activation of NHEJ and HR DNA repair pathway (54).
hSMG1 is a 3661 amino acid long, 410 kDa protein that belongs to the PIKK family of serine/threonine kinase. It is reported to form a surveillance complex with hUPF1, hUPF2, and hUPF3 and it majorly phosphorylates the C-terminal of hUPF1 at SQ motif (7). hSMG1 is functionally relatable to ATM kinase and is activated during genotoxic stressful conditions like exposure to UV radiations or IR radiations (55). SMG1 complex consists of two subunits, such as SMG8 and SMG9. SMG8 recruits SMG-1 to the mRNA surveillance complex and inactivation of SMG8 induces accumulation of Upf1-eRF1-eRF3 complex on mRNP (56). SMG1 and UPf1 transiently forms SURF i.e. SMG-1-Upf1-eRF1-eRF3 complex. Which facilitates the phosphorylation of hUPF1 at serine residue in SQ motif, and plays a critical role in nonsense-mediated mRNA decay (NMD) (57-58).
All PIKK family kinases (except TRRAP) have intrinsic serine/threonine kinase activity. The arrangement of catalytic residues in the C-terminal kinase domain (KD) is significantly similar in PIKKs. Indeed the kinase domains (KDs) of PIKKs share considerable sequence homology with PI3Ks. Owing to this sequence similarity, the reported PI3Ks high-resolution structures serve as the basis for understanding PIKK function. Figure 3 highlights the conserved residues in the kinase domain of PIKKs family. ATR, ATM and mTOR shares sequence identity of 27.1%, 27.8% and 27.2% respectively with DNA-Pkcs. hSMG, share sequence identity of 24.1% with DNA-Pkcs. Residues like Glu, Trp, Pro and, Lys are highly conserved and support ATP association with active enzyme of PIKK family kinases. Glu (i.e. D2475 in ATR and D2338 in mTOR) and His residue (i.e. His2477 in ATR and 2340 in mTOR) plays a key role in orienting and activating hydroxyl group of substrate for the nucleophilic attack, and electrostatic stabilization of the transition state. Most of the currently available type I kinase inhibitors target the ATP binding pocket (59). On the other hand, the ATP-binding pocket is largely conserved in human protein kinases and suffers from low kinase selectivity. Type I inhibitors require intense medicinal chemistry optimization program to modulate the cross selectivity. Further, to target the inactive or active kinases, type I½and type II ATP binders have been explored that bind to the DFG (Asp-Phe-Gly) in or out conformation motif in kinases. DFG motif is highly conserved among few protein kinases, targeting the DFG motif with small molecules has been widely studied to control the selectivity (60-62). In PIKKs the DFG motif is only conserved in mTOR and DNA-PKcs. In ATR, ATM and hSMG the DFG motif is replaced by DLG, DFN and DYN motifs respectively. Thereby, giving an excellent opportunity for the development of highly selective kinase inhibitors for PIKKs.
 Figure 3
Figure 3Multiple sequence alignment of PIKK family kinases (ATR, ATM, DNA-PKcs, and hSMG) kinase domain. Conserved residues are highlighted in red color.
Wortmannin is a fungal metabolite and the first PIKK inhibitors identified in the year 1996 (Figure 3). It is a broad-spectrum inhibitor that comparatively inhibits all the members of PI3K-related proteins such as ATR, mTOR, DNA-PKCs, and ATM (63). It is an irreversible inhibitor of PI3K kinase (IC50 =1-3 nM) binds covalently to a ε-amino group of lysine in the catalytic site. Surprisingly, wortmannin is also an irreversible of ATR (IC50 =1.8 µM), mTOR (IC50 =200 nM), DNA-PKCs (IC50 =1-3 nM), hSMG (IC50 =60 nM) and ATM (IC50 =100-150 nM) (63). Wortmannin enhanced the cytotoxic effect in response to DNA damage in human tumor cell lines such as HeLa, SW480, and MCF-7 (64). Although wortmannin showed potent cellular activity but failed as a therapeutic agent due to its toxicity and stability in the biological system.
Caffeine is a methylxanthine derivative found in tea, coffee, cola, and other products (Figure 4). It is a pan inhibitor of the PIKK family. Siu-Long Yao, demonstrated the anti-cancer activity of caffeine by selective sensitization of p53–deficient tumor cells to irradiation and induced apoptosis, suggesting that the radio-sensitizing is mainly due to inhibition of ATM/ATR kinase (65) and its influence on G1-S, and G2-M cell cycle arrest. Further, studies by Alessandra Blasina et al., highlighted the inhibition of Chk2 kinase by caffeine in radiation sensitized cells in vitro and that ATM kinase activity was directly inhibited by caffeine in vitro (66). However, lack of selectivity and high toxicity of caffeine in the in vivo studies have prevented their further biological evaluation.
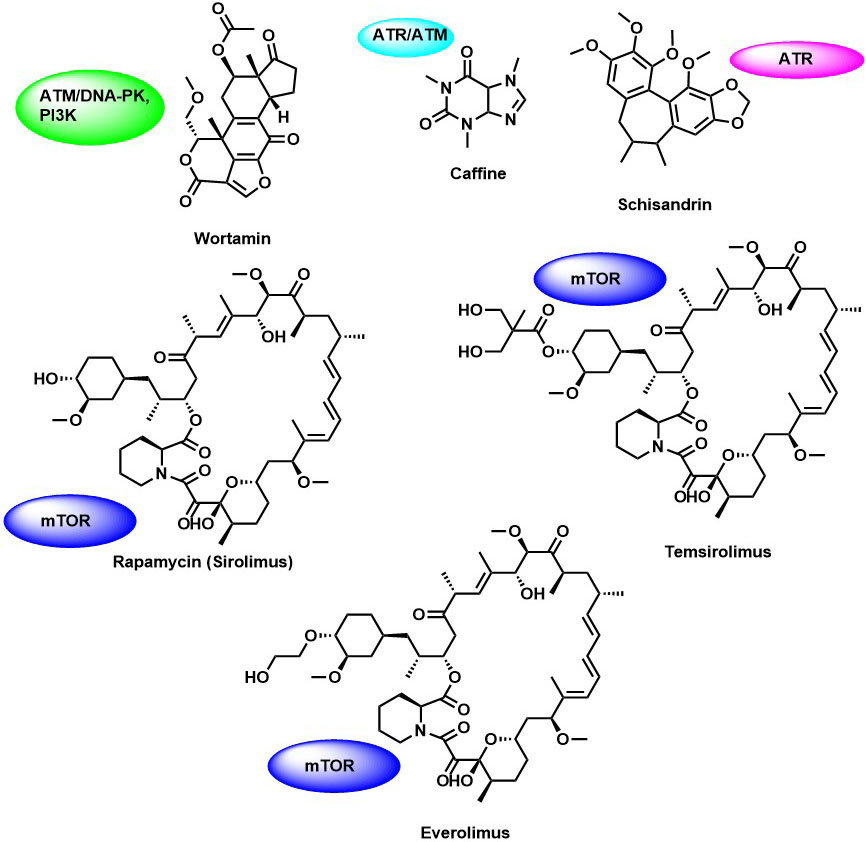 Figure 4
Figure 4Naturally derived PIKK family kinase inhibitors.
Schisandrin B (SchB), an active ingredient of Fructus schisandrae, has been identified as the first ATR kinase inhibitor by Nishida et al., (Figure 4). In their study, the treatment of A549 adenocarcinoma cells with Schisandrin B (SchB) after UV exposure resulted in the reduced phosphorylation level of ATR. Sch B reduced the kinase catalytic activity of immunoaffinity-purified ATR in a dose-dependent manner with IC50: 7.25 µM (67). Further, Sch B inhibited the phosphorylation of p53 and Chk1 kinases abrogated the intra S-phase and G2/M cell cycle checkpoint and induced cytotoxicity in combination with UV radiation in lung cancer cells (68).
Rapamycin is an antifungal macrolide metabolite produced by Streptomyces hygroscopicus found in the soil of Easter -island. It was reported that the compound possesses immunosuppressive and antiproliferative properties in mammalian cells (69). In all human tumors, many oncogenic pathways are linked to hyperactivation of mTORC1 function. Thereby, mTOR acts as an attractive drug target for cancer therapy. In a mammalian cell, Rapamycin acts through the allosteric mechanism by binding to the FKBP12 and interfere with FRB domain of mTOR and differentially inhibits mTORC1 kinases activity. Thereby it inhibits the phosphorylation of mTOR substrate S6Ks and 4E-BP1 by binding to mTORC1, but not mTORC2 (70). However, how the interaction with the FRB leads to the inhibition of mTOR kinase activity remains remain unclear. The cryo-electron microscopy study from Aylett et al.,(71) showed that rapamycin serves as a gatekeeper of the active substrate-binding site by interacting with the hydrophobic site in FKBP12 domain, and destabilizes the mTORC1 dimer and its activity. mTORC1 signaling mainly regulates cell growth, stimulating translation, and autophagy in response to hypoxia, cell nutrients, and amino acid levels. However, due to the poor solubility and pharmacokinetic profile rapamycin has limited its use for clinical studies. To overcome this problem rapamycin analogs (rapalogs) temsirolimus (72) and everolimus (73) have been developed with improved water solubility than Rapamycin. In 2007 and 2009, the Food and Drug Administration (FDA) approved these rapalogs, for the treatment of advanced renal cancer carcinoma (RCC) (Figure 4). Additionally, everolimus was used to treat patients with PNET, advanced non-small cell lung cancer, and hepatocellular carcinoma (NSCLC).
In 1993, Vlahst et al., first reported the2-(4-morpholinyl)-8- phenyl-4H-l-benzopyran-4-one (also known as LY2940002) as a specific inhibitor of Phosphatidylinositol 3-Kinase with IC50 = 0.43 µg/d 1.40 µM (74). On the basis of similarities between the catalytic subunit of Phosphatidylinositol 3-Kinase and DNA-PK, Izzard et al., and others carried out the inhibition studies of LY2940002 on DNA-PK and demonstrated that LY2940002 was also able to sensitize cells to the effects of IR (IC50 = 6 µM) (75). The crystal structure analysis of LY2940002 with porcine PI3K (p110γ) revealed that morpholine oxygen is required to form hydrogen bonding with backbone Val882 in the ATP binding pocket also, the ATP binding pocket of PI3K is very similar to that of the PIKK family kinases. These results led to the investigation of LY2940002 derivatives as PIKK family kinase inhibitors. In 2005, a diverse range of Benzopyran-2-ones, Benzopyran-4-one, and pyrimidoisoquinolin-4-one derivatives (Figure 5: A-I) were investigated for their inhibitory activity against the DNA-PK (IC50 values ranging from 0.19 to >10 μM) (76). In this study, all the fused chromones systems were able to inhibit PIKK family kinases in the presence of PI3K isoforms and proved to have a superior activity than LY2940002. NU7026 was identified as a potent inhibitor of DNA-PK with IC50 = 1.26 µM. Further, they also investigated the isosteric pyrimido(2,1-a)isoquinolin-4-one, which showed comparable activity with that of NU7026. Interestingly, structure-activity relationship studies on 2-position of pharmacophore template LY2940002 and NU7026 (74, 77) for DNA-PK inhibition has led to the identification of excellent DNA-PK inhibitor compound I (NU7163). Compound I showed ATP competitive inhibition of DNA-PK with an IC50 value of 0.19 μM. Effect of 2- methyl substitution on the morpholine ring was not detrimental due to the unavailability of the crystal structure of DNA-PK, but it greatly improved the activity against DNA-PK. An in-vitro study of NU7026 and I (5 μM and 10 μM) on HeLa cell lines showed an enhancement of cytotoxicity in combination with radiation therapy (76). In another study, Ian et al., explore the various functional groups at 6, 7 and 8th position of NU7026 and discovered two compounds, 2-N-morpholino-8-dibenzofuranyl-chromen-4-one (NU7427) and the 2-N-morpholino-8-dibenzothiophenyl-chromen-4-one NU7441) with excellent DNA-PK inhibition (IC50 DNA-PKcs =40 and 13 nM, respectively). In which substitution at the 8th position of Benzopyran-4-one markedly improved the inhibitor activity, and substitution at 6 th and 7th led to the loss of activity.
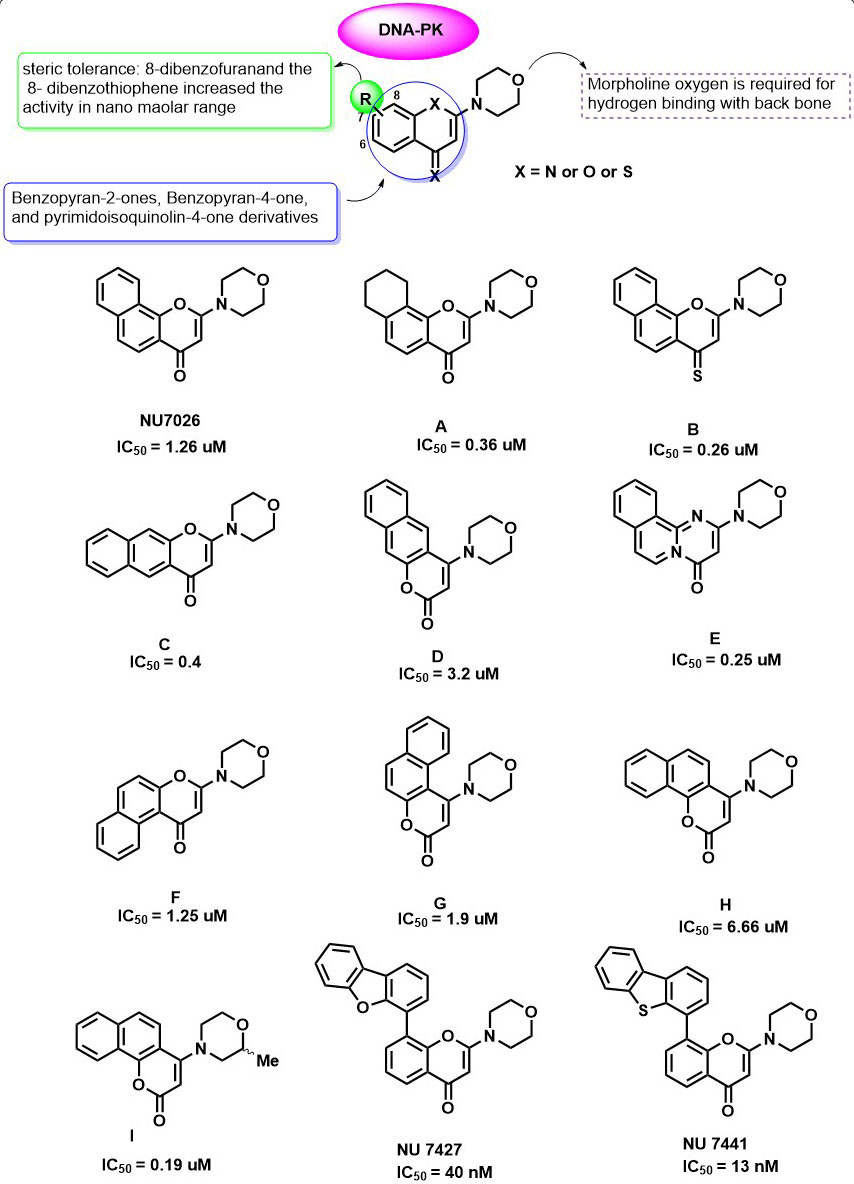 Figure 5
Figure 5Benzopyran-2-ones, Benzopyran-4-one, and pyrimidoisoquinolin-4-one derivatives as DNA-PK inhibitors.
Through screening of a small library of molecules developed for PIKK family kinase, Hickson et al., identified a novel 2-morpholin-4-yl-6-thianthren-1-yl-pyran-4-one (KU 55933) as ATP competitive inhibitor for ATM with IC50 of 13 nmol/L and a Ki of 2.2 nmol/L (78) (Figure 5). It displayed 100-fold selectivity over DNA-PK and other PIKK family members. KU-55933 successfully inhibited the IR dependent phosphorylation of various ATM substrates such as p53, γH2AX, NBS1, and SMC1. Also, it is known that KU-55933 suppresses cell proliferation and induces apoptosis by inhibiting the phosphorylation of Akt in response to insulin-like growth factor (79). Exposure of cells to KU-55933 along with chemotherapeutic agents (doxorubicin, etoposide, and camptothecin)/ IR radiation resulted in significantly sensitization and cytotoxicity.Further, optimization of KU-55933 has led to the discovery of novel thianthrenpyran-4-one derivative (KU-60019) with improved selectivity (Figure 6). The second-generation ATM inhibitor KU-60019 showed 10 times more potency than its predecessor KU-55933 with little to no nonspecific target effects at 1 µmol/L against a panel of 229 protein kinases (80).
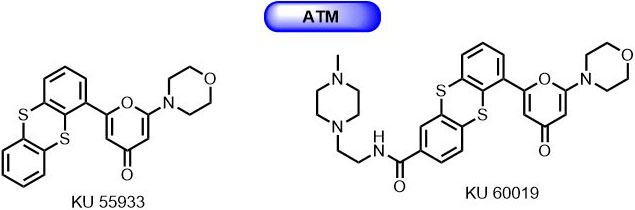 Figure 6
Figure 6Thiaanthrenpyran-4-one derivatives as ATM inhibitors.
It is known that quinolines have a broad spectrum of biological activities such as antimalarial, analgesic activity, anti-inflammatory, antineoplastic, antifungal, anti-bacterial, antiviral, antiprotozoal, cardiovascular, CNS effects, hypoglycemic,) and others. Quinolines nucleus has great significance in cancer therapy, they are “privileged” ATP-site binders to the protein kinases in the signaling pathway. Many of the protein kinases are inhibited by several quinoline derivatives like, MAPKS, PKC, CK2, EGFR, IGF, PDGFR, CSF, and TGFβ. In 2010, Liu et al., screened a chemical library to identify selective mTOR kinase inhibitors. Their screening led to the discovery of quinoline scaffold 6-(pyridin-3-yl)-N-(3-(trifluoromethyl)phenyl)quinolin-4-amine with moderate inhibition against mTOR (mTORC1 IC50 = 5 μM). It was further optimized to improve the selectivity and the cellular potency to mTOR and managed to obtain Torin1 compound having cellular IC50 (mTOR) =2 nM; IC50 PI3K =1800 nM (81). In another study, they further optimized Torin1 to improve its selectivity, solubility, metabolic stability and led to the discovery of Torin2 (Figure 7). Torin2, 9-(6-aminopyridin-3-yl)-1- (trifluoromethyl)phenyl)benzo(h)(1,6)naphthyridin-2(1H)-one, derived from Torin1, has better half-life and bioavailability. It has been classified as second-generation ATP-competitive mTOR inhibitor with IC50 of 0.25 nM in the HCT116 cell line, which also showed inhibition of ATR and ATM kinase in kinativ scanning (82). Torin2 exhibits strong cellular activity against ATR with EC50 of 35 nmoles/L (HCT-116 cell lines) (82-83). Structural analysis of Torin2 and mTOR complex by Haijuan Yan et al.,(4) revealed the important residues and functional group involved in the stabilization of complex. In mTOR-Torin2 complex, the tricyclic benzonaphthyridinone ring occupies the adenine site of ATP and forms a hydrogen bond with the ‘hinge’ region. In addition, benzonaphthyridinone ring forms stacking interaction with the indole group of Trp 2239 (Figure 8).
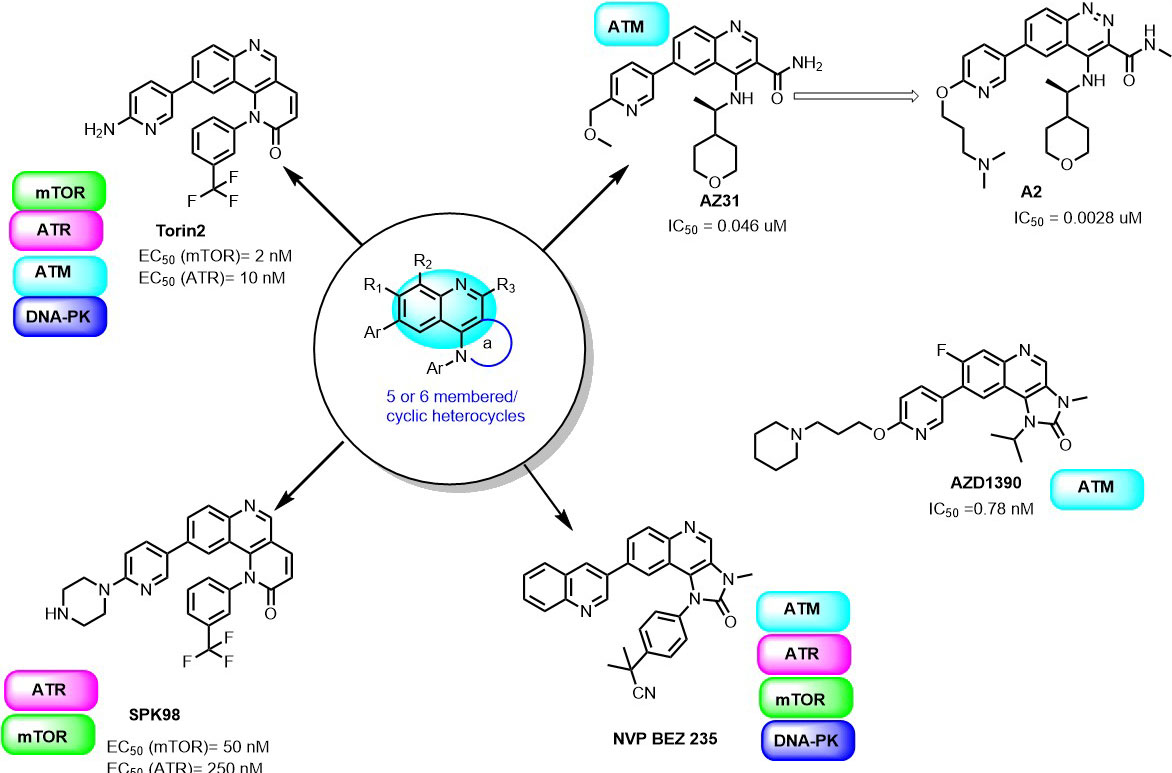 Figure 7
Figure 7Quinoline derivatives as PIKK inhibitors.
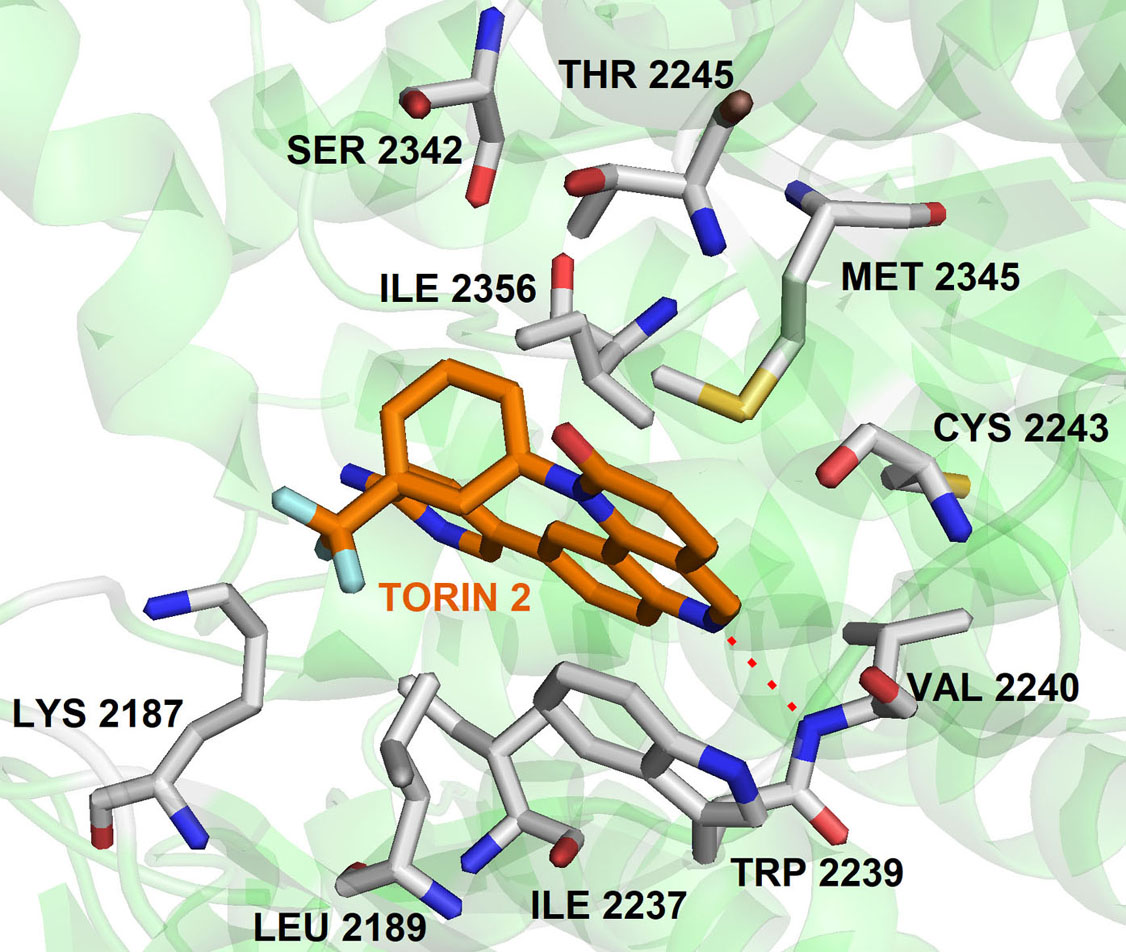 Figure 8
Figure 8The binding pose of compound Torin2 with mTOR. The protein structure was highlighted as the green color and interacting residues are represented as tube model (grey color). Torin2 was showed as a tube model (orange colour) (Adapted with permission from PDB ID: 4JSX).
Further, the examination revealed that the Torin2 trifluoromethyl group stabilizes Torin2 – mTOR complex by the spacing between Ile2163, Pro2169 and Leu2185 residues of N-lobe. Interestingly, the amino group of Torin2 extends to the ‘inner hydrophobic pocket’, an area at the back of the cleft that many PIKK kinase inhibitors contact. However, it failed to form hydrogen bonding with Asp2195, Asp2357 and Tyr-2225 residues giving an opportunity to develop next-generation Torin2 analogs to mimic this interaction (4). In our recent study, we explored Torin2 analogs as mTOR/ATR kinase inhibitor and led to the development of SPK98 with promising selectivity towards mTOR/ATR in HCT116 cells and enhanced the sensitization in combination with UV radiation (EC50 (mTOR) = 50 nM; . EC50 (ATR) = 250 nM) (84). NVP BEZ 235 is an imidazo(4,5-c)quinoline derivative identified as a dual ATP-competitive PI3K and mTOR inhibitor by Maira et.al, (85). NVP-BEZ235 successfully inhibited the dysfunctional activation of the PI3K pathway in cellular and in vivo studies. The docking analysis of NVP-BEZ 235 into the catalytic site of PI3K (homolog of ATM) showed that quinoline ring forms hydrogen bonding with Val 851 in the hinge region of ATP binding site and further stabilized by hydrogen bonding interaction from cyano group and quinoline nitrogen ring with the Asp933 and Ser774. NVP-BEZ235 demonstrated a measurably significant antitumor activity against the PTEN-null, U87MG, and PC3M tumor xenografts (85). In another study, Toledo et al., demonstrated the potency of NVP-BEZ 235 against ATR and ATM kinase in p53 deficient cancer cells when sensitized with UV/IR radiation (86-87). AZD1390 is another CNS penetrating quinoline substituted ATM inhibitor developed from AZD 1056. It is a potent ATM inhibitor with IC50 0.78 nM, currently undergoing pre-clinical trials (88). In 2016, 3-Quinoline carboxamide derivative developed by Astra Zeneca (89) showed high selectivity towards the ATM kinase in the presence of other PIKK kinases. In their structural elucidation analysis, they reported the crystal structure of compound 3QH (IC50 = 0.049 µM) with PI3Kγ, a known homology of ATM (PDB ID: 5G55).
In the crystal structure, the 3-Quinoline carboxamide derivative nicely fits into ATP binding site and stabilized by hydrogen bonding interaction between quinoline nitrogen and Val882 residue located in the backbone of ATP binding site (Figure 9). It demonstrates the selectivity of the quinoline ring towards the PIKK family kinases. This investigation gave the opportunity to perform SAR studies of compound 3QH on ATM kinase activity and led to the discovery of AZ31 with excellent ATM kinase selectivity (IC50= 0.046 µM) and better rodent oral ADME properties (89). In a recent study, they also explored 3-cinnoline carboxamide in place of 3-quinoline carboxamide derivatives as an ATM kinase inhibitor to improve the ATM kinase selectivity. They identified compound A2, a very selective ATM inhibitor (ATM cell IC50 0.0028 μM) with favorable physicochemical and pharmacokinetic properties in their in vivo and in vitro studies (Figure 7) (90). Additionally, compound A2 in combination with irinotecan showed tumor regression in the SW620 colorectal tumor xenograft model, which showed superior inhibition in comparison with tumors treated with irinotecan alone (90).
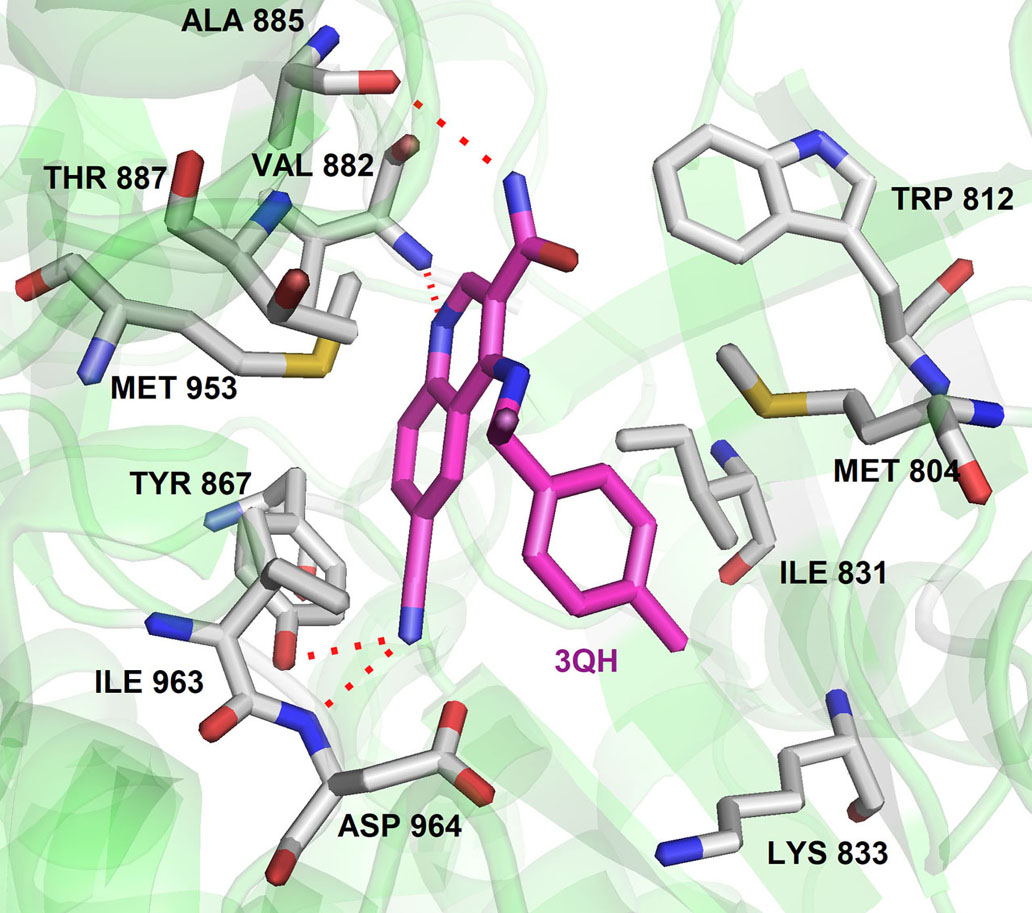 Figure 9
Figure 9The binding pose of compound 3QH with ATM homology (PI3Kγ). Protein structure highlighted as the green color and interacting residues are represented as tube model (grey color). 3QH was highlighted as tube model (magenta color) (Adapted with permission from PDB ID : 5G55) and hydrogen bonding was highlighted as dotted lines.
In 2010, a series of 2-aminopyrazine derivatives were studied against ATR kinase by the researchers at vertex pharmaceutical company (Figure 10). They filed several patents proposing 2-aminopyrazine as specific and selective ATP competitive ATR kinase inhibitors. In their study, they carried out detailed SAR studies of 2-Aminopyrazine derivatives, including their ATR enzymatic and cellular inhibition studies against HCT116 cell line in the presence of DNA damaging agents such as cisplatin. Most of the synthesized compounds were able to inhibit the full-length ATR kinase with Ki<100 nM. The SAR optimization of 2-amino pyrazine scaffold led to the identification of VE-821 compound with IC50 = 26 nM (Ki =6 nM) against ATR kinase (41). In addition, Remko et al., demonstrated that VE-821 increases the sensitivity of pancreatic cancer cells (PSN-1, MiapaCa-2, and primary pancM) to radiation and gemcitabine-induced phosphorylation of Chk1 (91). VE-821 selectively inhibited ATR kinase in response to hypoxia and induced G2/M arrest in the cancer cell (92).
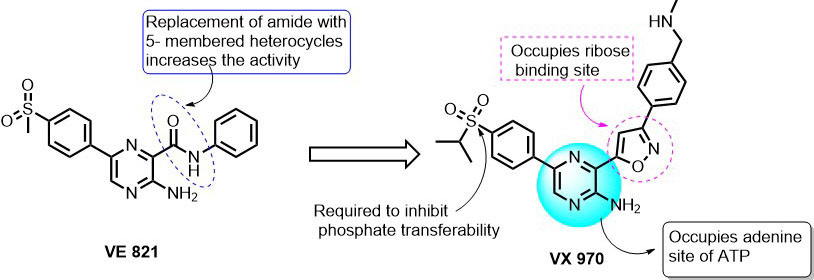 Figure 10
Figure 10Pyrazine amines as ATR inhibitors.
Further optimization of VE-821 resulted in a potent and selective ATP-competitive inhibitor i.e. VE-822 (VX-970) with an IC50 of 19 nM in HT29 cells. From the SAR studies, it was understood that the replacement of the amide group in 3-position of pyrazine with 5-member heterocyclic members like isoxazole, triazole, oxadiazole, pyrazole, dihydroisoxazole increased the inhibition towards ATR. Replacement with alkene, alkynes, ketone, and ethers has led to its inhibition at higher concentrations. On the other side, the substitution of CH3 in the sulfonyl group with various propyl, isopropyl, piperidine-3-yl, and piperidin-4-yl linkers showed great inhibitory effect along with the improved pharmacokinetic profile of 2-amino pyrazine series. The docking of VX-970 into the catalytic site of ATR revealed that pyrazin-2-amine group occupies the position of adenine (purine base) in ATP binding site. The 5-membered oxazole ring extends to the ribose binding region of ATP. Interestingly, the sulfonyl group of VX-970 occupies the position of the γ-phosphate group of ATP and is expected to prevent the γ-phosphate group transfer from ATP to substrate (93). In another study, published by Vertex Pharmaceuticals, VX-970 (VE-822) showed selective chemo-sensitization to a panel of non-small cell lung cancer cell lines, over normal cells, to multiple DNA damaging drugs, namely cisplatin, gemcitabine, and etoposide (94). In vivo investigation of VX-970 + cisplatin over a wide-range of the patient-derived primary lung, xenograft demonstrated that VX970 blocked ATR activity in tumors and dramatically enhanced the efficacy of cisplatin (94). These interesting results have led to the entering of VX-970 into phase I clinical trials (95). Preliminary clinical data showed that VX-970 can be used as single therapy/combinatorial therapy (in combination with Carboplatin) with early evidence of pharmacodynamics and antitumor activity. VX-970 is now being tested in phase 2 combinatorial/ monotherapy in patients with DNA repair defects (95).
NU6027 (2,6-diamino-4-cyclohexyl-methyloxy-5-nitroso-pyrimidine) is a first pyrimidine derivative studied against PIKK family member. It effectively inhibits the ATR cellular activity (IC50=6.7 μM) and improved cytotoxicity of hydroxyurea and cisplatin in A2780 cells (96). NU6027 inhibited G2/M cell cycle arrest and RAD51 focus formation following DNA damage.
Thereby, it enhanced the cytotoxicity of the major classes of DNA-damaging agents for effective cancer therapy. Importantly, NU6027 was found to be synthetically lethal when DNA single-strand break repair was impaired either through poly (ADP-ribose) polymerase (PARP) inhibition or defects in XRCC1. Hence NU6027 can serve as the novel lead for further drug development of pyrimidine derivatives against ATR (Figure 11). In another study, Gopalswamy et al. screened a panel of kinase library and discovered compound 1 as a hSMG1 inhibitor in sub-micromolar range ( IC50 = 0.45µM) (97). They further optimized compound 1 to reduce its activity against CDK1 by docking into the active site of hSMG1 homologous protein PI3Kγ. In the active site (PDB ID : 4FUL), compound 1 showed U shape conformation and was stabilized by hydrogen bonding interaction between the nitrogen of pyrimidine ring and the Val882 positioned in the backbone of ATP binding pocket. In addition, amino group and the carbonyl oxygen of compound1 form multiple hydrogen-bonding interactions with Val882 and Lys833 (Figure 12).
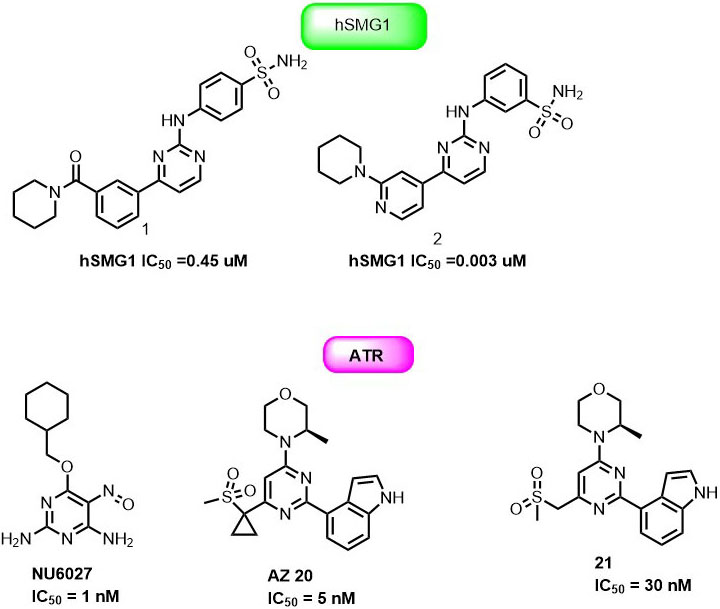 Figure 11
Figure 11Pyrimidine scaffolds as ATR and hSMG1 inhibitors.
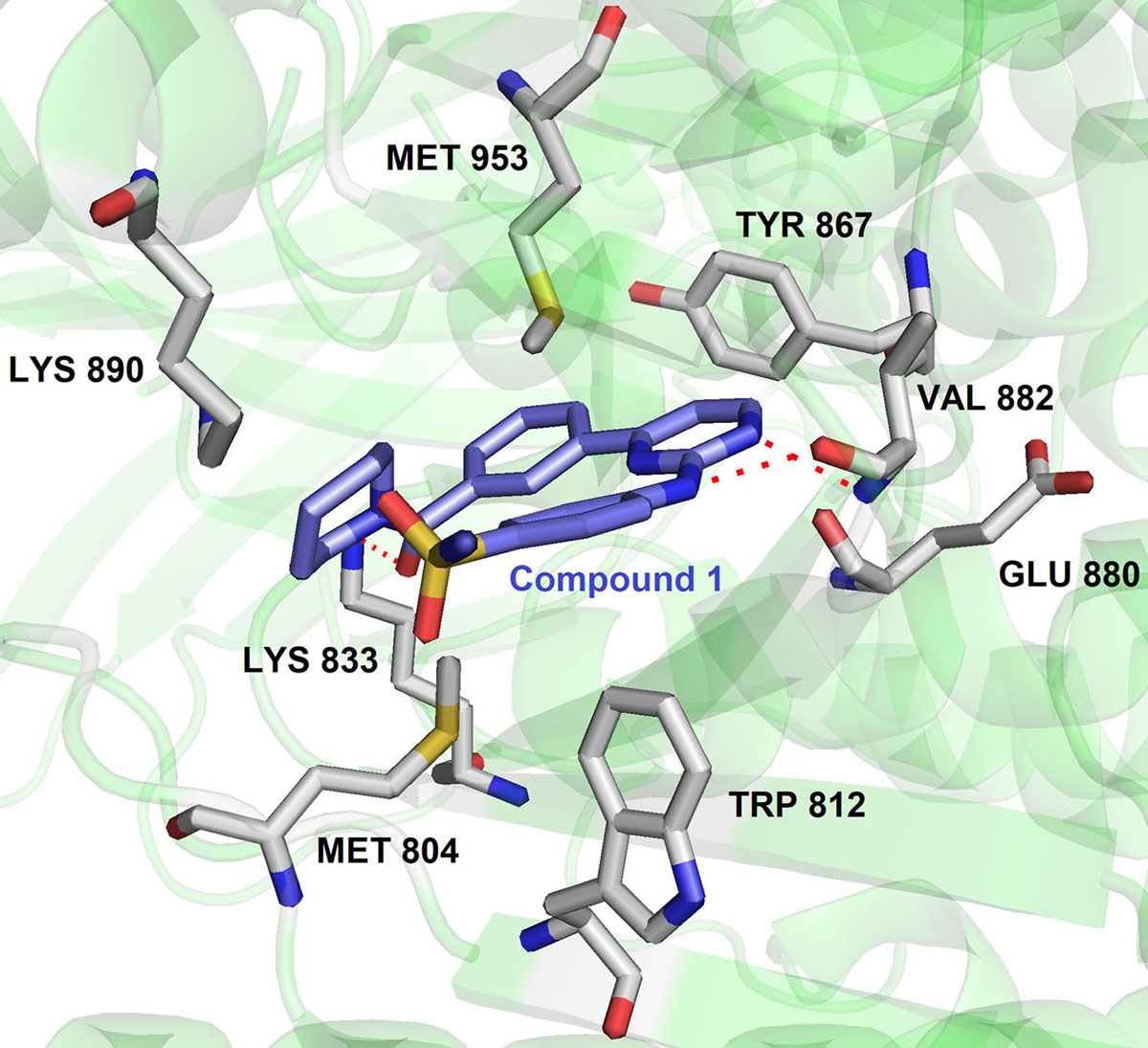 Figure 12
Figure 12Binding pose of compound 1 with hSMG. Protein structure highlighted as the green color and interacting residues are represented as tube model (grey color). The inhibitor was highlighted as a tube model (blue color) (Adapted with permission from PDB ID: 4FUL).
Remarkably, the p-sulphonamide group fails to make interaction with active residues in hSMG1. Further optimization of 1 led to the identification of 2 with excellent hSMG1 selectivity (IC50 =0.003µM) over CDK1 kinase. Changing the sulfonamide substitution from para to meta position led to the formation of hydrogen bonding interaction with the backbones of Gly884 and Ala885. This additional hydrogen bonding could be reasoned with excellent selectivity of compound 2 towards hSMG1 in presence of other kinases. These key features make pyrimidine derivatives a novel chemical scaffold for selective targeting of hSMG-1 binding site in order to improve kinome selectivity of other chemical scaffolds targeting hSMG1. A number of patent applications from AstraZeneca have been filed on ATR kinase. NU6027, AZ20 and 21 (Figure 11) were claimed as single agents or combination with other anticancer drugs for cancer therapy. AstraZeneca compounds exhibited IC50 = 3.97μg/ml in HT29 colon cancer cells but suffered from inhibition of CYP450 enzymes (98-99).
For the last two decades, there has been remarkable progress in the development of PIKK family kinase inhibitors. Several research groups have made enormous efforts in screening natural and synthetic libraries to targets these kinases. mTOR kinase has been targeted for decades due to its central role in regulating various cell processes in the cancer cell. Inhibitors like Temsirolimus, Everlimus, AZD8055, AZD2014, OSI-027, and INK 128/MLN0128 have entered into clinical trials. Rapalog inhibitors like Temsirolimus, Everlimus has been approved by FDA for renal carcinoma. Activation of ATR, ATM, and DNA-Pkcs under oncogenic stress highlights their role in DDR pathway of cancer cell. The development of specific inhibitors for ATR, ATM, and DNA-Pkcs has made prominent progress. VX970, NVP-BEZ235, Torin2, AZ20, and AZ31 are the diverse scaffolds that have entered the pre-clinical for the treatment of various human cancers (Table 1). VX970, AZ20, and AZ31 have selectivity sensitized cancer cell in the presence of DNA damaging agents such as antimetabolites, alkylating agents, topoisomerase inhibitors and platinating agents. VX970 an ATP- competitive ATR kinase inhibitor has made phase into I/II trials as part of mono or combinatorial therapy for the treatment of patients with solid tumors. NVP BEZ 235 is a dual ATP-competitive PI3K and mTOR inhibitor which effectively blocks the activation of the PI3K pathway in cellular and in vivo studies. Further, it displayed significant antitumor activity against the PTEN-null, U87MG and PC3M tumor xenografts. NVP-BEZ 235 reduced the ATR and ATM kinase activity in p53 deficient cancer cells when sensitized with UV/IR radiation (86-87). Torin2 and its analogs showed remarkable selectivity towards mTOR kinase in the presence of PIKK family kinases. 3- quinoline carboxamide and 3-cinnoline carboxamide derivatives emerged as ATM kinase inhibitors with improved metabolic stability. The role of hSMG in RNA surveillance pathways has made it as an attractive target in cancer therapy. hSMG directly phosphorylates the hUPF1/SMG-2 in the evolutionally conserved mRNA surveillance complex. The development of potential hSMG1 inhibitors for carcinomas are currently underway.
| Drug | Drug targets | Phase I or II Clinical trials | Ref |
|---|---|---|---|
| VE 821 | ATR | (100- 103) | |
| VX 970 | ATR | NCT02157792, |
(94), (101), (104), (105) |
| AZ20 | ATR | (106) | |
| AZD6738 | ATR | NCT03328273 |
(107-108) |
| Torin2 | ATR, ATM, mTOR, DNA-PKcs | (82-83) | |
| NU6027 | ATR, DNA-PKcs | (96) | |
| NVP-BEZ235 | ATR, ATM, mTOR, DNA-PKcs | (109) | |
| AZ31 | ATM | (110) | |
| AZD1390 | ATM | NCT03423628 | (88) |
| Compound A2 (Cinnoline carboxamide) | ATM | (90) | |
| CCI 779 (Temsirolimus) | mTOR | NCT00003712 |
(111-112) |
| RAD0001(Everlimus) | mTOR | NCT01419639 |
(113-115) |
| AZD8055 | mTOR | NCT01316809 |
(116-118) |
| AZD2014 | mTOR | NCT 03071874 |
(117) |
| OSI-027 | mTOR | NCT00698243 | (119-120) |
| INK 128/MLN0128 | mTOR | NCT01351380 | (121-122) |
| Compound 1 (Pyramidine derivative) | hSMG | (97) |
There have been several PIKK family inhibitors reported between micromolar to the nanomolar range, their strategic synthesis, and biological data gave an insight to establish safety, better pharmacokinetic and pharmacodynamics profile during early-stage drug development process of PIKK members.
Due to the unavailability of the high-resolution crystal structure of these atypical kinases, there has been limited/stagnant progress in the field of drug development by structure-guided drug design. Although recent advances in cryo-EM helped the scientist to study the structure and domain architecture of large PIKKs protein (2549 to 4128 amino acids), the kinase domain in PIKK family kinase is highly conserved and targeting the ATP binding site of these kinases exhibit cross-selectivity. The development of irreversible inhibitors and DFG motif (in or out) binders can overcome the problem of low selectivity. Another hurdle in the drug development process of PIKK family inhibitors is lack of fully active PIKK family kinases, their activation requires several other protein factors to activate PIKK family kinases. This has limited the screening of library of chemical compounds against PIKK family kinase.
These aspects have been the main challenge to develop next-generation PIKK inhibitors for cancer therapy. We can expect better inhibitors in the future only if we confront these challenges and work towards the betterment of medicinal chemistry optimization strategy keeping in mind the important structural aspect that plays a very important role in drug design.
The authors acknowledge DRDO, Ramanujan Fellowship and SERB for generous funding to SK and IIT Gandhinagar for overseas fellowship to AS. We also thank FIST grant for the kind support to establish single crystal X ray facility at IIT Gandhinagar.
PIKKs
Phosphatidylinositol-3 kinase-related kinases
Phosphatidylinositol 3-kinase
Ataxia telangiectasia mutated kinase
ATM- and Rad3-related kinase
DNA dependent protein kinase catalytic subunit
mammalian target of rapamycin
suppressor with morphological effect on genitalia family member
Transformation/transcription-associated protein
FRAP ATM TRRAP
FKBP- rapamycin binding
ATR-interacting protein
Topoisomerase binding protein I
eIF4E-binding protein 1
ribosomal S6 kinases
Protein Kinase C α
hydrophobic motif
DNA doble strand breaks
DNA single strand breaks
replication-associated protein A
DNA damage and response
Mre11-Rad50-Nbs1
checkpoint kinase 1
checkpoint kinase 2
Ultraviolet
Ionizing radiation
breast cancer suppressor protein 1
nonsense-mediated mRNA decay
Kinase domain
Adenosine triphosphate
Schisandrin B
poly (ADP-ribose) polymerase
Cyclin dependent kinase 1