



Frontiers in Bioscience-Landmark (FBL) is published by IMR Press from Volume 26 Issue 5 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.

1 Amity Institute of Biotechnology, Amity University Uttar Pradesh, Noida, Uttar Pradesh, India
Abstract
Human kinases represent a large family of enzymes with their primary function being the phosphorylation of various biomolecules. Kinases along with G-Protein Coupled Receptors (GPCRs) represent two of the most common protein targets in drug discovery. Kinases are classified by the substrate they phosphorylate namely, protein kinases, carbohydrate kinases and lipid kinases. These different classes have unique mechanism of action but show considerable overlap in their structural assembly and sequence of chemical modifications. Compounds can modulate kinase activity by interacting with the enzyme’s ATP binding site (orthosteric site) or the allosteric site. These modulators have been classified as Types I, II, III and IV depending on their mode of binding. Inclusion of atypical kinases and pseudokinases in the targetable kinome along with the recent approval of kinase-based therapeutics provides an impetus to the ever-growing field of kinase modulation. This review attempts to summarize the identification, historical stance, catalytic structure and subsequent development of kinases as significant drug targets with an emphasis on their catalytic machinery and modulation.
Keywords
- Kinase
- Kinase modulation
- Kinase Drugs
- Kinase review
- Kinase mechanism
- Review
Kinases are enzymes that transfer a phosphate group (usually from ATP) to different biomolecules in a process termed ‘phosphorylation’. Kinases can phosphorylate amino acids (protein kinases), lipids (lipid kinases) or sugars (carbohydrate kinases). They differ from ‘phosphorylases’ in that they transfer a phosphate group from a high-energy molecule like ATP to a substrate as opposed to transferring a ‘lone’ inorganic phosphate group to a substrate (1), (2). Many attempts have been made towards classification of kinases, most notably by Cheek et al. (3), (4). Kinases are essential for the regulation of many biochemical pathways ranging from blood glucose regulation to cell apoptosis. Hence, their dysregulation can lead to a plethora of conditions such as inflammatory diseases, arthritis, cancer etc (5). Because of this there has been a widespread interest amongst researchers for the last few decades to identify small molecules which can modulate the activity of these kinases. These modulators, majority of which are inhibitors, have found wide application for the treatment or management of many such conditions (6).
Burnett and Kennedy’s discovery of enzymatic phosphorylation of proteins in 1954 (7) and the subsequent determination of PKA (protein kinase A) mediated signaling pathway (8), (9) paved the way towards the understanding and exploitation of kinases. The first prototype-kinase inhibitors to be discovered were quercetin (10)- a plant flavonoid polyphenol and staurosporine (11)- an alkaloid isolated from Streptomyces staurosporeus. These compounds either had low potency (for instance, quercetin showed a low micromolar potency) or exhibited poor selectivity (staurosporine is known to inhibit many different kinases (12)). The early 1990s witnessed the identification of various important kinase mediated signaling pathways such as; MAPK/ERK pathway (Ras-Raf-Mitogen activated protein kinase/Extracellular signal regulated kinase pathway) (13), (14), JAK-STAT (Janus Kinase-signal transducer and activator of transcriptional proteins) pathway (15), (16) and PI3K pathway (Phosphatidylinositol-3-kinase pathway) (17). Structure-based design of kinase modulators was made possible by the elucidation of the crystal structure of the kinase catalytic domain of PKA (cAMP-dependant protein kinase A) by Knighton et al. (18). In 1999, Rapamycin became the first FDA (US Food and Drug Administration) approved kinase inhibitor (19) for its use as an immunosuppressant, although Fasudil was already approved in Japan as early as 1995 for the treatment of cerebral vasopasm (20). The breakthrough drug Imatinib gained FDA approval in 2001 for the treatment of chronic myelogenous leukemia (CML) under the trade name Gleevec® (21). Although Fasudil was approved before Imatinib, the latter is widely considered as the first-ever approved synthetic kinase inhibitor because Fasudil’s mode of action was not determined at the time of approval and partly due to the fact that Fasudil was never granted approval by FDA or EMA (European Medicines Agency).
Imatinib’s approval provided the much needed impetus towards the exploration of kinase modulators which led to the discovery of MAPK/ERK inhibitors (2003) (22), identification of GNF-2 and its analogues as kinase inhibitors with a novel inhibitory mechanism (2006) (23) and also the approval of various other kinase modulators such as Sorafenib (24), Trametinib (25), Afatinib (26), Ibrutinib (27), (28), Idelalisib (29), (30)etc. As of March 2019, PKIDB (Protein Kinase Inhibitors Database) (31) a regularly updated online database of protein kinase inhibitors (PKIs) mentions 53 protein kinase inhibitors that have been approved for human therapeutic use.
Protein kinases can be broadly classified according to the amino acid (s) they phosphorylate; serine/threonine kinases, tyrosine kinases, dual-specificity kinases (Ser/Thr and Tyr) and kinases which phosphorylate other amino acids such as histidine, lysine etc. (32). Regardless of the difference in size, specific amino acid they phosphorylate, number and shape of subunits, sub-cellular localization etc., all protein kinases possess a homologous catalytic domain termed ‘Protein Kinase Domain’.
This catalytic domain functions to transfer a γ-phosphate group of ATP to an amino acid. The core of the domain (~270 residues) is highly conserved even amongst evolutionarily distant species. The entire core resembles a bilobal structure having a small lobe (N-lobe) and a large lobe (C-lobe). These lobes contain many conserved subdomains which assemble together to work as functional units (33). Seminal works of Hanks, Hunter, Manning, Taylor, Knighton etc (18), (32), (34), (35) have attempted to classify, group and exhibit the internal mechanism of this highly conserved core. Hanks et al. classified the catalytic core in terms of 12 subdomains; subdomains 1 to 4 are present in the small lobe, subdomains 6 to 10 in the large lobe while subdomain 5 traverses through both the lobes (fig1). Many conserved residues such as the DFG (Aspartic acid-Phenylalanine-Glycine) and APE (Alanine-Proline-Glutamic acid) motifs of the ‘activation loop’ have also been well characterized (36). The twelve conserved subdomains form certain functional assemblies that serve a common function but are composed of residues which are non-contiguous in the primary structure of the protein. (33) Hanks and Hunter were also able to identify 12 nearly invariant conserved residues present in the catalytic core. It is to be noted that these 12 conserved residues are present asymmetrically in the 12 conserved subdomains mentioned by the same authors. This implies that some subdomains do not contain any conserved residue while some may contain more than one. The composition and function of the 12 conserved subdomains are shown in Table 1. Table 1 also mentions the 12 conserved residues by using cAMP-dependent protein kinase, also known as Protein Kinase A (PKA) as the reference (PDB entry 1ATP). The functional units formed by the 12 subdomains are present in Table 2.
| Subdomains | Key features | Conserved Residue (s) (PKA as reference) | Function |
|---|---|---|---|
| I | 2 β-strands linked via a ‘P loop’ with (GxGxxG) typical sequence motif | Glycine-50 |
‘P loop’ is a glycine rich stretch of residues that acts as an ‘ATP-binding region’ |
| II | A β-strand with an invariant lysine residue | Lysine-72 | Lysine interacts with the phosphate groups of the ATP |
| III | An α-helix with a conserved glutamic acid residue | Glutamic acid-91 | Glutamic acid forms a salt bridge with the conserved lysine in Subdomain II to form a stable ATP pocket |
| IV | A central β-strand | - | Contributes to the core of the small lobe |
| V | A β-strand (hydrophobic) in small lobe linked to an α-helix in large lobe, also contains ‘Gatekeeper’ residue | - | Traverses the 2 lobes and contributes to the ATP-binding pocket while the gatekeeper influences accessibility of the ATP-binding pocket |
| VIa | A long α-helix | - | Parallels the α-helix of Subdomain IX which contains a conserved aspartic acid residue |
| VIb | Catalytic loop with the general sequence (xRDLKxxN) | Aspartic acid-166 |
Aspartic acid acts as the catalytic base (hydrogen acceptor of Ser/Thr or Tyr) |
| VII | 2 β-strands linked via a loop which contains the ‘DFG motif’ | Aspartic acid-184 |
Aspartic acid of the DFG motif chelates a Mg2+ ion (between β- and γ- phosphates) in ATP while the phenylalanine fits into a hydrophobic pocket |
| VIII | Contains the ‘APE loop’ along with the ‘P+1 loop’ | Glutamic acid-208 | Glutamic acid of the APE loop forms a salt bridge with an Arginine of Subdomain XI to provide stability to kinase core while ‘P+1 loop’ helps recognize substrate |
| IX | A hydrophobic α-helix | Aspartic acid-220 |
Anchors the catalytic and regulatory spines |
| X and XI | Contain 3 α-helices | Arginine-280 ( XI) | Forms the core of the large lobe and is involved in substrate recognition |
| Functional Unit | Key Features | Function | References |
|---|---|---|---|
| Activation loop | Situated between DFG motif (Subdomain VII) and the APE loop (Subdomain VIII) | Functions as a switch to turn the kinase ‘on or off’. It serves to position the DFG motif, catalytic loop and P+1 loop where they can perform catalysis | 121,122 |
| Catalytic spine | Includes the adenine ring of ATP, a few residues from the N-lobe and many hydrophobic residues from the C-lobe | It is the region where the catalytic activity takes place. It is anchored to N-terminus of the α-helix in Subdomain IX | 33,119,123,124 |
| Regulatory spine | Includes 4 non-contiguous residues; 2 conserved Leucines from N-lobe and conserved Phenylalanine and Tyrosine from C-lobe | Conformation of this spine determines the active or inactive state of the kinase. It is anchored to a conserved aspartic acid in α-helix of Subdomain IX | 124–126 |
| Gatekeeper |
It is a residue in Subdomain V and is positioned deep within the ATP-pocket. It is present between the 2 spines | It controls the accessibility of the ATP-binding pocket and is generally a bulky residue like Leucine, Phenylalanine etc. Residue mutations can cause activation of kinase via autophosphorylation | 127,128 |
The active conformation of the kinase is generally termed as ‘closed’ while ‘open’ refers to the inactive conformation. This terminology stems from the conformation a kinase takes up when active, which is more compact; hence ‘closed’. The active conformation of the majority of kinases is very similar to each other but the variation one observes in their inactive conformations due to the misalignment of one of the essential functional units or subdomains is worth noting.
Being central to the energy needs of all organisms, carbohydrates are key players in energy metabolism. Activation of carbohydrates via phosphorylation is important for their utilization and transformation. Carbohydrate kinases perform this crucial function (37). The importance of this reaction and the enzymes associated can be gauged from the fact that carbohydrate kinase mutations very often lead to various medical conditions such as juvenile cataract (galactokinase deficiency) (38), hyper-insulinism (glucokinase deficiency) (39), hemolytic anemia (glucose-6-phosphate deficiency) (40) etc. Carbohydrate kinases belong to 5 different and evolutionarily distinct classes, although their function and catalytic mechanism remains the same. These classes are Hexokinases, ROK kinases (repressor, open reading frame, kinase), Ribokinases, GHMP kinases (galactokinase, homoserine kinase, mevalonate kinase, phosphomevalonate kinase) and Phosphatidylinositol kinases. These classes differ in their substrate specificity, choice of phosphate donors and structural features as detailed in Table 3. Phosphatidylinositol kinases are considered as both, carbohydrate and lipid kinases (41), (42).
| Carbohydrate Kinase Class | Common substrates | Phosphate donors |
Key structural features | References |
|---|---|---|---|---|
| Hexokinase | Glucose, Mannose, Fructose | ATP (ITP) | Monomeric or dimeric (bilobal and asymmetric) | 129,130 |
| ROK Kinase | Glucose, Allose, Fructose, |
ATP (Polyphosphate) | Dimeric or Tetrameric (monomers are bilobal and asymmetric) | 131,132 |
| Ribokinase | Ribose, 2-deoxy-D-ribose, adenosine | ATP, ADP (GTP) | Generally dimeric (monomers are bilobal and asymmetric) | 133,134 |
| GHMP Kinase | Galactose & its derivatives, |
ATP (GTP, ITP) | Generally dimeric (monomers are bilobal with equally sized domains) | 135,136 |
| Phosphatidylinositol kinase (considered as lipid kinases also) | Phosphatidylinositol and its phosphates | ATP (GTP) | Usually heterodimeric, Large and multi-domain |
137 |
It is interesting to note that inspite of having diverse protein folds, the general scheme of catalysis remains the same amongst various classes of carbohydrate kinases. The carbohydrate to be phosphorylated is appropriately positioned by hydrogen bonds between the unreactive hydroxyls (i.e., hydroxyl group which would not be phosphorylated) and the residues within the substrate binding site. The reactive hydroxyl is deprotonated usually by a conserved aspartic acid residue to form a nucleophilic (O-) anion. This deprotonated hydroxyl performs a nucleophilic attack upon the donor phosphate group (usually ATP) to form the phosphorylated product. This reaction goes through a penta-coordinated transition state which is stabilized by a divalent cation or positively charged residues like lysine (41).
Lipids and their derivatives take part in a wide variety of essential processes such as cell growth, differentiation, cytoskeletal transformations, signal transduction, motility, platelet function, intracellular trafficking etc (43). Hence their dysregulation leads to many diseases like cancer, inflammatory disorders, diabetes etc (44), (45). Similar to carbohydrates, lipids are phosphorylated by lipid kinases so that they can be utilized in various biochemical reactions. A unique aspect of lipid kinases is that most of them are membrane bound proteins. These kinases are categorized into 3 major classes; Phosphatidylinositol Kinases (PIKs), Sphingosine Kinases (SKs) and Diacylglycerol Kinases (DGKs). The PIK class, in particular, is well studied due to its direct relation to tumor growth (PI3K/AKT/mTOR pathway). Meanwhile SKs and DGKs share many similarities in structure and regulatory mechanism (46). Table 4 shows various classes of lipid kinase alongwith their common substrates, phosphate donor and key structural features.
| Lipid Kinase Class | Sub-classification | Phosphate donors | Key structural features | References |
|---|---|---|---|---|
| Phosphatidylinositol kinases (PIKs) | 3 subfamilies |
ATP | Usually heterodimeric, |
137,138 |
| Sphingosine kinases (SKs) | 2 mammalian isoforms |
ATP | Generally monomeric in solution |
139 |
| Diacylglycerol kinases (DGKs) | 2 prokaryotic variants |
ATP |
Generally dimeric in solution |
140,141 |
Although the lipid substrate differs in these classes, the general structure of a lipid kinase contains a few common features, such as, a region for association or stabilization with the plasma membrane or sub-cellular membrane. This region anchors the catalytic machinery to certain locations such as endoplasmic reticulum or to certain vesicles. Another region is responsible for the recognition and orientation of the phosphate donor, ATP or in some cases cytidine tri-phosphate (CTP). This nucleoside binding site recognizes and orients the phosphate donor. Slightly different modes are adopted for ATP binding by the different classes but overall it involves π-stacking interactions between adenine ring and various residues, hydrogen bonding to the hydroxyls of the ribose group, Mg2+ ion being chelated by a conserved aspartic acid or glutamic acid residue and hydrogen bonds being made to the α,β,γ-phosphates of ATP. The binding and orientation of the lipid substrate is governed by another region in the kinase. This lipid binding region/cavity interacts with the lipid’s hydrophilic portion (via hydrogen bonds to amino acids like aspartic acid and serine present at the protein surface) and hydrophobic portion (via hydrophobic interactions with residues situated in the interior of the protein) (41). In Sphingosine kinases for instance, this binding region forms a tunnel into which the lipid substrate is inserted. (47) The orthosteric site is generally present in an extended cleft between the two domains in the bilobal structures of SKs and DGKs. In PIKs the orthosteric site is contained within a region termed as the ‘catalytic domain’ (48).
Lipid kinases employ a very similar catalytic scheme to that of carbohydrate kinases. Lipid binding region interacts with the substrate and positions it appropriately. The reactive hydroxyl is then deprotonated by a catalytic base which is generally a conserved residue such as an aspartate in Sphingosine kinases, glutamate for Diacylglycerol kinases or histidine in case of Phosphatidylinositol kinases. This deprotonated hydroxyl anion then attacks the γ-phosphate of the ATP present in the nucleoside binding site. This site binds ATP and orients it for the nucleophilic attack by the reactive hydroxyl of the lipid substrate. The final phosphorylated product is formed with the transition state being stabilized by positively charged neighboring residues like arginine and lysine in case of PIKs. (49)
Kinase modulation occurs through different approaches (50), (51). Some compounds target the ATP-binding site and/or a nearby pocket while others may exploit an altogether distinct site of the kinase. These different approaches for kinase inhibition or activation stem from the ability of kinases to adopt different conformations thereby exposing previously unreachable or ‘buried’ motifs/pockets. Kinase modulators are classified into 4 types; depending on their mode of binding to the kinase and the way they achieve the regulation;
• Type-I modulators bind to the ATP-binding site and directly compete with ATP. They generally bind to the ‘DFG-in’ conformation of the kinase.
• Type-II modulators bind to the ATP-binding site as well as a nearby pocket which is exposed due to the kinase adopting a ‘DFG-out’ conformation. This site is known as the extended pocket. These are also competitive with ATP.
• Type-III modulators bind to a site different from the ATP-binding site known as the allosteric site. They stabilize or destabilize a particular conformation of the kinase depending upon the function they perform (activation or inhibition). They may or may not be ATP competitive.
• Type-IV modulators bind to another protein which is the interaction partner of the kinase thereby modulating the kinase via its downstream regulation.
Type-I and type-II modulators are collectively termed as orthosteric while type-III is considered allosteric. However, it must be noted that different authors have used slightly varying classifications for kinase modulators. The definitions for type-III and type-IV kinase modulators vary in published literature (52), (53). Also some sources indicate ‘type-V’ modulators as bivalent inhibitors that bind to two different kinase domains/portions (6). Such compounds would be considered as type-III modulators as per the classification mentioned above.
A large number of kinase inhibitors, especially the ones discovered initially are orthosteric in nature, i.e., they bind to the ATP-binding site of the kinase and compete directly with ATP. A reason for this could be the fact that most of them were discovered via enzyme assays which considered only the activity of the enzyme, which in-turn would pertain to the orthosteric-site of the enzyme. Orthosteric modulators are the most common type of kinase modulators although the trend is rapidly shifting towards allosteric modulators.
A striking feature of most orthosteric protein kinase modulators is their relation to the ‘DFG-motif’ and the conformations it allows to be sampled by the kinase (54), (55). The ‘DFG-out’ and ‘DFG-in’ conformations are essential for the binding of many type-I and type-II drugs to protein kinases. The DFG-motif is an essential element of the activation loop. The aspartate of the DFG-motif coordinates the magnesium ion (Mg2+) between the β- and γ- phosphates of the ATP. This chelation is necessary as it positions the γ- phosphate of the ATP for transfer to the substrate. The phenylalanine of the DFG-motif generally places itself into a hydrophobic pocket between two residues, one from small lobe and the other from large lobe. The packing of this phenylalanine is necessary for the proper positioning of the aspartate of the DFG-motif. This arrangement when the phenylalanine is packed inside the hydrophobic pocket is termed as ‘DFG-in’ conformation and represents the ‘active conformation’ of the enzyme. The ‘DFG-out’ conformation, when the phenylalanine slides out of the hydrophobic pocket and displaces the aspartate, is the ‘inactive conformation’. In the ‘DFG-out’ conformation the aspartate is not able to chelate the Mg2+ ion (54). Many kinases do not possess the ability to sample a DFG-out conformation, for example, cyclin dependent kinases (CDKs) and protein kinase C family (PKCs), and hence cannot be modulated via a type-II mechanism (56). Type-I inhibitors interact with the orthosteric site in the DFG-in conformation and compete directly with ATP. Dasatinib (57) and bosutinib (58) are examples of such drugs. Type-II inhibitors bind to the DFG-out conformation which is the inactive conformation and stabilize it (56).
Fasudil is a type-I protein kinase modulator which inhibits the kinase ROCK (Rho-dependant Kinase). ROCK is an important enzyme of the AGC-kinase family and is involved in cytoskeletal structure assembly, cellular motility, vasoconstriction, vascular remodeling etc. It phosphorylates MBS (Myosin binding subunit) domain present on myosin phosphatase thereby increasing its activity which in-turn induces vasoconstriction via smooth muscle contraction. Inhibition of this cascade by Fasudil can potentially treat pulmonary hypertension, internal hemorrhaging, stenosis and certain neurodegenerative disorders. Fasudil binds to the cleft formed between the N-lobe and C-lobe where the ATP-binding site is present. Its isoquinoline ring mimics adenine of ATP while the homopiperazine ring takes up the space where the ribose group and the phosphates bind. (59), (60) (fig2- A). It directly competes with ATP and shows good selectivity towards ROCK1 over the similar PKA (protein kinase A). This selectivity stems from the type of steric interactions that the 7-membered homopiperazine ring makes with the residues of the phosphate binding loop.
Kinase modulators belonging to the carbohydrate or lipid classes have a considerably limited coverage in published literature. Even fewer have been granted approval for public use (6). The mechanism of action of these modulators is quite similar to the protein kinase modulators and fits the previously mentioned classification of type-I and type-II modulators. Most of these modulators bind to the orthosteric site and are directly competitive with ATP. Amongst carbohydrate kinase modulators, much work has been done on hexokinase inhibitors which serve as anti-parasitic drugs against organisms like Trypanosoma (causative organism of African sleeping sickness) (61), (62) and Clonorchis (human liver fluke) (63). These inhibitors utilize the fact that the organisms in question are reliant on glycolysis for their energy requirements, thereby making the selective inhibition of parasitic hexokinases an effective strategy. However, no carbohydrate kinase modulator has been approved till date.
Development of lipid kinase modulators has been focused towards designing inhibitors of class-I PIKs due to their role in the PI3K/AKT/mTOR pathway and subsequent involvement in tumor growth (64), (65). A well studied lipid kinase inhibitor is the fungal metabolite wortmannin (66), which is an irreversible PI3K inhibitor. Wortmannin covalently modifies a conserved lysine residue (Lys-802) in PI3K by forming an enamine which leads to enzyme inhibition. Massive lead optimization efforts of wortmannin to improve its pharmaceutical properties and therapeutic indexes have led to compounds such as PX-866, which has shown anti-tumor properties in a variety of cancers when used alone or in conjugation with other chemotherapeutics. Another inhibitor which is also one of the first synthetic compounds designed against PI3K is LY294002 (67) (68). Unlike wortmannin, which is an irreversible inhibitor, LY294002 is a typical ATP competitive orthosteric site inhibitor. A discussion about PI3K inhibitors should involve the mention of Idelalisib (69). Developed by Gilead Sciences, Idelalisib, is the only FDA approved lipid kinase inhibitor as of March-2019 (6). It binds reversibly, in ATP-competitive manner with class-I PI3K isoform ‘P110δ’ (delta isoform) and is currently being used in patients with chronic lymphocytic leukemia (CLL).
A well-studied example of a type-II modulator is one of the first kinase inhibitors to be granted FDA approval, namely Imatinib. Imatinib (Gleevec®) is a Bcr-Abl (Breakpoint Cluster Region - Abelson murine leukemia viral oncogene homolog 1) kinase inhibitor and is a potent drug for management of Philadelphia chromosome positive (Ph+) conditions like Chronic Myelogenous Leukemia (CML). Imatinib binds to the target kinase, Abl kinase when it is present in its inactive conformation i.e., DFG-out conformation. It binds to the orthosteric site as well as an extended nearby pocket which gets exposed when the phenylalanine of the DFG motif ‘pops-out’ of the hydrophobic pocket. The structure of Imatinib resembles a linear association of various 6-membered heterocyclic groups and substituted phenyls. The terminal pyridine and pyrimidine rings occupy the site where the adenine ring of the ATP binds and forms a π-π stacking interaction with 2 separate phenylalanine residues (Phe317 and Phe382). Methionine-318 and Threonine-315 form important stabilizing hydrogen bonds with the nitrogen atom of the terminal pyridine ring and the hydrogen atom of the 2-amine nitrogen connecting the pyrimidine ring to the central methyl-phenyl ring respectively. The methyl-piperazine end of Imatinib along with the adjacent benzamide fit into the ‘extended hydrophobic pocket’ which gets exposed when the enzyme takes up the DFG-out conformation (70). The importance of this ‘extended pocket’ nearby the orthosteric site is proven by the fact that resistance to Imatinib often occurs due to mutations in this region (71).
Having established the importance of the DFG-motif, it is crucial to understand what factors contribute towards the ability of the kinase to adopt such in/out conformations. Two residues have a significant role to play in a kinase’s ability to sample such conformations, one of them being the ‘gatekeeper’ residue and the other being the ‘xDFG’ residue. The gatekeeper residue in particular has a special importance for Imatinib as it is responsible for the most common cause of Imatinib resistance. A mutation known as ‘Bcr-Abl/T315I’ which involves the mutation of a threonine to isoleucine as the gatekeeper residue is generally the major cause of Imatinib resistance (72). The isoleucine is unable to contribute towards stabilizing a hydrogen bond with Imatinib and also hinders the proper binding of the drug via a steric clash. This mutation also confers resistance to other inhibitors like Dasatinib, Bosutinib and Nilotinib. The ‘xDFG’ residue is the amino acid preceding the DFG-motif. Mutations at this position also affect the ability of the kinase to adopt a DFG-out conformation. Hence it is important for the activity of type-II inhibitors (73).
Of the 518 protein kinases encoded by the human genome, a 478-member superfamily of eukaryotic protein kinases (ePKs) contains similar conformation around the orthosteric-site, showing the highly conserved nature of the catalytic machinery of these proteins (35). The homologous nature of the catalytic site poses a major issue when it comes to designing compounds selective towards a single kinase (18). Broad targeting of many kinases simultaneously can be an effective strategy against cancer and related conditions as it fulfills the task of down regulating or blocking many kinases in a signaling cascade at once thereby collectively inhibiting tumor proliferation (74). However, this property severely limits the implications of such compounds outside of oncology due to toxicity and side-effects associated with binding to kinases outside the target space (75). Resistance is another major issue that plagues kinase modulators, both inhibitors and activators. (76) Resistance occurs when certain mutations in the target kinase inhibits the drug binding to its site. For instance, the appearance of Abl kinases which contain single point mutations in amino acid sequence near the orthosteric site has severely limited the activity of imatinib and various other Abl-kinase inhibitors. Yet another fallacy of the ATP-site targeting kinase modulators is a result of the structure and chemical nature that the catalytic site possesses. The ATP-binding site is composed of mostly hydrophobic residues and is flat and extended in structure. Drugs designed with the same pharmacophoric features exhibit undesirable physicochemical properties such as poor solubility, modulation of hERG activity (electro-cardio potential) and have generally large and elongated structures (77).
These issues of selectivity, resistance and poor physicochemical properties necessitate a change of approach towards targeting these kinases, which have led to the discovery of ‘allosteric’ modulators. These compounds modulate kinases by binding to sites distinct from the ATP-binding site of the kinase (78). Such a mode of binding negates many of the shortcomings of the ATP-site targeting molecules, as the ‘allosteric sites’ are not nearly as conserved as the ATP-binding site. The mechanism of action that these allosteric modulators employ is distinct from the simple competitive inhibition of the orthosteric site for the target kinase. Table 5 compares orthosteric and allosteric inhibitors highlighting the advantages conferred by the latter. The mode of action employed by these different modulators can be found in Table 6 along with some representative examples.
| Property in question | Orthosteric modulators | Allosteric modulators | References |
|---|---|---|---|
| Selectivity | Due to the highly conserved regions present within kinase families; selective targeting is a serious issue | Since the allosteric sites are much less homologous amongst kinases; much more selective modulation can take place | 142–144 |
| Resistance | Mutations within or nearby the orthosteric site can often cause resistance towards type-I & type-II modulators via inhibition of ligand binding | Although not completely immune to resistance, allosteric modulators in combination with drugs that bind to other sites can negate such ill effects | 74,145,146 |
| Physicochemical properties | Since the ATP-site is mostly hydrophobic in nature, compounds derived show poor pharmaceutical properties such as poor solubility, large structures etc. | Allosteric sites are much more flexible in their composition and allow a variety a pharmacophores with comparatively better physicochemical properties | 147,148 |
| Functional diversity | Orthosteric modulators are obligate inhibitors and hence only allow the disruption of kinase by preventing phosphorylation | Allosteric modulators can be inhibitors or activators and hence can be used for a wider control over the kinase. For instance the use of Glucokinase activators against diabetes | |
| Target validation | Due to the high degree of homology, target validation or designing tags is not appropriate by using these compounds | The specific nature of allosteric modulators make them useful for target detection and bio-activity studies | 149 |
| Chemical Space | Over the past 2 decades, most ATP-competitive scaffolds have been claimed via IPR (Intellectual Property Rights) thereby hindering further progress | Allosteric sites, however, are much more varied and dynamic in nature providing ample opportunities for creative development of novel drugs |
| Modulator type | Mode of action | Examples (Target kinase) | References |
|---|---|---|---|
| Type-I (inhibitors) | Direct competition with ATP in the DFG-In conformation | Gefitinib (EFGR kinase) | 150 |
| Type-II (inhibitors) | Binding to ATP-site and the inactive DFG-Out conformation. |
Imatinib (Abl-kinase) | 56 |
| Type-III inhibitors | Binds to the allosteric site in a way that stabilizes the inactive state of the kinase |
GNF-2 (Abl-kinase) |
151 |
| Type-III activators | Destabilizing the inactive state of the enzyme |
DPH (Abl-Kinase) |
153 |
| Type-IV modulators (inhibitors/activators) | Modulating the regulatory partner of the kinase thereby showing indirect control over the enzyme | AMG-3969 (GKRP) | 101 |
Allosteric modulation (allostery, as it is often called) of kinases occurs quite often in-vivo. Allostery generally occurs as a part of the feedback response in most signal cascades and catalytic pathways (79). The end-products of a metabolic pathway regulate it via feedback inhibition, such as in carbohydrate metabolism, cell cycle cascades, cell growth signaling pathways etc. Many examples of natural allosteric modulation are known such as, activation of CDK2 (Cyclin Dependant Kinase 2) by cyclin (80) or the inhibition of aspartate kinase by threonine and lysine (81). Allostery also occurs via proteins or regulatory proteins dedicated for this precise function. Glucokinase Regulatory Protein (GKRP) regulates the activity of Glucokinase in-vivo by binding to it allosterically (82). Allosteric modulation of kinases involves proper positioning of the activation loop through the conformational changes that occur at the allosteric site.
A striking feature of Type-III modulators is their ability to modulate kinases using both, activation and inhibition, in contrast to only inhibition, exhibited by Type I and Type II modulators. Discovery of kinase Type III agonists is related to the identification of its antagonists. It is plausible to reason that an inhibitor which binds to the allosteric site and stabilizes the inactive conformation could be modified such that it stills binds to the allosteric site but now ‘destabilizes’ the inactive conformation (83). Such an approach has led to the discovery of Abl-kinase agonists which bind to the allosteric site (myristoyl binding site) and activate the kinase (84). Kinase activation via an effector molecule is not uncommon in nature, for instance activation of Aurora Kinases A/B by TPX2 (Microtubule nucleation factor) and INCENP (Inner Centromere Protein). Generally, synthetic type-III modulators inhibit or activate the kinase by driving it towards a specific conformational state i.e., inactive or active state. In contrast to type-II modulators, the binding of type-III modulators is independent of the binding of ATP, as confirmed by crystal structures showing the kinase bound to both (85). This underlines the truly distinct nature of the allosteric site when compared to the extended ATP-site.
Two examples of allosteric modulation of protein kinases, namely CHK1 (Checkpoint Kinase 1) and Abl-kinase by synthetic molecules have been discussed. A common feature of many cancer types is their down-regulation of tumor suppressor p53 in order to ‘override’ cellular machinery towards tumor growth by preventing an important check system. However, p53 is also a part of DNA damage response (DDR) along with CHK1 in most cells (86). CHK1 is a member of the CAMKL family and serves to function as a regulator of the ‘S-G2’ checkpoint during cell cycle (87). With p53 downregulated, cancer cells must rely exclusively on CHK1 for addressing the DNA damage caused by radiotherapy or chemotherapeutic agents. Thus inhibition of CHK1 can sensitize p53-deficient cells towards DNA damaging interventions and thereby increasing the efficacy of such treatments. Earlier efforts have focused on ATP-competitive inhibitors of CHK1 (88)– (91), however, these came along with the the usual issues present in type-I inhibitors; selectivity and marked difference between in-vivo and in-vitro response. The identification of an allosteric site on the C-lobe surface opens new avenues towards targeting CHK1. This site, distant from the ATP-binding site by 18Å comprises of a shallow hydrophobic pocket. (92) Subsequent allosteric CHK1 inhibitors with varied scaffolds such as carbamate, semicarbazide and thioquinazolinones and good efficacy (92), (93) have been designed. Fig2 (B1/B2) shows a semicarbazide-based CHK1 inhibitor bound to the allosteric site along with its interaction diagram.
Philadelphia positive Chronic Myelogenous Leukemia (Ph+/CML) accounts for around 15% of all leukemias found in adults. CML prognosis has been improved drastically by the emergence of Bcr-Abl inhibitors. These inhibitors act upon the Bcr-Abl kinase (or simply Abl-kinase) to prevent the excessive phosphorylation of certain cell components which may lead to cancer. Imatinib, Dasatinib and Nilotinib are examples of orthosteric Abl-kinase inhibitors. As mentioned earlier, a mutation known as ‘Bcr-Abl/T315I’ present in the Abl-kinase causes widespread, multi-drug resistance in patients which renders the above-mentioned drugs obsolete. Progress was made towards solving the issue of resistance with the serendipitous discovery of GNF-2 via a cytotoxicity screen involving pyrimidine-based library. GNF-2 is an allosteric inhibitor of wild-type Abl-kinase which binds to the ‘myristate binding site’ present in the C-lobe of the kinase. Figure 3- A compares the binding site of GNF-2 to Imatinib. Also GNF-2 does not inhibit the kinase domain itself, rather stabilizes the inactive conformation of the enzyme in conjugation with its SH-domains (Src-Homology domain). It is important to note that that GNF-2 shows no activity against the T315I-mutant of Abl-kinase. How is it then an effective alternative during resistance? True inhibitory action against a mutant Abl-kinase is observed when GNF-2 is used in conjugation with other ATP-competitive inhibitors such as Imatinib. GNF-2 along with other type-I or type-II inhibitors not only show activity against a mutant kinase but also prevent the emergence of new mutants. Optimization has led to the development of GNF-5 which is an improved analogue of GNF-2.
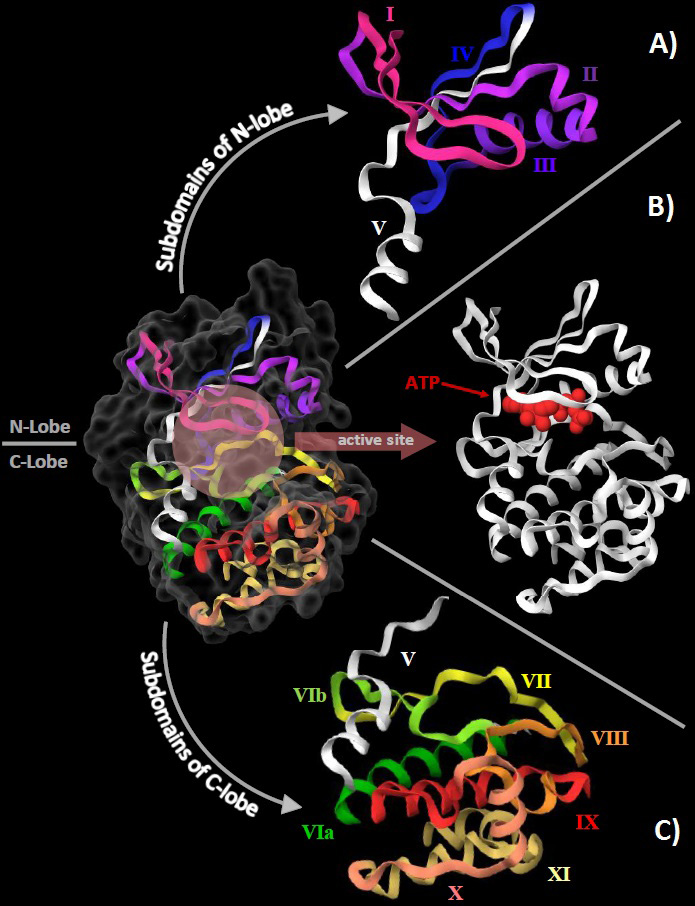 Figure 1
Figure 1The structural organization of the eukaryotic protein kinase domain using the crystal structure of cAMP-dependent protein kinase (PDB:1ATP) as reference. A. The 4 subdomains of N-lobe are represented with roman numerals from I to IV. Subdomain V (white) traverses between the 2 lobes. B. The orthosteric/active site is shown here at the cleft between the 2 lobes with a molecule of ATP (red) already bound to it. C. The 7 subdomains of the C-love are shown here with roman numerals from VIa to XI with subdomain V (white).
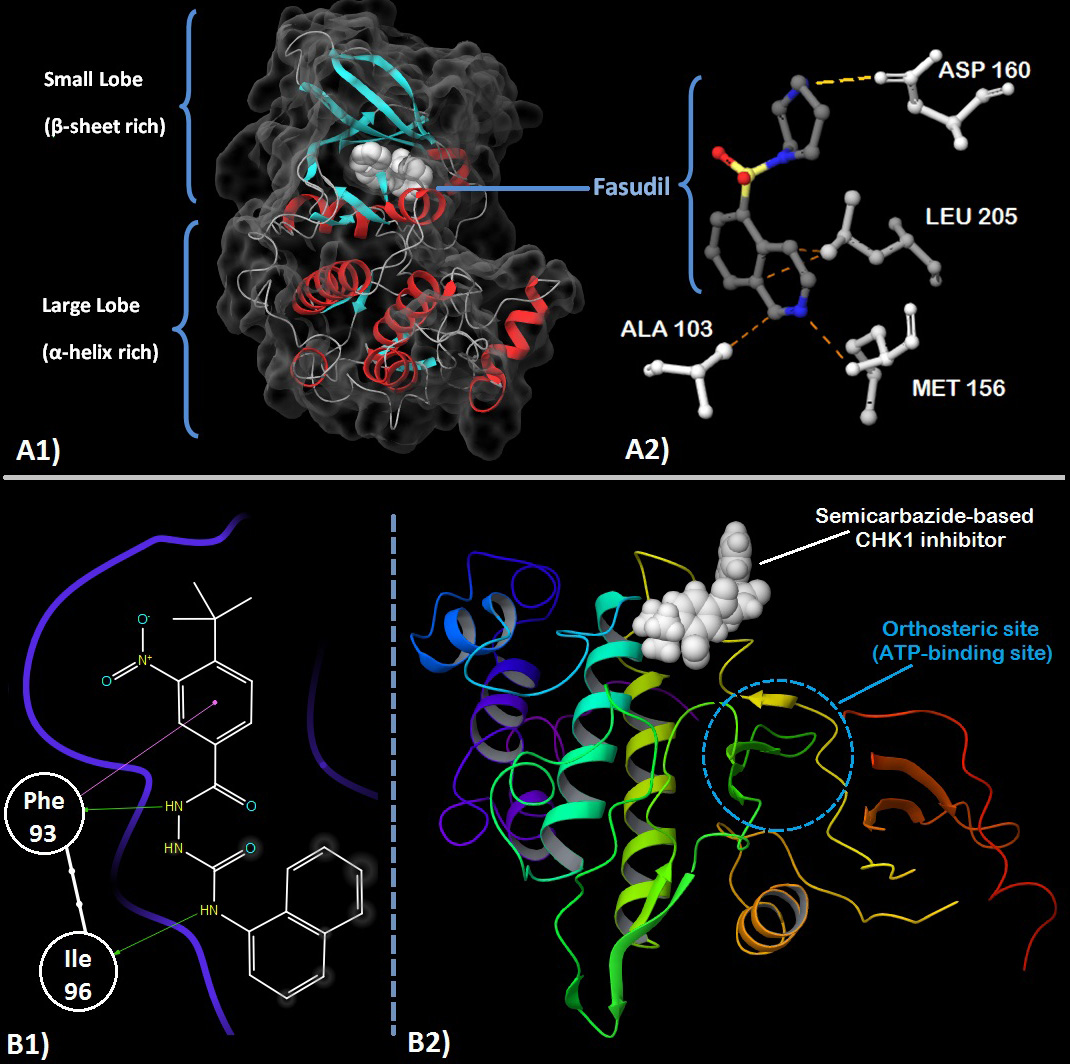 Figure 2
Figure 2A1. Crystal structure of ROCK1 showing the small and large lobes and Fasudil (white) at the orthosteric site. Secondary structure with α-helices (red), β-sheets (cyan) and turns/loops (grey) can be seen with the solvent accessible surface contours (dark grey). A2. Fasudil’s (grey carbons) interaction with residues (white) present in the orthosteric site of ROCK. Methionine 156 is involved in hydrogen bonding with the nitrogen of the isoquinoline ring while Asparatic acid 160 forms a hydrogen bond with the nitrogen of homopiperazine ring. Alanine 103 and Leucine 205 provide stabilizing hydrophobic interactions with the orthosteric site. (PDB: 2ESM). 59 B1. 2D-ligand interaction diagram of an allosteric semicarbazide CHK1 inhibitor. The phenyl group of Phe 93 is involved in a pi-pi stacking interaction with the nitro-substituted benzene ring of the inhibitor while its carbonyl oxygen forms a hydrogen bond with the hydrazide group of the semicarbazide group. Ile 96 forms a hydrogen bond with the nitrogen atom of the 1-amino substituent of the naphthalene ring. B2. The 3D view shows the inhibitor bound at the allosteric site which is clearly distinct & distant (~18Å) from the orthosteric site. (PDB: 3JVS) 92
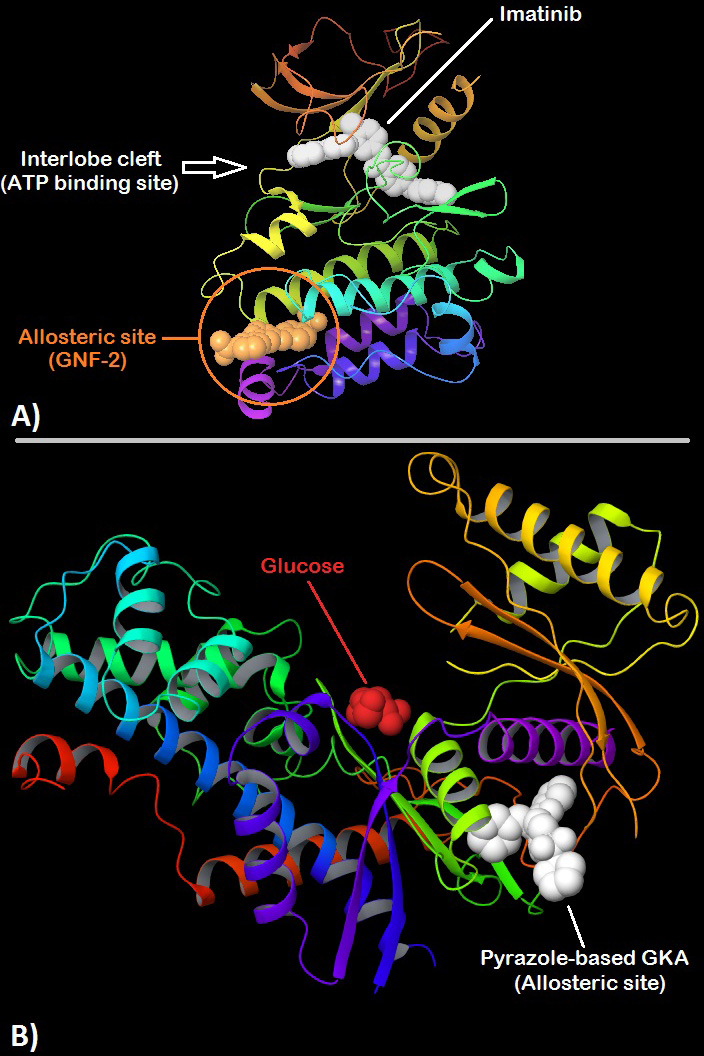 Figure 3
Figure 3A. 3D structure of ABL kinase showing Imatinib (white) bound to the orthosteric site present at the interlobe cleft while GNF-2 is shown bound to a distant (~30Å) allosteric site also called the myristate binding site in the large lobe. (PDB:3K5V) 151 B. 3D structure of human Glucokinase with bound glucose (red) nearby the orthosteric site at the interlobe cleft and a pyrazole-based glucokinase activator (white) bound to the allosteric site which is present posterior to the interlobe cleft at a distance of about ~25Å. (PDB:5V4X) 156.
Allosteric modulation of carbohydrate kinases via synthetic compounds has not received the same attention as that of protein kinases. Natural allosteric modulation of carbohydrate kinases is well understood due to their role in critical pathways of energy metabolism. Most often this modulation is a part of the feedback response. Ribokinase class of the carbohydrate kinases has developed very elaborate and complex allosteric modulation mechanisms. Phosphofructo Kinase (PFK), a type of Ribokinase, has regulatory domains present on the kinase surface where allosteric modulators/effectors may bind. Eukaryotic PFKs, for instance, have 2 such regulatory domains and over 20 known allosteric effectors.
Another example is the eukaryotic Hexokinase-I & -III which have a specific regulatory domain where allosteric effectors bind. The binding of these effectors can have an inhibitory effect (such as binding of the end-product Glucose-6-Phosphate) or an activation effect (binding of phosphate & citrate analogues). The importance of this type of allosteric regulation shall be evident from the fact that many types of cancers that rely heavily on glucose metabolism over-express eukaryotic Hexokinase-II. Unlike Hexokinase-I & -III, the type-II hexokinase does not have the domain for allosteric regulation thereby preventing appropriate regulation leading to tumor proliferation. This over-reliance on glycolysis by certain cancers is known as the ‘Warburg effect’. Studying allostery in the case of carbohydrate kinases has enhanced our understanding of energy metabolism and the intricate regulation of these pathways.
Apart from functional analyses and structure-activity-relationship studies of carbohydrate kinases, allosteric regulation via small synthetic molecules has also been researched. Much of these efforts have been directed towards developing allosteric ‘activators’ of Glucokinase for their potential use in the management of type-II diabetes (94). Glucokinase, an isoform of Hexokinase, plays the role of a glucose sensor in pancreas and liver thereby stimulating the release of insulin and glucose uptake for glycogen synthesis (95). This makes Glucokinase an attractive target for anti-diabetic therapy. Crystal structure of Glucokinase has shown the presence of an allosteric site to which small molecules may bind to act as allosteric effectors (96). A well-known Glucokinase Activator (GKA) is Piragliatin, an experimental drug by Roche that has shown promising results in phase-II clinical trials (97). Glucokinase, like most kinases, can exhibit an active or inactive conformation. The mechanism of action of these GKAs involves the stabilization of allosteric site which in-turn stabilizes the active conformation of the enzyme. This stabilization shifts the equilibrium such that the enzyme is more likely to be present in the active state hence increasing the sensitivity of Glucokinase towards glucose. There is also strong experimental evidence that GKAs actually promote the dissociation of GKRP from Glucokinase at low glucose concentrations (98), (99). The combined result of these effects is a systematic enhancement of glucose sensitivity by GKAs in liver and pancreas which represents a potential therapy for type-II diabetes management. Although the long term usage of piragliatin was not successful due to the development of hypoglycemia in a few patients. Novel strategies such as the discovery of hepato-selective and peptidic GKAs may help overcome these challenges (100). Fig3- B shows the structure of human Glucokinase with bound glucose (nearby the orthosteric site) and a pyrazole-based GKA bound to its allosteric site.
A different method of regulation is exhibited by type-IV modulators via indirect control over the kinase. These compounds modulate the activity of the target kinase by disrupting/stabilizing its interactions with its regulatory/accessory partner. Such a method of kinase modulation is not well explored but has certainly produced promising results (101), (102). The obvious advantage of such compounds is that they need not be orthosteric or allosteric to the target kinase and hence can have a higher range of diversity in their structures. These modulators may specifically bind to the kinase, to its interaction partner or bind in an unspecific manner to either of the two. Also, since they affect kinase-protein interactions and not the kinase itself, they are capable of controlling a specific pathway/cascade the target kinase is involved in and preserves its other functions. This significantly reduces any unwanted inhibition of the kinase elsewhere in the metabolism and hence guards against the kinome-wide knock-out effects which are generally associated with conventional kinase inhibitors. One recent type-IV modulator is AMG-3969 which is a disruptor of Glucokinase-GKRP interaction. GKRP is a regulatory protein which sequesters Glucokinase in the nucleus thereby preventing it from participating in glucose phosphorylation in the cytosol. By disrupting the Glucokinase-GKRP complex, AMG-3969 bypasses the inhibitory effect of GKRP which normalizes the blood glucose levels in diabetic individuals (101), (103). As mentioned earlier a similar effect was seen in the case of GKAs; there however, the disruptive effect was only residual in nature. The Glucokinase-GKRP disruption seen in the case of GKAs was due to the fact that GKAs stabilized the ‘active’ conformation of Glucokinase which has low affinity towards GKRP. As a result, GKRP association was unlikely to occur with GKAs. However, AMG-3969 disrupts the formation of Glucokinase-GKRP complex regardless of which conformation is sampled by Glucokinase hence making it a true type-IV modulator.
An insight into the basic structural and functional units of protein, carbohydrate and lipid kinases helps us to understand the architecture of this huge enzyme family. The catalytic mechanism of these kinases provides a deeper understanding of their mode of action. Kinases, especially, protein kinases, form an important class of drug targets, second only to G-protein coupled receptors. Modulation of kinases has yielded many drugs against various forms of cancer, cerebral vasospasm, pulmonary arterial hypertension, rheumatoid arthritis, organ rejection etc. Kinase modulators can be orthosteric or allosteric depending upon their site of interaction with the enzyme. Orthosteric modulators are obligate inhibitors which represent the most common form of kinase modulators present. They target the ATP-site and/or a nearby hydrophobic pocket. However, orthosteric modulators also pose several problems such as that of resistance, selectivity, undesirable physicochemical properties etc. Allosteric modulators interact with the kinase at a site distinct and often distant from the ATP-site. Allosteric modulators have a considerable advantage as they negate many of the previously mentioned shortcomings of orthosteric modulators. With features such as the ability to inhibit or activate the kinase, overcome selectivity due to the more varied nature of the allosteric pocket, provide better physicochemical properties etc., allosteric modulators have attracted a lot of scientific attention in the recent years.
Expansion of target space opens new avenues for drug discovery. Hence an ever expanding human kinome offers more possibilities of employing different methods for modulation. targeting different members of the kinase superfamily which have been previously neglected due to intensive studies being performed on a chosen few kinases104. Two different classes of proteins are now being investigated as new drug targets within the kinome realm; atypical kinases and pseudokinases. Atypical kinases are enzymes with kinase activity but low sequence similarity to the ePK domain (eukaryotic protein kinase domain). Although discovered in the early 1990s, atypical kinases have only recently being thoroughly examined as potential drug targets105–107. Pseudokinases are, by definition the opposite of atypical kinases; they are proteins which have considerable homology with the catalytic domain but do not exhibit any kinase property. Although pseudokinases lack the phosphoryltransfer activity of kinases, they have recently been discovered to have a multitude of diverse functions from signal transduction to tumor suppression108–110. The inclusion of atypical and pseudokinases has considerably increased the target space that kinases have to offer.
Large-scale ventures involving public-private partnerships have also emerged to systematically work towards kinome expansion111. Such partnerships aim to find high-quality chemical probes with a systematic approach of first performing in-vitro SAR (structure-activity relationship) studies followed by cell-based biomarker modulation and finally in-vivo pharmacokinetic exposure and biomarker validation. Such an approach can reduce redundancies while sharing the cost and risk of the entire process. With over 9 kinase modulators approved by drug regulatory authorities in the last couple of years, kinase research is proving to be an actively growing and rewarding area of research112–118 with a promising future.
We would like to acknowledge the Department of Biotechnology, Ministry of Science and Technology, Government of India for supporting our research under the grant id: BT/PR19310/BIC/101/709/2016.
ATP
Adenosine Tri-Phosphate
Cyclic Adenosine Mono-Phosphate
Aspartate-Phenylalanaine-Glycine motif
Protein Data Bank
Protein kinase B
Mammalian Target Of Rapamycin
Human Ether-a-go-go-Related Gene
Glucokinase
Glucokinase Activator
Glucokinase Regulatory Protein
Targeting Protein for Xklp2