





















Frontiers in Bioscience-Landmark (FBL) is published by IMR Press from Volume 26 Issue 5 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.

1 Department of Biochemistry, Biotechnology and Bioinformatics, Avinashilingam Institute for Home Science and Higher Education for Women, Coimbatore, 641043, Tamil Nadu, India
2 Redox Regulation Laboratory, Department of Zoology, CBSH, Odisha University of Agriculture and Technology, Bhubaneswar-751003, India
Abstract
Genetic and epigenetic modifications in DNA contribute to altered gene expression in aging and cancer. In human cancers, epigenetic changes such as DNA methylation, histone modifications, micro RNAs and nucleosome remodelling all control gene expression. The link between the genetics and epigenetics in cancer is further shown by existence of aberrant metabolism and biochemical pathways in cancer or mutation in genes that are epigenetic players. Reversal of these epigenetic changes has been clearly shown to have therapeutic value in various forms of lymphoma and preleukemia and similar results are appearing for the treatment of solid tumors. In this review, we discuss the functional effects of epigenetic changes inducible by hypoxia, the epigenetic alterations in cancer and how they contribute to tumor progression and their relevance to epigenetic therapy.
Keywords
- Epigenetics
- Cancer
- Hypoxia
- DNA Methylation
- Histone Modifications
- miRNAs
- Chromatin.
The human genome project has been one of the most important scientific achievements in modern history. It has ushered in a new era in the field of life science research. However, among the project’s many great discoveries, surprising findings such as only particular subsets of genes being able to be expressed at a particular location and time, led to the realization that knowledge of DNA sequences is insufficient to understand phenotypic manifestations. The mechanism by which DNA, or the genetic code, is translated into protein sequences is not merely dependent on the sequence itself but also on a sophisticated regulatory system that interplays between genetic and environmental factors. These mechanisms comprise the science of epigenetics, and the control of genes through various chemical interactions for the basis of at least part of the regulatory system overseeing the expression of the genetic code (1).
Eukaryotic genomic information is modulated by a variety of epigenetic modifications that play both a direct role in establishing transcription profiles, modulation of DNA replication and repair processes and also indirect effects on the aforementioned processes through the organization of DNA architecture within the cell nucleus. Nowadays, the role of epigenetic modifications in regulating tissue-specific expression, genomic imprinting or X chromosome inactivation is widely recognized. In addition, the key role epigenetic modification during cell differentiation and development has been highlighted by the identification of a variety of epigenetic alterations in human disease. Particular attention has been focused on the study of epigenetic alterations in cancer, which is the subject of intense multidisciplinary efforts and has an impact not only in understanding the mechanisms of epigenetic regulation but also in guiding the development of novel therapies for cancer treatment. In addition, a number of genetic disorders such as Immunodeficiency-Centromere Instability-Facial anomalies (ICF) or Rett syndromes are directly associated with defects in elements of the epigenetic machinery. More recently, epigenetic changes in cardiovascular, neurological and autoimmune disorders as well as in other genetically complex diseases have also started to emerge. All these examples illustrate the widespread association of epigenetic alterations with disease and highlight the need of characterizing the range and extension of epigenetic changes to understand their contribution to fundamental human biological processes (2).
The history of epigenetics is linked with the study of evolution and development. But during the past 50 years, the meaning of the term “epigenetics” has itself undergone an evolution that parallels our dramatically increased knowledge of the molecular mechanisms underlying regulation of gene expression in eukaryotes. Our present definitions of epigenetics reflect our understanding that although the complement of DNA is essentially the same in all of an organism’s somatic cells, patterns of gene expression differ greatly among different cell types, and these patterns can be clonally inherited. This has led to a working definition of epigenetics as “the study of mitotically and/or meiotically heritable changes in gene function that cannot be explained by changes in DNA sequence” (3, 4). More recently added to this definition is the constraint that initiation of the new epigenetic state should involve a transient mechanism separate from the one required to maintain it (5). Until the 1950s, however, the word epigenetics was used more broadly (and less precisely) to categorize all of the developmental events leading from the fertilized zygote to the mature organism—that is, all of the regulated processes that, beginning with the genetic material, shape the final product (6).
Epigenetics is formally defined as a heritable change in gene expression or chromosomal stability by utilizing DNA methylation, histone covalent modification or non-coding RNAs without a change in DNA sequence (7). The term “epigenetics” was originally used to denote the poorly understood process by which a fertilized zygote developed into a mature, complex organism. The definition of epigenetics was changed to focus on the ways of heritable traits, with the knowledge of mechanisms of gene expression that can be connected not with changes in the sequence of nucleotide, but with DNA chemical modifications, or of the structural and regulatory proteins bound to it. New discoveries about the role of these mechanisms in early development may make it advantageous to return to the indigenous definition of “epigenetics” (8).
Waddington introduced the term epigenetics in 1942 (9) as a refinement of his conception of an “epigenetic landscape” (10). He used the term to describe the class of internal and external interactions between the environment and the genes leading to the development of phenotype. In molecular epigenetics the term “epi” is interpreted as meaning “over,” as in the molecular process sitting over and operating on the genes; However, Waddington knew nothing about molecular processes as sitting over the genes, Avery's identification of DNA as the genetic material wasn't published until 1944 (11) and Waddington could only theorize about the processes involved. His theoretical work was of a piece with his experimental work on environmental influences on the development of phenotype in Drosophila (see (12)) an excellent overview of Waddington's life and work), His view was that there was a landscape of choices facing an organism and the initial constraints and starting point were set by genes, but during development environmental and physiologic forces, increasingly came into play. These forces would then operate along with, and in interaction with genes and each other over time and push (structure) the organism into typically deeper canals resulting in the organism's eventual phenotype. The interactive process—canalization—meant that individual organisms that might have identical genetic make-up could develop radically different phenotypes (13). His view, perhaps predated in some ways by Lamarck (though Waddington wasn't a Lamarckian (13)), was an initial clear statement of a mechanistic theory of gene X environment (GxE) interaction. His conceptualization had profound influences on different fields, especially developmental fields, which strive to specify the nature of the environment and its underlying physiologic and later neurophysiologic effects in interaction with genes on the eventual phenotype of the organism.
The precision of the term ‘‘epigenetics’’ shaped by these findings to become the study of gene expression modifications that do not involve in DNA nucleotide sequences changes (14). Hence, gene regulation of the epigenetic layer controls both normal cellular processes and abnormal events related to disease, notably cancer (15).
For cancer initiation and progression, changes in cellular function by the accumulation of mutations have been recognized as secondary for many years. Inherited or sporadic mutations, activation of oncogenes or the inactivation of tumor suppressor genes, changes in the epigenome (both DNA and histones) may result in the beginning and the development of cancer. To define long term changes in cancer that alter the physiology of a subset of cells in a tissue independent of change in DNA sequence is increasingly used. Epigenetic markers can act in response to alterations in physiological conditions, which can be drivers of the progression of cancer, additionally to gene mutations and epigenetic markers are similarly dynamic. Additionally alterations in DNA methylation, histone modifications and global reprogramming of epigenetic marks are known to occur during malignancy (16).
In cancer deregulated transcription of proto-oncogenes and tumor suppressors plays central role. Distal cis-regulatory elements that are decorated by specific epigenetic marks are known as enhancers, and it is crucial for the regulation of the expression of tissue-specific genes. Enhancer sequence mutations, enhancer-promoter communication alteration, and epigenetic enzymes mis-regulation and transcription factors that bind enhancers lead to enhancer malfunction, which are frequently answerable for a cancer deregulated transcription program (17). The fundamental mechanism leading towards carcinogenesis is the activation of oncogenes or the deactivation of tumor suppressor genes has long been accepted. By the epigenetic phenomena like nucleosome remodelling by histone modifications, DNA methylation and miRNAs mediated targeting of various genes, various biochemical pathways that are necessary towards tumorigenesis are regulated. The alliance of epigenetics in cancer has further strengthened by the existence of mutations in the genes controlling the epigenetic players. For targeted anti-cancer drug therapy, this combination has opened up newer avenues with many pharmaceutical industries focusing on enlarging their research and development pipeline with epigenetic drugs (18), one of example in clinical trial drugs for targeting epigenetic in cancer is for the treatment of haematological malignancies, compound – EPZ-5676 is currently in clinical trial for targeting the enzyme DOT1L (19, 20).
Cancer is a disease caused due to multiple reasons but predominantly caused by modulation in gene expression, where the complex networks ruling homeostasis in multicellular organisms are deranged, which allows cells to grow without reference to the needs of an organism as a whole. The clear sets of cellular control pathways are pretentious and paralysed in nearly all types of cancers (19). Mutational activation of oncogenes or inactivation of tumor suppressor genes (TSG’s) supports the key cellular pathway alterations on the genetic basis of cancer.
Epigenetic alterations regulating heritable changes are critical for the development of all human cancer (20, Table 1, Figure 1). In the epigenetic alterations abnormal patterns of DNA methylation, disrupted patterns of histone post-translational modifications (PTM’s), and alterations in chromatin composition and organization can be observed. These changes in the epigenome occur largely due to disrupted epigenetic machinery. Epigenetic machinery comprises of DNA coiled with histones in a nucleosome. Signalling gene (oncogenes) mutations are often dominant in many human cancers and drive the formation of cancers. Eg: RAS.
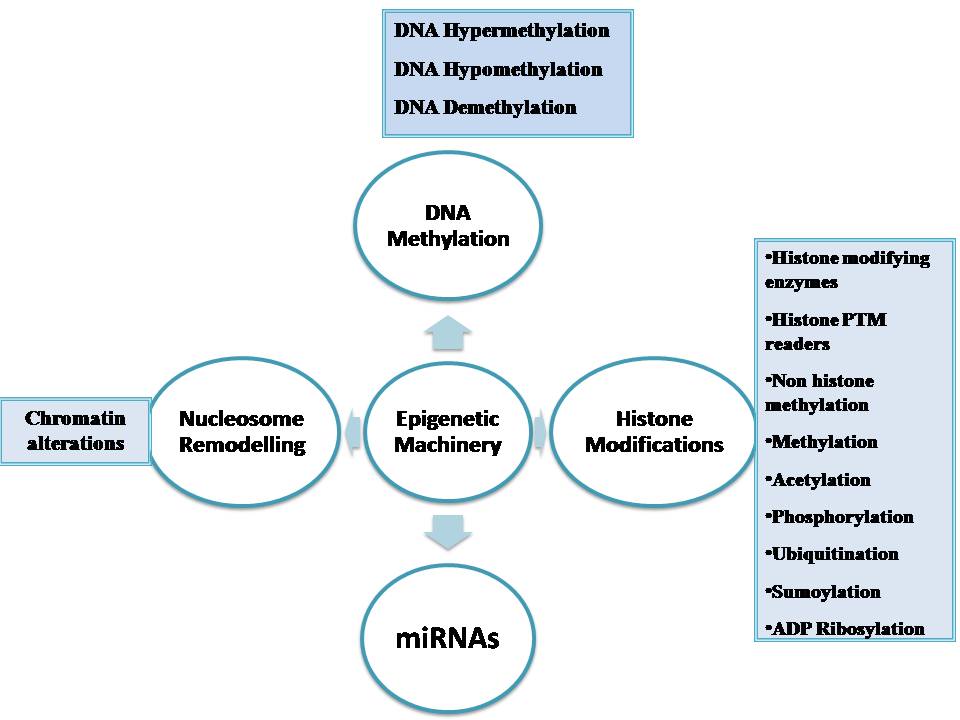 Figure 1
Figure 1The epigenetic machinery.
| Pathway | Epigenetic alteration |
|---|---|
| Self-sufficiency and self-dependant for growth event related signals | Methylation of RASSFIA gene |
| Not sensitivity to antigrowth related signals | Down-regulation of TGF- ß receptors |
| Tissue invasion and metastasis related events | Methylation of E-cadherin promoter |
| Unlimited replication capacity | Silencing of p16 or pRb genes by promoter methylation |
| Continuous angiogenesis and related cellular pathways | Silencing of thrombospondin-1 |
| Strength to evade apoptosis | Methylation of DAPK, ASC/TMS1, and HIC1 |
| Capacity to repair DNA | Methylation of GST Pi, O6-MGMT, MLH1 |
| Genomic instability monitoring cellular pathways | Methylation of Chfr |
| Protein ubiquitination functions regulating mitotic control genes | Methylation of Chfr |
Epigenetic mechanisms are essential for normal development and maintenance of tissue-specific gene expression patterns in mammals (21).Chromatin is made of repeating units of nucleosomes, which consist of 146 base pairs of DNA wrapped around an octamer of four core histone proteins (H3, H4, H2A and H2B) (22). Epigenetic mechanisms that modify chromatin structure can be divided into four main categories: DNA methylation (Figure 2A), covalent histone modifications (Figure 2B), non-covalent mechanisms such as incorporation of histone variants and nucleosome re-modelling (Figure 2C), and non-coding RNAs including microRNAs (miRNAs) (Figure 2D). These modifications work together to regulate the functioning of the genome by altering the local structural dynamics of chromatin, primarily regulating its accessibility and compactness. The interplay of these modifications creates an ‘epigenetic landscape’ that regulates the way the mammalian genome manifests itself in different cell types, developmental stages and disease states, including cancer (23-28). The distinct patterns of these modifications present in different cellular states serve as a guardian of cellular identity. Here, we will discuss the important aspects of the key epigenetic mechanisms present in normal cells.
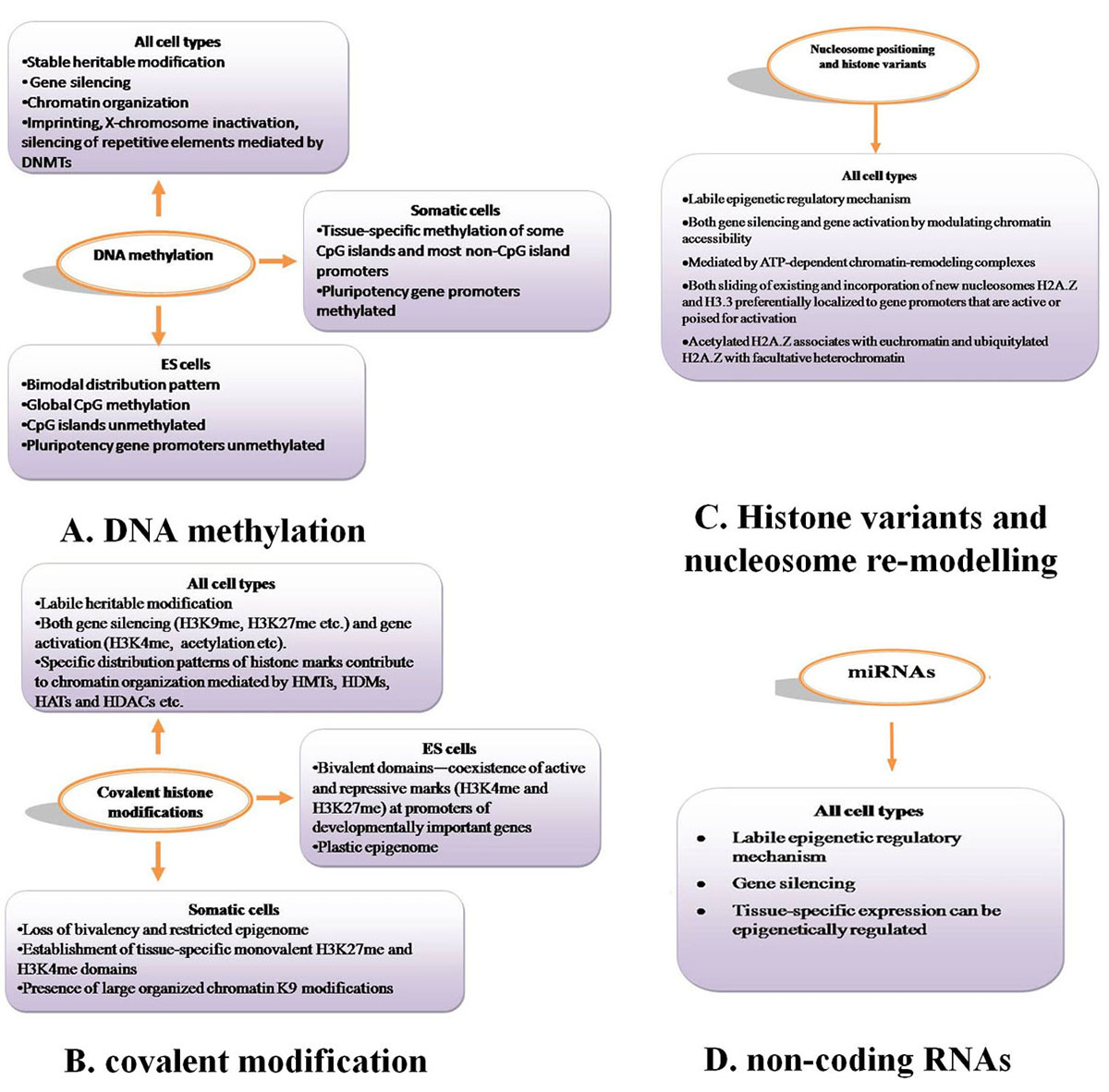 Figure 2
Figure 2Epigenetic mechanisms involved in regulating gene expression and chromatin structure in normal mammalian cells. A. DNA methylation, B. covalent modification. C. Histone variants and nucleosome re-modelling, D. non-coding RNAs.
Epigenetic mechanisms including DNA methylation (Figure 2A), covalent histone modifications (Figure 2B), nucleososme positioning (Figure 2C) and miRNAs (Figure 2D) are essential for normal mammalian development and regulation of gene expression. These epigenetic modifications display unique properties and distribution patterns in different mammalian cells. The distinct combinatorial patterns of these modifications, collectively termed the epigenome, are key determinants of cell fate and gene activity. ES cells maintain a more plastic epigenome required for developmental processes. In contrast, the epigenome of differentiated tissue displays a relatively restricted structure that is stably maintained through multiple cell divisions.
Malignant cancer emerges from normal healthy cells in a multistep process that involves both genetic and epigenetic lesions. Both genetic and environmental inputs participate in driving the epigenetic changes that occur during human carcinogenesis. Malignant cancer cells arise from normal cells via a multistep process that involves both genetic and epigenetic change. Similar to genetic lesions, epigenetic lesions can be diverse in nature, serving to alter the structure and function of the genome thereby participating in a cell’s acquisition of limitless uncontrolled growth and the phenotypic hallmarks of the malignant cancer cell. In general, the degree of epigenetic difference between cancer cells and normal cells greatly exceeds the epigenetic differences that are seen between normal cells of different phenotypes and even different germ layers (e.g., fibroblasts and epithelial cells). Since epigenetic mechanisms are a primary determinant governing normal cell identity, this comparison underscores how epigenetically different cancer cells are from normal cells. Mutation and altered expression of proteins involved in the writing or reading of the epigenetic code are two mechanisms that help produce aberrant epigenetic changes seen in not only cancer, but other human diseases as well. The complexity and the frequency of the epigenetic changes seen in cancer cells, however, seem to defy explanations that rely on a single event. Instead, it appears that pathologic epigenetic change during carcinogenesis results from myriad genetic mutations and environmental inputs which perturb the manifold nodes of epigenetic regulation (29).
Tumorigenesis is a complex and multifactorial progressive process of transformation of normal cells into malignant ones. It is characterized by the accumulation of multiple cancer-specific heritable phenotypes, including persistent proliferative signaling, resistance to cell death, evasion of growth suppression, replicative immortality, inflammatory response, deregulation of energy metabolism, genomic instability, induction of angiogenesis, and activation of invasion ultimately resulting in metastases (30). The acquisition of these cancer-specific alterations may be triggered by the mutational and/or non-mutational (i.e., epigenetic) events in the genome which, in turn, affect gene expression and the downstream phenotypes listed above (30, 31). Furthermore, it has been suggested that epigenetic alterations may play as important or even more prominent role in tumor development (32). Epigenetic events , most prominently manifested by stable and heritable changes in gene expression that are not due to any alteration in the primary DNA sequence ( 33) , signify the fundamental molecular principles in which genetic information is organized and read ( 35) . Epigenetic modifications include change in methylation patterns of cytosines in DNA (35, 36), modifications of the proteins that bind to DNA (35, 36), and the nucleosome positioning along DNA (33). These epigenetic marks are tightly and interdependently connected and are essential for the normal development and the maintenance of cellular homeostasis and functions in adult organisms, particularly for X-chromosome inactivation in females, genomic imprinting, silencing of repetitive DNA elements, regulation of chromatin structure, and proper expression of genetic information (39). The epigenetic status is well-balanced in normal cells, but may be altered in many ways in cancer cells. Additionally, growing evidence indicates that a number of lifestyle and environmental factors may disrupt this epigenetic balance and compromise the stability of the epigenome in normal cells leading to the development of a wide range of pathologies, including cancer (40).
Aging is defined as the unavoidable time-dependent alleviation in both functional and structural integrity of organ physiology. Aging and its associated complications such as overweight, smoking, drinking alcohol and telomerase shortening are considered as one of the major risk factors for cancer development and progression (41, 42). As a result of ultra-modern health care, increase in hygienic knowledge, better nutritional habit (43) and conscious lifestyle, the process of aging is somehow observed to be controlled. Therefore, life expectancy is now noticed to be elevated in many developed and developing countries, for example, 84.118 years in Japan, 83.468 years in Singapore, 82.864 years in Sweden, 81.892 years in the UK and Hong Kong and Macau being topped the list having >84.19 years life expectancy. On the other hand, it leads to a shift in the proportion of people from young to a more aged one. Aging and cancer have a very close relationship, being the former believed to be one of the important causes of the later (44). Mechanisms of both aging and cancer are also found to be common in some cases. Such mechanisms include the role of genomic instability, telomere attrition, epigenetic changes, and loss of proteostasis, decreased nutrient sensing and altered metabolism. So, it is suggested to target both with same or similar strategies even with the same or similar drugs, for example to supress micro RNA that are common in both. However, unraveling clear molecular events sharing both the cellular disorders are anticipated to target them with the same or similar strategies or drugs (45).
Owing to the observed tight association between aging and cancer, it is noticed that both share epigenetic control over their entire process of development and progression. Various epigenetic mechanisms are influenced by several external factors such as environment, pollution, lifestyle and quality and quantity of diet. They are also believed to play a pivotal role in gene expression (46). Dietary supplements such as antioxidants (lycopene, curcumin and vitamin E and A etc) can influence various cellular events associated with aging and cancer as well. Especially, sulforaphane present in cruciferous vegetables and epigallocatechin-3-gallate found in green tea is examined to influence several epigenetic events such as inhibition of the enzyme DNA methyltransferase, histone modifications through enzymes such as histone deacetylase, histone acetyltransferase inhibition and non-coding RNA expression. The above epigenetic pathways found to control both the formation and progression of various neoplasms. Due to the key role in epigenetic modulation, such diets are referred to as epigenetic diets. On the other hand, they can control both the processes of cellular longevity and carcinogenesis through specific key genes that encode telomerase. Therefore, caloric restriction can modulate both aging and cancer-associated events, notably, high caloric diet can up-regulate both the events. So, epigenetic diets that are rich in genistein, sulforaphane, and epigallocatechin-3-gallate are believed to have many health benefits in terms of influencing epigenome positively (42).
DNA methylation is established by DNMTs, which catalyze the adding of methyl group in C5 (carbon 5) position of cytosine to produce C5-methyl-cytosine (5mC). So far, two types of DNMTs have been defined: de novo methyltransferases and maintenance methyltransferases (Figure 3A and 3B). De novo methyltransferases create hemimethylated CpG dinucleotide sites in double-strand DNA, and are responsible for setting up the pattern of methylation. Maintenance methyltransferases add methylation to DNA when one strand is already methylated, and are responsible for maintaining the methylation pattern that had been established by the de novo methyltransferases. According to the catalytic features for methylation, DNMTs are classified into three families: DNMT1, DNMT2, and DNMT3. Generally speaking, DNA 5mC in the genome of mammalian somatic cells is found almost entirely within CpG dinucleotide. It has been proposed that within housekeeping promoters, CpG methylation should be rare at CpG islands, while this modification could be highly prevalent in repetitive sequences of promoters and enhancers, the genes of which are regulated so that they may be stabilized or locked in a silent state (47).
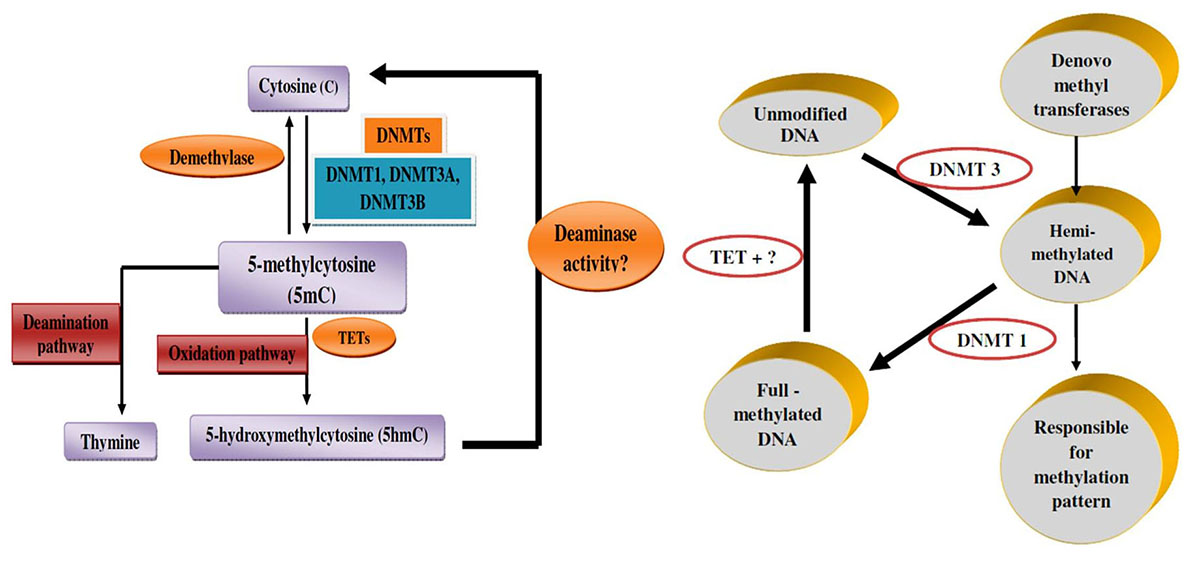 Figure 3
Figure 3Mechanism of DNA methylation. A. Mechanism of DNA methylation (A) DNMTs add methyl group in C5 (carbon 5) position of cytosines to produce 5mC, while TETs catalyze 5mC to 5hmC, then some other factors turn 5hmC back to cytosine. B. Genomic DNA methylation is established by DNMT3 as hemi-methylated templates, and maintained by DNMT1 to full-methylated DNA.
There are many ways that gene expression is controlled in eukaryotes, but DNA methylation is a common epigenetic signalling tool that cells use to lock genes in the off position (48). DNA methylation represents a crucial mechanism for stable gene expression in mammals. The inclusion of a methyl group to the 5' position of cytosine residues inside a CpG dinucleotide sequence context known as DNA methylation. In the genome methylation has a bimodal pattern of distribution, generally, most regions are extremely methylated (85% to 100%) whereas (0% to 5%) of CpG islands are unmethylated (49, 50). In the methylated fraction; many genes, including those only expressed in specific tissues, are located. Whereas genes with CpG island promoters (mainly with housekeeping function) are constitutively unmodified (51).
CpG islands are known as Clusters of CpGs (the predominant target for DNA methylation) which are located at the 5′ ends of many human genes. Almost all CpG islands are unmethylated, in tissues even when the related genes are not expressed. Inspite of that, DNA hypermethylation happens at numerous CpG islands, in cancer as well as in the global DNA hypomethylation (7).
A good deal is known about how the DNA methylation patterns are maintained in in vivo. Originally it was shown that when in vitro, methylated DNA templates are introduced into somatic cells in culture, they retain the exact methylation pattern of the original substrate regardless of sequence, and even after many cell divisions (52, 53). This proposed that during the process of replication, there should be a mechanism for actually copying the position of methyl moieties. The basis for this lies in the symmetry of CpG dinucleotide- each CpG on single strand has a CpG complementary to it on the opposite strand, and methylated sites are most often modified on both strands of the DNA. During the replication process, hemimethylated sites are generated by the synthesis of the new strand. During the process of replication, however, synthesis of the new strand generates a hemimethylated site. This is, then, specifically recognized by the enzyme Dnmt1 (DNA methyltransferase 1) (54) which then methylate the new CpG, thereby copying the methyl group from the native strand in a semi conservative manner (55). Because the Dnmt1 enzyme has a high preference for hemimethylated sites, CpG sites that are not methylated on the parent strand do not serve as good substrates, thus, preserving their unmodified state on the newly synthesized DNA (56).It is now recognized that the specificity for this main reaction does not only depend on the Dnmt1 properties itself, yet it is aided by additional proteins associated with the replication fork (57). As anticipated, in the complex, either knockdown of Dnmt1 or other proteins will lead to overall, nonspecific demethylation in dividing cells (58, 59).
In gene regulation, the mechanism of copying DNA methylation and histone posttranslational modification (PTM) patterns following DNA synthesis likely plays an important role. During replication, the passage of the DNA polymerase complex disrupts nucleosome placement. The indigenous chromatin structure should then be recreated on the newly synthesized daughter DNA molecules (60). Since DNA methylation takes part in creating unreachable chromatin conformations and setting histone modification patterns, (61-64), for preserving DNA methylation patterns, the alive of an autonomous covalent mechanism considerably helps in this reassembly process. Taken with each other, this system serves as a global, long-term repression pathway. In this scheme, most DNA regions, which are mostly methylated at CpGs, are naturally put in a comparatively closed conformation, whereas CpG islands are kept open and in therapy, this kind of gene regulation can be switched. Thus global repression is possible without the need to identify specific sequence element at each individual gene. Global repression may lead to a reduction in transcription. However, this represents only one of the factors that control the multi-cascade process of gene regulation (51).
Gene silencing is always associated with promoter methylation, boosting the feasibility that aberrant methylation might cause silencing and be part of the transforming process. When methylation is advertised to occur at known tumor suppressor genes a strong mechanistic pathway is suggested as a potential role in tumorigenesis (66).
DNA hypermethylation of RB gene (retinoblastoma) controls cell cycle which is one of the first epigenetic lesions to be involved in carcinogenesis and is combined with the loss of RB expression (67, 71).
In carcinogenesis, the case of RB methylation remains one of the able arguments in favor of a causal role for aberrant methylation; RB gene is commonly active in the precursor cells of tumors and promoter methylation seems to have the same consequence as the genetic mutation of the gene (68).Another tumor type in which this happens is microsatellite unstable colon cancer, by germ line mutation of the DNA mismatch repair (MMR) protein MLHI the inherited forms of the disease are commonly caused (65).
Almost 15% of cases of sporadic colon cancer lack MMR gene mutation although still display microsatellite instability, in these cases, MLH1 promoters have methylated and lack expression of the gene (67, 70).
This by the treatment with the demethylating agent 5-aza-2' –deoxycytidine, the MLH1 repression is reported to be reversed in cell lines showing this abnormality (69). The p16INK4a/CDKN2A promoter aberrant methylation has been shown to be present in both human squamous cell carcinomas and in the early stages of neoplastic transformation (71, 72). Similarly, methylation of GSTP1 (π-class glutathione s-transferase) in prostate carcinogenesis, is an early event and it is also found in premalignant lesions (73).
Likewise during the development of specific tumors in colorectal carcinogenesis, hypermethylation of chromosome 17p region, corresponding to the location of the tumor suppressor of p53 has been demonstrated to antecede its allelic loss, suggesting that methylation may not aimlessly mark chromosome regions that are altered (74).
It has been presumed that in malignant transformation, aberrant methylation plays an important role, based on these examples, particularly when methylation has been demonstrated to appear early in the tumorigenic process. Cells with a particular advantage over others, either by causing their increased proliferation or refiance to apoptosis may be provided by the methylation induced silencing of tumor suppressor genes. Because of premalignant cells, clonal expansion could result in the hyperproliferative phenotype which is characteristic of the early stages of tumorigenesis (75).
Genes such as RB, MLH1, and VHL are methylated, in tumor and also mutated commonly and suggesting that hypermethylation of CpG island during tumorigenesis (76). DNA hypermethylation has been used to subdivide tumor types and to distinguish them from non-malignant tissue (77). A CpG island methylator phenotype (CIMP) has been nominated as tumor subgroups with high levels of DNA methylation, and is mostly associated with worse prognosis (78).
Alterations of DNA methylation may contribute to oncogenesis, the initial discovery suggested that the cytosine base in DNA can be methylated to become 5-methylcytosine (5mC), consistently referred to as the 5th base. Over the past 40 years, there have been numerous studies exhibiting that alterations in the 5mC distribution patterns can distinguish cancer cells from normal cells. Partly three considerable routes have been recognized by which CpG methylation can contribute to the oncogenic phenotype. The first is by general hypomethylation of the cancer genome. Second, focal hypermethylation at TSG promoters may happen. Third, direct mutagenesis of 5mC-containing sequences by deamination, UV irradiation, or exposure to other carcinogens is achievable (Figure 4). Above mechanisms for cancer, suggest that the evolution of human cancer is altered at epigenetic homeostasis mechanisms which are central (20).
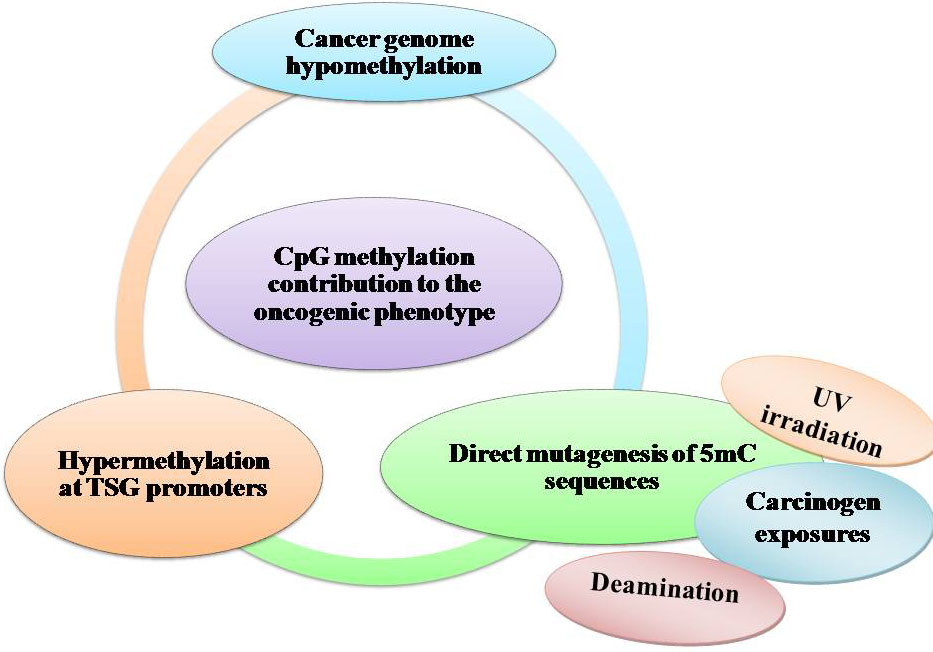 Figure 4
Figure 4DNA methylation and cancer by external agents.
In cancer cells, the most prominent and earliest identified change in DNA methylation patterns were regional decreases in this modification, now recognized as a global DNA hypomethylation by genome-wide analyses (79, 80). Although all of the consequences of these losses still need definition, DNA demethylation potentially contributes to genomic instability and increases in aneuploidies, both of which are classic hallmarks of cancer (80). Actually, deletion or reduction of the maintenance DNA methyltransferase, Dnmt1, results in raised mutation rates, aneuploidies, and tumor induction, a clear indication that DNA hypomethylation plays an effective role in developing chromosomal fragility (81, 82, 83 and 80).
Loss of DNA methylation is accompanied by the activation of transcription, permitting transcription of repeats, transposable elements (TEs), and oncogenes (Figure 5). As authenticated by the expanded frequency of chromosomal recombination at certain genomic regions (hot spots) the activation of repeats may predispose the genome of a cell to recombination or may express the proto-oncogenes which are nearby. Indeed, during the transposition process, transposable elements activation is another potential source of mutations. In the genome, most of the CpGs apart from CpG-rich regions are methylated 80% and in cancer, 40%–60% is the average CpG methylation levels. To map the patterns more precisely, researchers are allowed in advanced mapping technologies. Such studies have divulged that DNA hypomethylation can be fixed in blocks of 28 kb–10 Mb, covering about one-third of the genome (84, 85, 86 and 87).
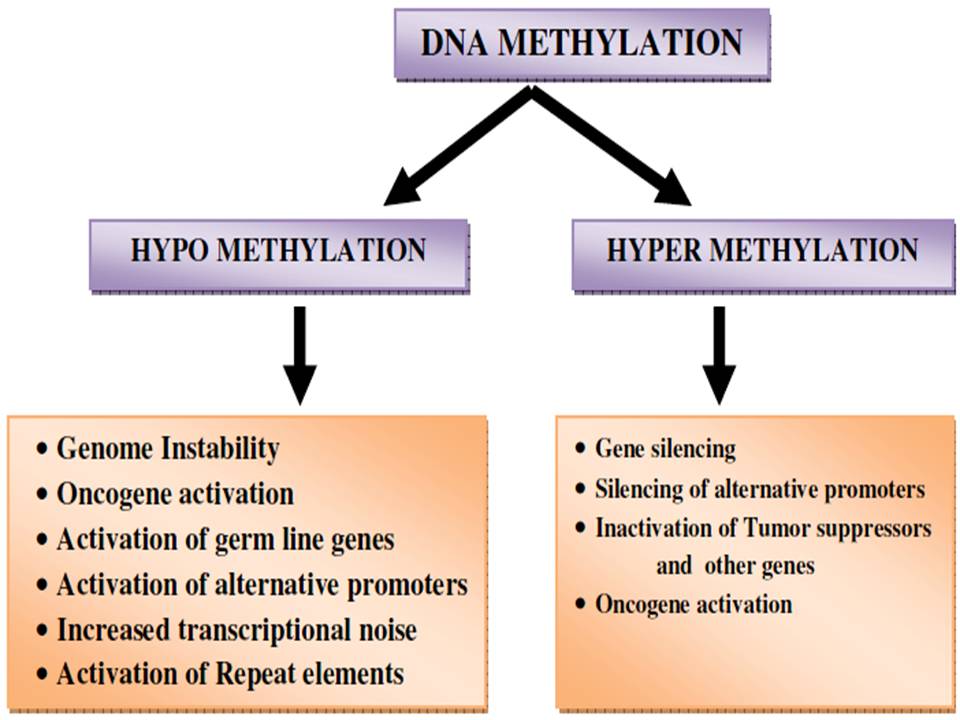 Figure 5
Figure 5Epigenetic alternation by DNA methylation.
The definite mechanism by which DNA methylation is lost from the cancer epigenome is not understood. For example, typically in cancer, a best action is that many regions of DNA hypomethylation could be integrally tied to broad shifts in chromatin organization. The broad epigenomics changes, in turn, could, in some instances, are the, consequences from mutations in chromatin regulators that influence DNA methylation homeostasis, such that the active or passive action of removing DNA methylation is promoted. This could happen, for example, as discussed below and in other articles, by the deregulated activation of ten-eleven translocation (TET) family members or the partial loss of function of the DNA methyltransferase (DNMT) proteins (20).
In cancer, DNA methylation change can be integrally combined with the transcriptional silencing, providing a different mechanism for the inactivation of genes with tumor suppressor function by mutation (20, Figure 6).
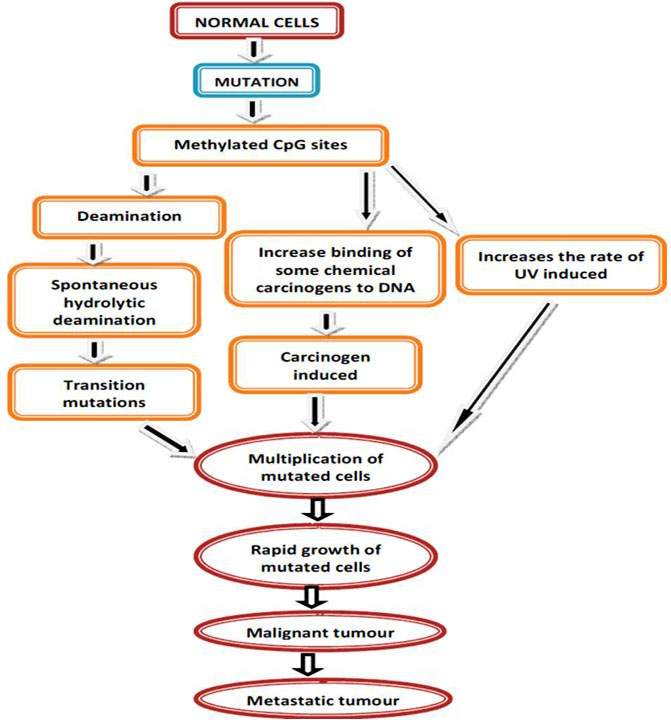 Figure 6
Figure 6Mutation mediated DNA methylation.
Abnormal hypermethylation of CpG islands at 5′ regions of cancer-related genes (i.e., hypermethylation) is well-chronicled DNA methylation change in cancer. About 60% of all gene promoters have CpG islands which are not DNA methylated at normal development or in adult cell renewal systems. Lack of methylation is fundamental, and active, or ready to be activated, the expression status of CGI genes (CpG island genes) in open chromatin states (20, 88). The fact that in cancers (~5% - ~10% of CGI genes), the methylated CpG island promoters are so frequent and are known to contribute to the carcinogenesis directly. The epigenetic therapy has led to new possibilities where epigenetic changes are targeted for therapeutic reversal (89, 90, 91, 92, 93, 94, and 95). It should be noted that 5mC commonly happens in the gene body of active genes and functional ramifications in this region may frequently be opposite to presence of this modification in promoters. In this manner rather than being associated with repression of transcription, gene body DNA methylation may assist the progress of transcriptional elongation and enhance gene expression (96, 97, and 88).CpG-island-specific DNA hypermethylation often occurring at gene promoters, which locks the affected gene into an inactive state. Loss of DNA methylation (hypomethylation) occurs genome-wide and is often observed at repetitive regions of the genome (98) (Figure 7).
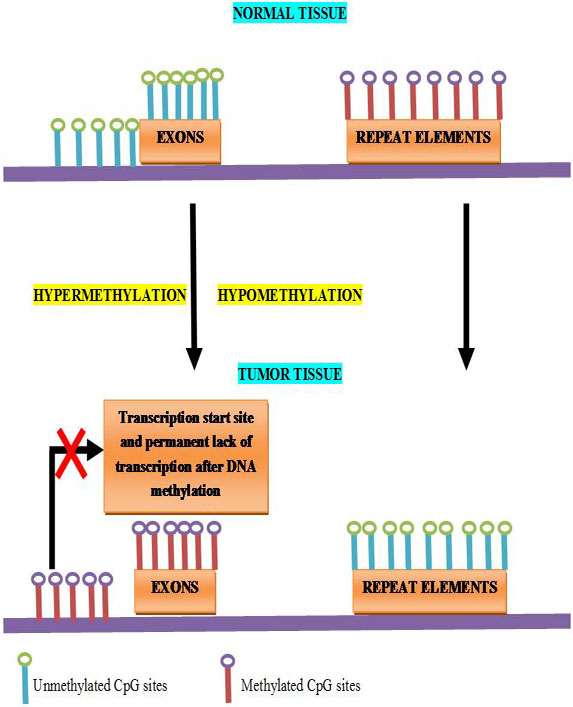 Figure 7
Figure 7Schematic outline of the most relevant DNA methylation changes observed in human cancers.
DNMT3A somatic mutations occur in certain patients with acute myeloid leukemia (AML) may predispose them to a loss - of gene body DNA methylation (99). Mutations in the TET enzymes may be related to a DNA hypermethylation with altered cellular metabolism, relating to IDH1 and IDH2 isocitrate dehydrogenase enzymes, which could involves in cancer. α – ketoglutarate produced by these enzymes are cofactor for the TET hydroxylases. Increase in the formation of abnormal metabolite, by mutations in IDH1/2, 2-hydroxy glutarate is, formed from α – ketoglutarate, hence with Leukemias and brain tumors an increased frequency of DNA hypermethylation can be observed. TET and IDH mutations are mutually exclusive underscores for the requirements of constant demethylation in ensuring the correct level of cellular 5mc in cancer. In the hematopoietic system, importantly an IDH mutation appears to drive tumorigenesis since it blocks the response of a cell to differentiation cues and, hence, skews lineage. Importantly, the experimental drug can change the abnormal DNA methylation patterns, to reinstate an element of cellular differentiation responses; it appears to be related with IDH mutations, showing therapeutic promise for treating these types of cancer (20).
DNA methylation has been postulated, in contribution to cancer development as despite evidence for regional hypermethylation. Global levels of 5-methylcytosine have actually been found to be 5-10% less in tumors compared to normal cells (100, 101). The methylation changes have been suggested to occur specifically between the stages of hyperplasia and benign neoplasia where the DNA was found to be significantly hypomethylated in both benign polyps and malignant tissues when compared to normal tissue (102). Therefore, before the lesions became malignant, methylation patterns were altered, proposing that they could be a key event in tumor evolution. The cause of global hypomethylation is unknown in cancer, but the outcome, in due course, maybe due to deregulation of other genes important for growth control or increased expression of the oncogene. For the demethylation of DNA assorted mechanisms have been proposed; due to the impotence of the maintenance methyl transferase, passive demethylation may happen, to complete the methylation step that would normally be guided by hemimethylated DNA post replication (Figure 8). This is thought to be during preimplantation, in the case of maternal pronucleus which undergoes passive demethylation, most likely expected to sequestration of the oocyte-specific form of DNMT1 (DNMT10) in the cytoplasm (103). Rapid demethylation of the paternal pronucleus appears, by TET3 due to the oxidation of 5-ethylcytosine to 5-hydroxy methylcytosine (104).
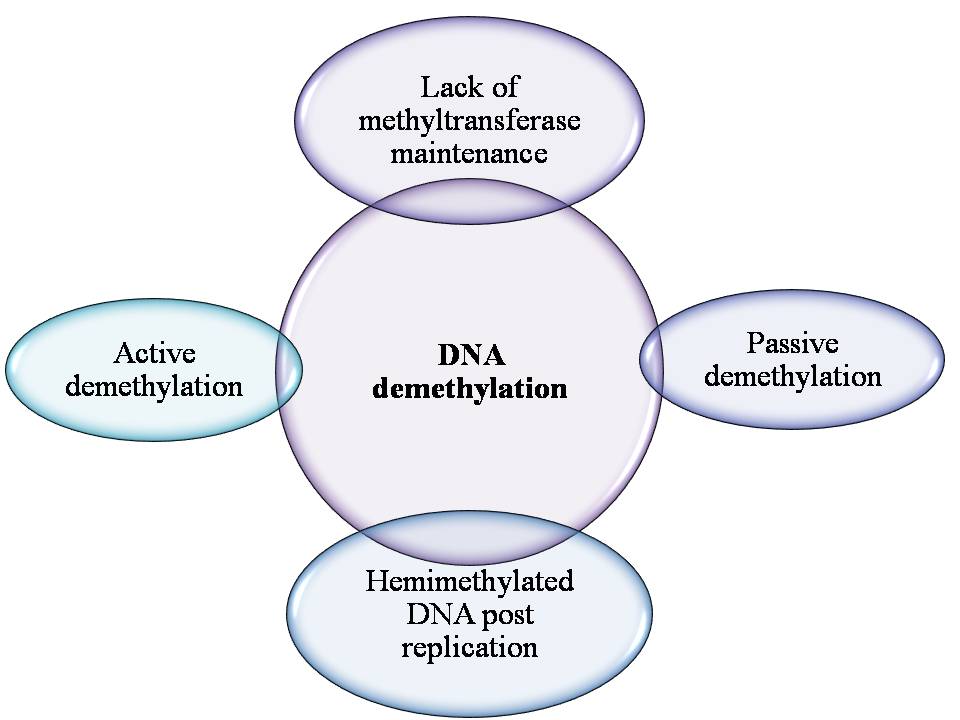 Figure 8
Figure 8Mechanism of DNA demethylation.
There is evidence that the maintenance of methyltransferase DNMT1 does not restore methylation to cytosine’s, in the newly synthesized daughter strand; if the diagonally opposite cytosine is hydroxyl methylated (105) resulting in replication-dependent passive dilution of 5-methylcytosine. In cultured human cells and the adult mouse brain, active DNA demethylation has been demonstrated to involve TET1 catalysed hydroxymethylation persued by AID/APO-BEC- mediated deamination of 5-hydroxymethyl cytosine, with the resulting base mismatch being removed by the base excision repair pathway (106, 107). TET proteins further oxidize 5-hydroxy methylcytosine to 5- formyl cytosine and by thymine DNA glycosylase (TDG) the 5- carboxyl cytosine can be excised and by the base excision repair pathway it has been repaired. In a study, the methylation status of a number of genes, DNA methylation and demethylation cyclic process have shown to occur approximately every hour, which has been examined when the cells were released from a synchronizing block, (108, and 109).
Different possibilities including a dynamic, replication – independent response to alterations in physiological conditions such as hypoxia; are accepted, this was a surprise discovery. The components of the base excision repair pathway and in TDG, a mechanism has been proposed, that these were enlisted to the promoter at the origination of each transcriptionally productive cycle and a reduction in TDG expression impaired demethylation and reduced transcriptional activity. In conflicting to expectations, loss of DNA methylation is mainly associated with loss of function of the TET2 methylcytosine dioxygenase. TET2 is mutated in approximately 15% of myeloid cancers, resulting in impaired hydroxylation. By the oncometabolite 2-hydroxy glutarate, the function of TET2 is also inhibited, generated by mutant IDH1 in acute myeloid leukemias. The downregulation of TET expression has been reported with reduced levels of 5-hydroxy methylcytosine in breast and liver cancers. DNA methylation patterns may be modified, by altered expression or activity of epigenetic regulators such as TET (7).
Chromatin remodelling involves various histone covalent modifications such as acetylation, phosphorylation, and methylation (110). By many chromatins associated protein complexes, the transcriptional state can also be regulated which are either involved in enhancing the promoter activity or fine-tuning and some of these respond to DNA modifications and histone arose altered contexts (Figure 9 and 10). Specific residues is very crucial in maintaining genome integrity, gene expression and evasion of cancer in the histone methylation balance in particular (111, 112, and 113).
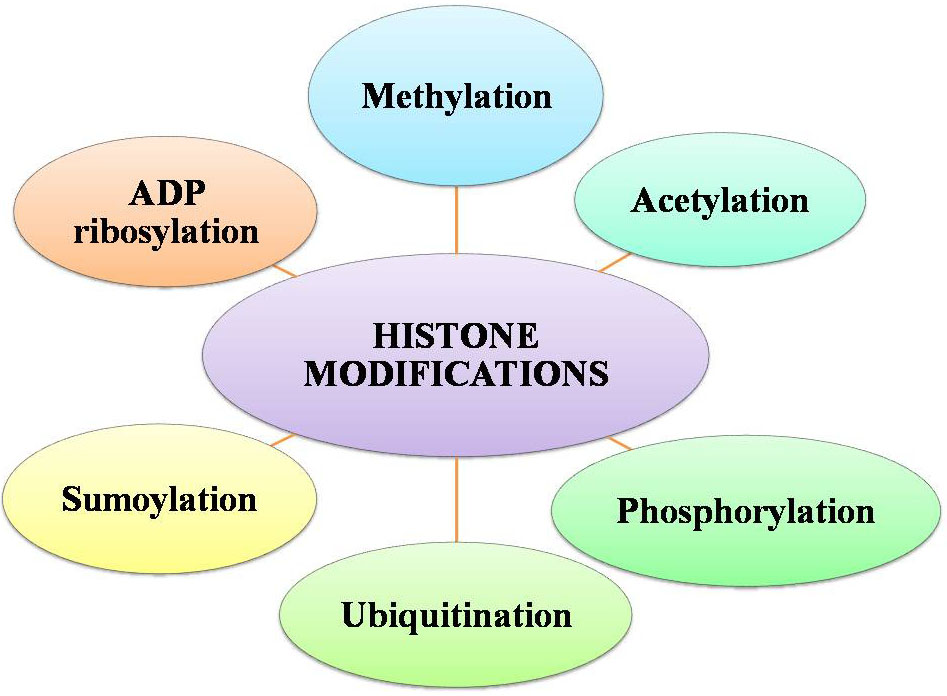 Figure 9
Figure 9Factors for histone modifications.
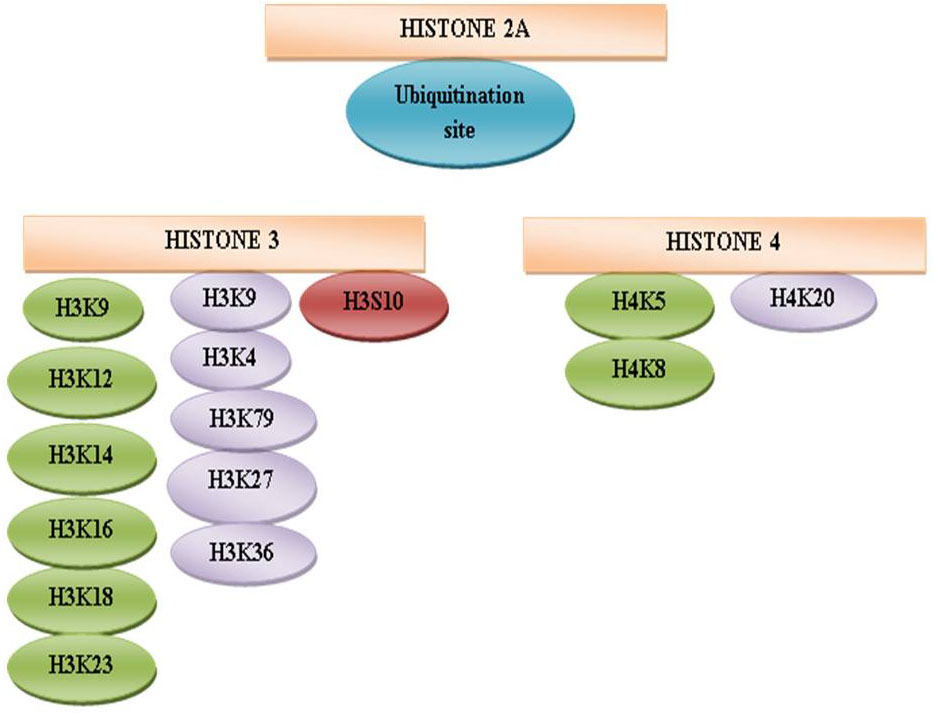 Figure 10
Figure 10Schematic representation of histone modification sites.
The protruding, charged N-terminal amino acid tails of core histones (especially H3 and H4) are hot spots for elaborate post-translational modifications, including methylation (114), acetylation(115) ,phosphorylation (116) , ubiquitination (117), sumoylation(118) and ADP ribosylation (119), (120) (Figure 9). The methylation sites are represented in violet color at H3K4, H3K9, H3K27, H3K36, H3K79, and H4K20 (121). The acetylation sites are shown in green color at amino acid H3K9, H3K14, H3K18, and H3K23 and H4K5, H4K8, H4K12, and H4K16 (122). The phosphorylation site is indicated in brown color at H3S10 (123). An ubiquitination site is randomly designated in H2A (124), H2B. The misregulation of the histone methyltransferases (HMTs) and the histone demethylases (HDMs) has been combined with a variety of cancer types including breast, prostate, lung and brain (125, 126, 127, 128, and 129). Categorically, the HTMs and the HDMs play pivotal roles in regulating multiple tissues methylation status of four lysine residues K4, K9, K27 and K36 on histone H3. Histone modification patterns have also been used similar to DNA methylation patterns, to anticipate diagnosis in multiple cancers. The reduced levels of H3K9ac, H3K9me3 and H4K16ac are corresponding with frequency of non-small cell lung cancer (130).
In prostate cancer, lower levels of H3K4me2 and H3K18ac were combined with poor prognosis. Loss of H3K9me3 has been beginning in patients with acute myeloid leukemia in the core promoter regions of genes. The prognosis of patient in acute myeloid leukemia was additionally able to predict the global H3K9me3 patterns. These cancers have deletions, somatic mutations, and amplifications which all lead to changes in HMTs and the HDMs enzymatic activities. For example, EZH2 (enhancer of zeste homolog 2), the catalytic SET domain mediates the H3K27 (H3K27me3) which is a repressive histone mark trimethylated protein that forms part of PRC2 (polycomb repressive complex 2). EZH2 has been reported to be up-regulated, in metastatic prostate cancer; relative to localized disease or benign prostatic hypertrophy, in prostate cancer development proposing a potential involvement and its overexpression also correlates with breast cancer aggressiveness and poor prognosis. By silencing EP-CAM, the H3K9 methyltransferase G9a promotes lung cancer invasion and metastasis. It is also known that histone H3K9 methylation was influenced by the hypoxia in tumors, as well as the chromatin remodelling factors by increasing G9a protein stability. It should be noted that here, it is the switching off of gene expression that drives tumor progression when the case was in consideration of the role of DNA methylation. However, there is an equal possibility for genes to be switched on through the enzyme changes that alter the epigenome, which is deleterious it would seem that it is the pivotal trigger for the development of tumors by switching off of genes through altering the inherent stable balance in cells (7).
In order to conserve methylation balance, several histone demethylases exist which demethylate specific residues, i.e. the reverse action of the methyltransferases on various histone residues. Two classes of HDM families identified to accomplish demethylation which uses definite biochemical reactions. Lysine specific demethylase 1 (LSD1) was the first enzyme identified to demethylate H3K4me1 and H3K4me2 and later found to also demethylate H3K9me1 and H3K9me2 (131, 132).
For demethylating the substrates, LSD1 is known to utilize flavin adenine dinucleotide (FAD) dependent amine oxidation reaction and appears to be a very promiscuous protein, having the ability to interact with many proteins and to be involved in multiple biological functions. It should be noted that from the use of cofactor, a potential linkage between metabolic state and gene expression arises, and this may be critical to ensure that it does not destabilize the epigenome. Several proteins that have a catalytic JMJC domain includes in the second class of demethylases. Histone residues are demethylated by these enzymes through a dioxygenase reaction which depends on Fe (II) and alpha-ketoglutarate as cofactors. It is interesting again to note the pivotal role of a metabolite which proposes that the assimilation of diverse cellular processes and the environment in which the cell resides is deciding on characterizing the pattern of genes that will be expressed or repressed. JMJC domain-accommodating demethylases such as JHDM3A have the capacity to demethylate trimethylated histone H3K9 andH3K27 residues, unlike LSD1 (133, 134).
More recently, the enzymatic activity affected by the deregulation and mutations has been found for the HDMs. In liver and lung cancers the H3K27 demethylase JMJD3 is found to be down-regulated, while in multiple tumor types, inactivating somatic mutations in the UTX gene are regularly found. Some of these HDMs have been generated in knock-out mouse models and consequence in definite phenotypes including numerous that are lethal, indicating that proper expression of HDMs is critical for development (7).
Other than histones, several proteins have been recognized to be methylated by the HMTs and also demethylated by the HDMs (135, 136, and 137). One of the first non histone substrates identified to be methylated by several HMTs including set9, smyd2, and G9a was tumor suppressor protein P53 (135, 136, and 137) and by LSD1 it also demethylated (137). The transcriptional activity of p53 is specifically regulated by depending on which lysine residue is methylated. By HMTs, methylation of non-histone proteins has been shown to consequence in a range of outcomes ranging from functional activation to repression or degradation (138, 139, 140, and 141).
By stabilising G9a, hypoxia persuades methylation of the chromatin remodelling protein pontin. To hyper activate a subset of HIF-α target genes, methylated pontin has a relation with p300 histone acetyltransferase and HIF-α (141). In hypoxia dependent manner, methylation of another chromatin remodelling protein Reptin increased by G9a. Reptin methylation results in negative regulation of a clear subset of HIF- α target gene, different from pontin methylation(172).Currently, two non-histone substrates of EZH2 have been reported both of which represses its transcriptional activity. By EZH2, GATA4 is methylated which lessen its interaction with its coactivator p300. Some group has shown that by EZH2, methylation of the nuclear receptor ROR α, results in more polyubiquitination and proteasomal degradation most important to decreased transcriptional activity (140). In turn, this causes the loss of ROR α tumor suppressor activity, which eventually leads to the advancement of more aggressive tumors. Not only the histone methyltransferases interact with various non-histone proteins; and found that JMJD1A, one of the HDMs interact with several proteins, perhaps targeting them for demethylation. Consequently, protein methylation net status appears to have a broad range of biological functions. In spite of the fact that the dynamic nature of this non-histone methylation seems to be mainly just as it is the case for histones, demethylation of these proteins has not been studied broadly(7).
Over the activity of a family of so-called nucleosome remodeling ATPases, the eukaryotic chromatin remains flexible and dynamic to acknowledge to environmental, metabolic, and developmental signals. Constant with their helicase ancestry, these enzymes experience conformation changes as they bind and hydrolyze ATP. Simultaneously they interact with DNA and histones, which change histone–DNA interactions in target nucleosome. Their exertion may guide to complete or incomplete disassembly of the nucleosome, the exchange of histones for variants, the assembly of the nucleosome, or the movement of histone octamers on DNA. Remodelling may give DNA sequences approachable to collaborating proteins or, conversely, encourage packing into tightly folded structures. In every aspect of genome function, remodelling processes engage. Remodelling activities are frequently integrated with other mechanisms such as histone modifications or RNA metabolism to assemble stable, epigenetic states (142).
The eukaryotic genome is packaged into the nucleus in the form of chromatin. Beyond a mechanism for packaging, chromatin has evolved as a means for dynamically regulating the genome. At its most basic description, chromatin consists of histone proteins in complex with DNA. Modification of the histone proteins and DNA plays a major role in regulating chromatin structure, and together they form an extensive signaling network. The modification state of chromatin has been found to be responsive to the environment and the metabolic state of the cell, and there is now evidence that some histone and DNA modifications are heritable. Moreover, dysregulation of chromatin signaling pathways underlies a wide range of diseases and disorders, providing a link between the environment and nutrition, gene regulation, and human health and susceptibility to diseases (143).
Considering the importance of chromatin in regulating eukaryotic gene expression and maintaining genome stability, it is perhaps not wholly unexpected that recent genome-wide sequencing studies have uncovered cancer-associated mutations in genes encoding chromatin regulatory factors and enzymes (144, Figure 11).
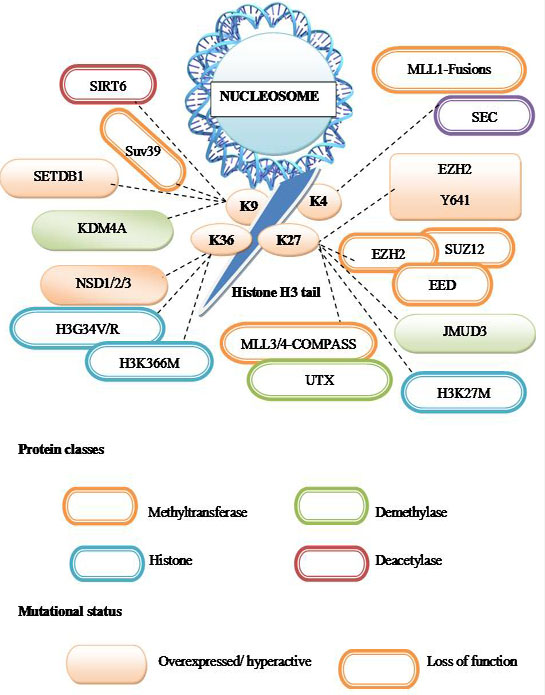 Figure 11
Figure 11Chromatin proteins mutated in cancer.
A summary of cancer mutations that affect post translational modifications of the histone H3 N-terminal tail. Proteins classes are indicated by the outline color; orange – methylation, blue- histone, green-demethylase, brown-deacetylase. Whereas mutational status is indicated by fill color- over expressed/hyperactive, outline color indicates – loss of function. Dashed lines indicate the residue of histone H3 that is expected to be modified due to the indicated cancer mutations.
In the regulation of gene expression, chromatin epigenetic modification plays a main role. Mostly in the sequence CpG and in vitro methylated promoters, DNA is methylated post-synthetically on cytosine residues are known to be generally inactive when transfected into eukaryotic cells (145). By a family of DNA methyltransferase (DNMTs), DNA methylation is catalyzed. Reciprocal methylation of the new DNA strand complementary to hemimethylated DNA was maintained by the DNMT1, and that is produced as a result of semi-conservative DNA replication. DNA methyltransferase is known to be DNMT3a and DNMT3b which is being able to methylate the completely unmethylated DNA duplex in vivo (146, 147). More recently it has been shown that, by a family of Fe2+, 2-oxoglutarate dependent methylcytosine dioxygenases known as TET proteins, 5-methylcytosine can be oxidized to 5-hydroxymethylcytosine (148), by a mechanism that appears to include base excision repair processes, which effectively results in the subsequent removal of the repressive methyl group. Other DNA modifications are also described such as methylation at sites other than CpG (149, 150), and the generation of formyl and carboxyl derivatives of DNA (151).
Beginning discussions that obtained from those that studied transgenerational phenomena concentrated on the classical set of DNMTs. Nevertheless, modifications of epigenetic go beyond DNA methylation. The chromatin histone proteins are also altered in their transcriptional states and N-terminal residues are often related to particular histone modifications (152, 153). The number and complication of the possible amalgamation of these have grown very quickly in recent years (111), but a simplified generalization could be that active genes are associated with acetylation of H3 and H4 histones and methylation of the lysine-4 residue of histone H3 (H3K4). Inactive genes are regularly hypo acetylated and may also be methylated on the lysine-9 (H3K9) or lysine-27 (H3K27) residues of histone H3 (154).
Most of the studies tend to focus either on the DNA or histone modifications and it is clear that in order for a gene to be transcribed there is an interaction between the methylated DNA and the modified histones. Many enzymes have been recognized that methylate, demethylate, acetylate, deacetylate, phosphorylate, ubiquitinate or sumoylate histones. In these enzymes, there is sacking and specificity which is needed to deliver the full range of potential histone post-translational modifications. DNA methylation patterns and modification of histones have been established to be different when normal tissues and tumors derived from them are compared. By their epigenetic status ultimately all gene expression is controlled and it is not astonishing, hence in tumorigenesis, epigenetic change may play a key role. Epigenetic modifications mediated by the enzymes have been found to be mutated in cancers, which add to an indirect manner in which tumors develop as the alteration in the modifier can influence the gene expression patterns. This also suggests that for therapy, epigenetic modifiers may act as novel targets. Mutations of DNMT3a have been noticed in 22% of cases of acute myeloid leukemia (AML) where they are related to a poor outcome (155).
Similarly, in ~15% of myeloid cancers, the methylcytosine dioxygenase TET2 is mutated. In mutant mice, Tet2-deficiency causes myeloproliferation, suggesting a role in stem cell function (156). In multiple human cancers, the H3K27 demethylase UTX is mutated and the highest frequency (~10%) being in multiple myeloma (157). The finding of gene mutations that alter chromatin proposes that the disruption of epigenetic control has a very notable role in the promotion of cancers. Secondary roles are the specific proteins which bind correctly to modified histones. Alteration in their structure can also drive the development of tumours (7).
MiRNA production begins in the cell’s nucleus and involves a series of RNA processing steps (Figure 12). Intergenic miRNA genes are commonly clustered and, along with those located in the introns of protein-coding genes, are transcribed by RNA polymerase II. These transcripts, known as pri-miRNAs, are capped, polyadenylated and are usually several thousand bases in length. Pri-miRNAs are then cleaved by an RNase III enzyme Drosha in association with its cofactor Pasha (in flies) or DGCR8 (in humans) to generate, 70– 90 nt long precursor miRNA (pre-miRNA) which folds into an imperfect stem–loop hairpin structure. These pre-miRNAs are transported to the cytoplasm by exportin 5, where they are further processed by Dicer to form a transient 22 nt mature double stranded (ds) miRNA (miRNA duplex). One strand of this duplex is preferentially incorporated into a miRNA-associated RNA induced silencing complex (miRISC). The mature miRNA guides RISC to target mRNAs containing a sequence partially complementary (miRNA target site) to the miRNA (158) (Figure 12).miRNA genes are generally transcribed by RNA polymerase II (Pol II) within the nucleus to form large capped and polyadenylated pri-miRNA transcripts. These pri-miRNA transcripts are processed by the RNase III enzyme Drosha and its cofactor, DGCR8, to a pre-miRNA precursor product. The pre-miRNA is then transported to the cytoplasm by exportin 5. Subsequently, another RNase III enzyme, Dicer, processes the pre-miRNA to generate a transient, 22 nucleotide miRNA: miRNA* duplex. This duplex is then loaded into the miRNA-associated RNA-induced silencing complex (miRISC), which includes the Argonaute proteins, and the mature single-stranded miRNA is preferentially retained in this complex.
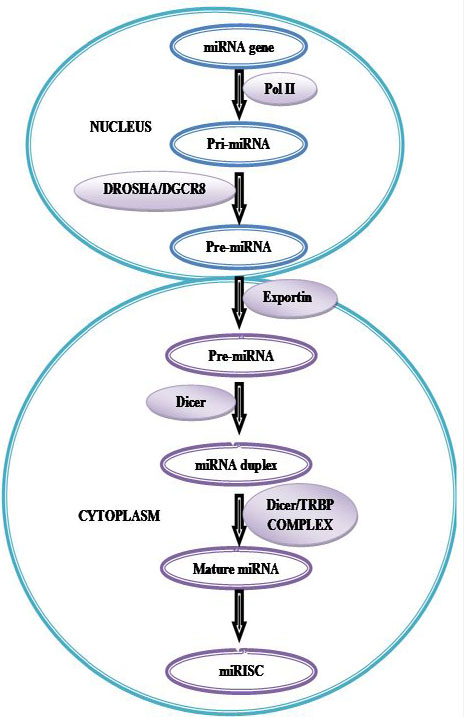 Figure 12
Figure 12Biogenesis of microRNAs (miRNAs).
A large number of studies have demonstrated that miRNAs are key regulators of a variety of fundamental biological processes such as development, cell proliferation, apoptosis, fat metabolism, haematopoiesis, stress resistance, neural development, death and, importantly, tumourigenesis (159, Figure 13).
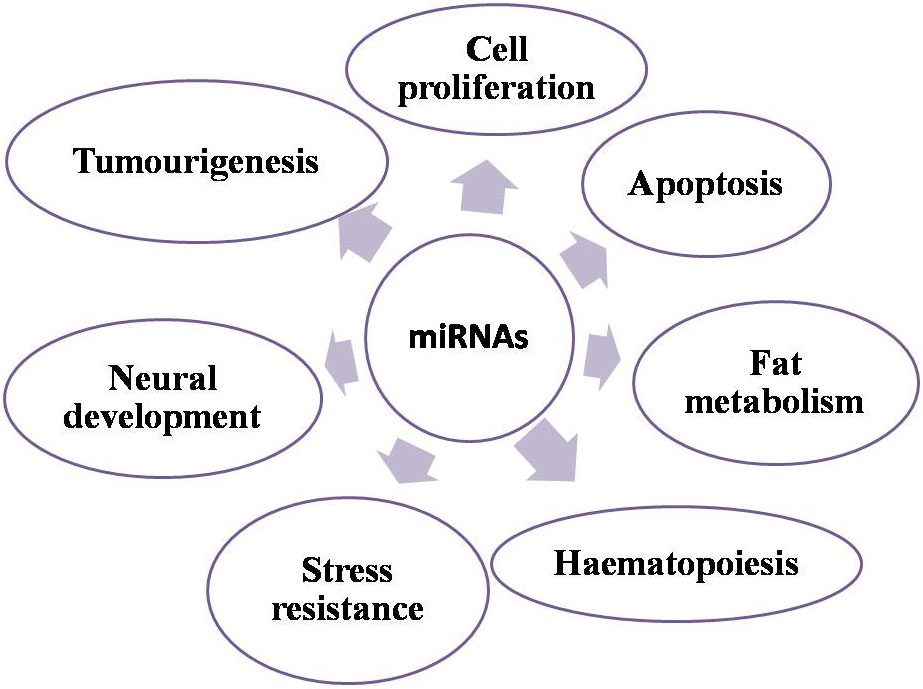 Figure 13
Figure 13Biological roles of miRNA.
Accumulating evidence demonstrates the importance of miRNAs in cancer. In contrast to the tight regulation during development and in normal tissues it is now well established that miRNAs are misregulated in cancer. MiRNAs that are overexpressed in cancer may function as oncogenes, and miRNAs with tumour suppressor activity in normal tissue may be downregulated in cancer (160, Figure 14).
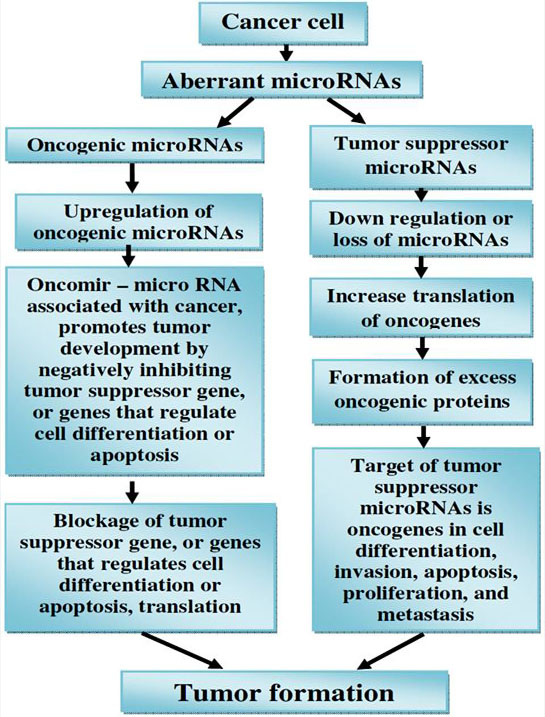 Figure 14
Figure 14microRNAs (miRNAs) as tumour suppressors and oncogenes.
Downregulation or loss of miRNAs with tumour suppressor function may increase translation of oncogenes and hence formation of excess oncogenic proteins, leading to tumour formation. On the other hand, upregulation of oncogenic miRNAs may block tumour suppressor genes and also lead to tumour formation.
MicroRNAs (miRNAs) consist of short noncoding RNA species, which regulates post transcriptional gene expression. Recent studies have demonstrated that epigenetic mechanisms, including DNA methylation and histone modification, not only regulate the expression of protein encoding genes, but also miRNAs, such as let7a, miR9, miR34a, miR124, miR137, miR148 and miR203. Conversely, the expression of important epigenetic regulators, are controlled by another subset of miRNAs including DNA methyltransferases, histone deacetylases, and polycomb group genes. This intricate network of feedback between epigenetic pathways and miRNAs appears to form an epigenetics–miRNA regulatory circuit and to organize the whole gene expression profile. Normal physiological functions are interfered with, contributing to various disease processes, when this regulatory circuit is disrupted (44, 161).
Previous literature has suggested that miRNAs are epigenetically regulated and in cancer deregulation of miRNAs has been extensively studied. In the cell most of the miRNAs are involved in regulating cell cycle progression, apoptosis, differentiation and other critical process and alterations in numerous cancer types are implicated them through epigenetic pathways (162). Recent research has clearly documented the role of miRNAs in all the hallmarks of cancer(161).For example at the chromosome location 13q14.3 the miR-15 and miR-16 was identified which is mostly deleted in chronic lymphocytic leukemia leading to aberrant expression of anti-apoptotic genes (163). Even though studies have recognized the over expression of miR-9 in brain, hypermethylation of miR-9 loci is apparent in numerous tissue including colon, neck and lung carcinoma (164). Additionally, the locus of miR-9-1 is heavily methylated both in invasive ductal carcinoma and the intra-ductal component of invasive ductal carcinoma of breast (165). Additionally, a recent study has indicated that miR-9 gene CpG island methylation was greatly higher in gastric cancer tissue (166). Moreover, in the metastasis of esophageal squamous cell carcinoma, the role of miR-9 has been established via suppressing E-cadherin (167). In the development of cancer, members of the miR-148/152 family consisting of miR-148a, miR-148b and miR-152 play a significant role. Growing evidence has recognized that miR-148/152 family members as potential oncogenes and tumor suppressor genes. In the plasma of multiple myeloma patients, studies have reported the upregulation of miR-148a leading to poor survival (168).
Furthermore, in hepatocellular carcinoma, the up-regulation of miR-148b was also observed (169).At the same time, especially in breast cancer, the studies have indicated the anti-tumor effect of miR-148a, whereby targeting MMP-13 it was able to halt the proliferation and migration of breast cancer cells (170).
Due to methylation occurring at the CpG islands of miR-148/152 family member genes, the expression of miR-148/152 family members is reduced. Literature suggests that over expression of DNMT1 in gastric cancer caused hypermethylation of miR-148a gene leading to its inactivation (171).
Moreover, in carcinogenesis, TGFβ signaling pathway plays a key role and is a target of miR-148 family members. By DNA methylation, epigenetic inactivation of the miR-148 family which leads to enhanced signaling of TGFβ leading to tumor growth and metastasis (170). The production of various target proteins associated with cell cycle progression and apoptosis is controlled by miR-34a, by DNA methylation occurring in the CpG island next to its transcriptional start site, miR-34a is inactivated, which is a frequent observation in various malignancies (171).
Additionally, Kwon and colleagues demonstrated that in human cholangiocarcinoma cells the expression of miR-34a is epigenetically silenced and suggesting its tumor-suppressive role (172). Hypermethylation of miR-34b/c, in soft tissue sarcomas (STS), is very frequently noticed in its late clinical stages (173). In some cancers caused by CpG island methylation, downregulation of miR-137 has been observed (174, 175, and 176).
Gathering evidence has recognized that miR-137 ectopic expression significantly lowered the levels of Cdc42 and Cdk6, and in lung cancer cells leading to cell cycle arrest at the G1 phase (177). Most frequent miRNA in the brain is miR-124 and a deviant expression leads to central nervous system related malignancies (178).Including glioblastomas, in numerous cancers, a diverse mode of miR-124 expression has been observed. Recent report suggests that miR-124 acts as a tumor suppressor and by targeting STAT3 it might be useful in treating human glioblastomas (179).Furthermore, studies have identified that the hepatitis C virus (HCV) induction of DNMT, in HCV related intrahepatic cholangiocarcinoma, led to the suppression of miR-124 (180). In non- Hodgkin’s lymphoma a greater frequency of miR-124-1 gene hypermethylation was observed. miR-200 is recognized as a cell’s autonomous suppressor of epithelial to mesenchymal transition (EMT) and metastasis (181). Reports suggest that in numerous cancer it has been identified that the finger E-box binding homeobox transcription factor 1 (ZEB1) is involved in EMT. Studies have identified that in colorectal cancer cells, miR-200 over expression inhibits ZEB1 mediated metastasis. Indeed it has been demonstrated that by CpG island hypermethylation of miR-200 silencing, causes the transition between EMT and vice versa leading to tumorigenesis (18).
In cancer progression, tumor hypoxia is an example of how epigenetic reprogramming happens. As a result of the limitation of oxygen diffusion in avascular primary tumors or their metastases, in solid tumors; hypoxia occurs (7).
The effectiveness of radiation and chemotherapy significantly reduced by persistent hypoxia and leads to poor outcomes. This is mostly due to prosurvival genes increase, which suppresses apoptosis such as c-myc, AMPK, GLUT1, and BNIP3 and enhance tumor angiogenesis, epithelial-to-mesenchymal transition (EMT), invasiveness and metastasis (183, 184, 185, 186, 187, 188).On examining the transcriptional targets of HIFs (hypoxia-inducible factors), ample of tumor hypoxia research has been centered. Oxygen regulated α subunit (HIF-1 α or HIF-2 α) and constitutively expressed β subunits (HIF-1 β) are comprised by HIF-1 α which is a heterodimeric transcription factor. An oxygen responsive transcription factor is HIF-1 α, which mediates adaptation to hypoxia (189, 190).HIF- α is stabilized and translocates to the nucleus, under low oxygen concentrations, leading to specific target gene expression through binding of HIF-1 β to a hypoxia response element (HRE) (191). The hypoxia-inducible factor 1 transcriptional activator complex (HIF-1) is involved in the activation of the erythropoietin and several other hypoxia-responsive genes. The HIF-1 complex is composed of two protein subunits: HIF-1beta/ARNT (aryl hydrocarbon receptor nuclear translocator), which is constitutively expressed, and HIF-1alpha, which is not present in normal cells but induced under hypoxic conditions. The HIF-1alpha subunit is continuously synthesized and degraded under normoxic conditions, while it accumulates rapidly following exposure to low oxygen tensions (192). Hundreds of genes, supervised by HIF- α, is involved in many biological processes including tumor angiogenesis, invasion, glycolysis, metabolism and survival and hence dramatically in these conditions, changes in the functioning of cells. Hypoxia not only activates gene expression but also involved in gene repression. While some of these genes, by the recruitment of specific repressors such as DEC1 and snail, they are known to be transcriptionally down-regulated (193).
In hypoxic conditions, it has been shown that the expression of G9a and EZH2 are raised, leading to global hypermethylation of H3K9 and H3K27 respectively (7). By hypoxia these repressive modifications were elevated in the promoter regions of tumor suppressor genes such as RUNX3 and MLH1 which correlated with their silencing, potentially promoting tumor progression (194, 195). In tumor the activity of G9a is deregulated, in hypoxic conditions methylation of the non-histone proteins Reptin negatively or positively maintains the transcription of a particular set of genes involved in tumor metastasis (141, 182). Examples of gene regulated by Reptin are VEGF, BNIP3 & PGK1 (196).
Cancer is usually recognized as a disorder initiated by changes in DNA. However, the high genetic changeability seen in cancer cells leads to difficulties to understand these alterations. This makes further complicacy for the treatment of cancer. Therefore, a trend has been noticed to identify and understand the mechanism of cancer caused by non-genetic factors. Following such trends, the involvement of oxygen is identified as one of the important non-genetic factors to be targeted for cancer therapy. Especially, targeting activated hypoxia-inducible factor 1 (HIF-1) that plays an important role in cancer development in tumor mass provides a new window for cancer therapy. Such novel views on the involvement of oxygen as one of the nongenetic factors that may lead to the altered oxygen metabolism and production of active oxygen species is believed to be crucial for the therapeutic attempt (197). It may be noted that reactive oxygen species and oxidative stress resulted by environmental conditions and habitual bad lifestyle is one of the major consequences or causes of aging and associated diseases including cancer (198, 43, 199, 200, 201, 41).
In normoxia, proteasomal degradation of HIFs prevents HIF- α binding to a hypoxia response element (HRE) and transcriptional activation does not occur (Figure 15). To maintain homeostasis the expression of other genes can be regulated by methylation at histones H3K9 and H3K27 by G9a and EZH2 respectively (Figure 16).
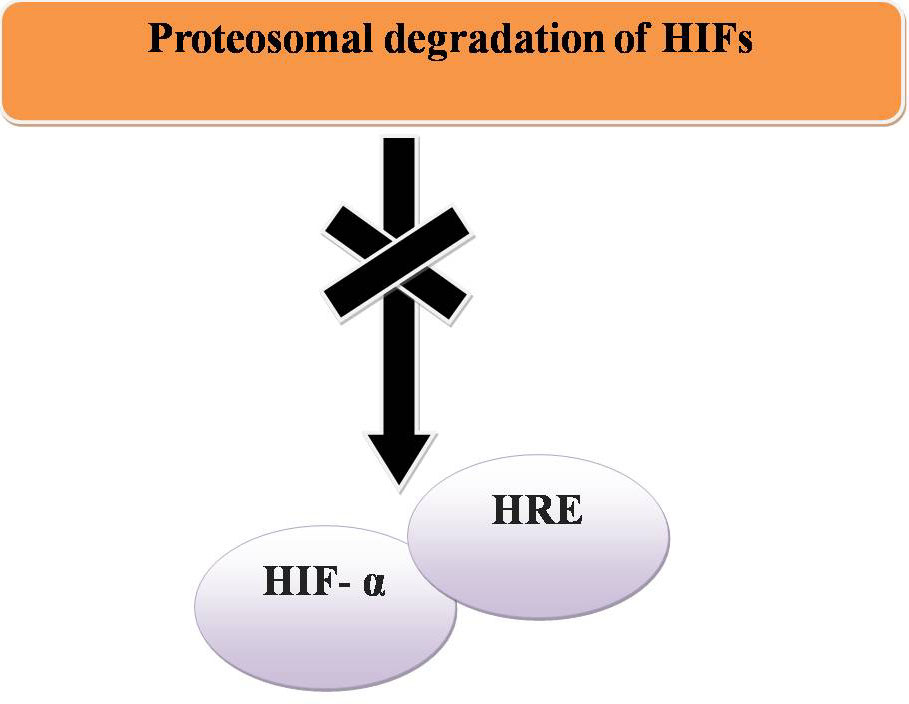 Figure 15
Figure 15Normoxia and cancer.
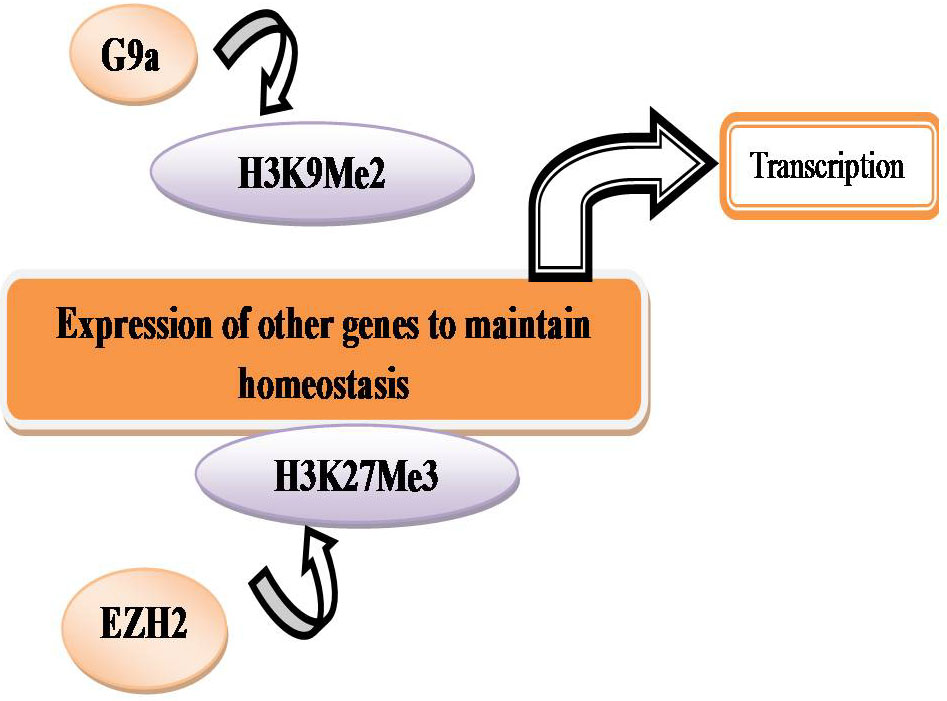 Figure 16
Figure 16Histone methylation and expression of genes.
In hypoxia HIF- α is stabilized and is able to bind to HREs and activate transcription (Figure 17). By co-regulators the transcriptional activity of HIF 1-α can be altered. In hypoxia, G9a methylates chromatin remodeling complex proteins such as Reptin and pontin. By HIF- α at a subset of HIF- α target genes, methylated Reptin negatively regulates transcriptional activation by recruiting a transcriptional co-repressor (7). Conversely, by increasing the recruitment of a transcriptional co-activator, pontin methylation potentiates HIF- α- mediated transcription at other distinct subsets of HIF- α target promoters (Figure 18). In hypoxia, the expression of histone methyltransferases such as G9a and EZH2 is raised, which leads to silencing of tumor suppressors through the histones H3K9 and H3K27 hypermethylation (7, Figure 19).
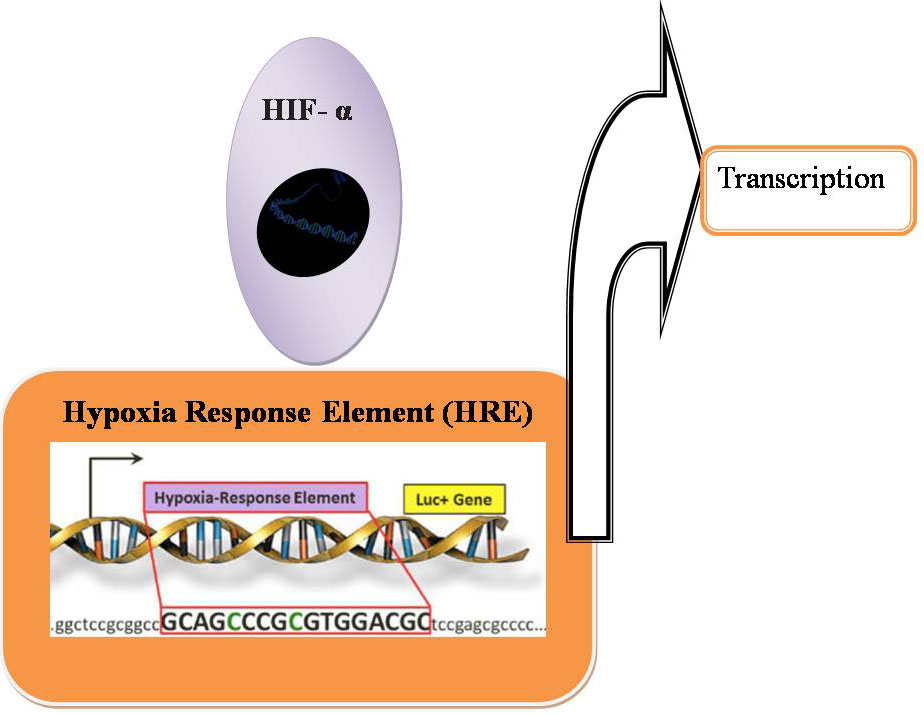 Figure 17
Figure 17Hypoxia inducible HIF expression.
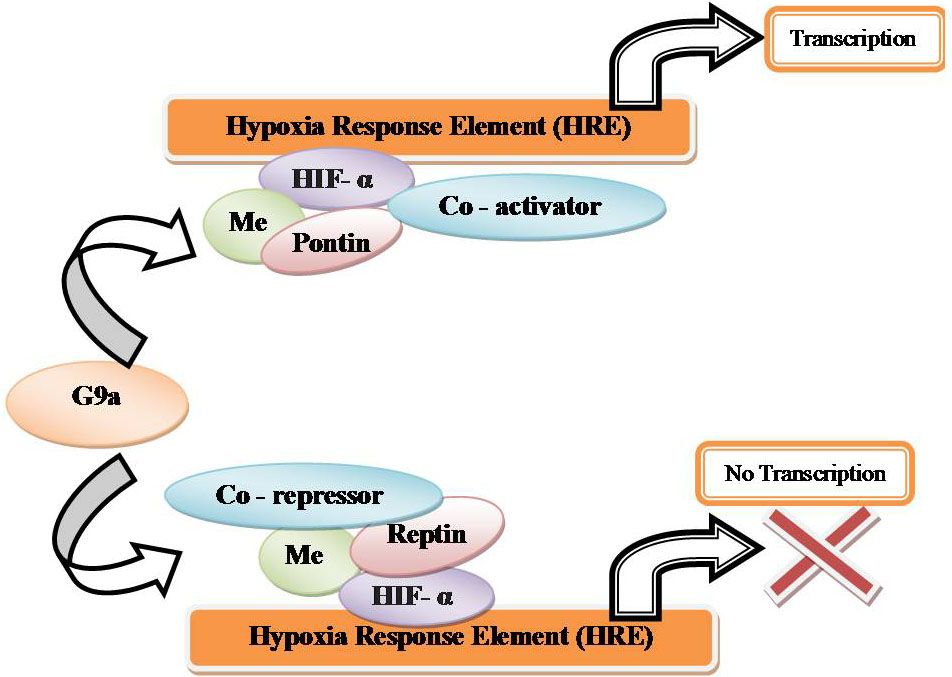 Figure 18
Figure 18Co-activators and co-repressors under hypoxia inducible gene expression.
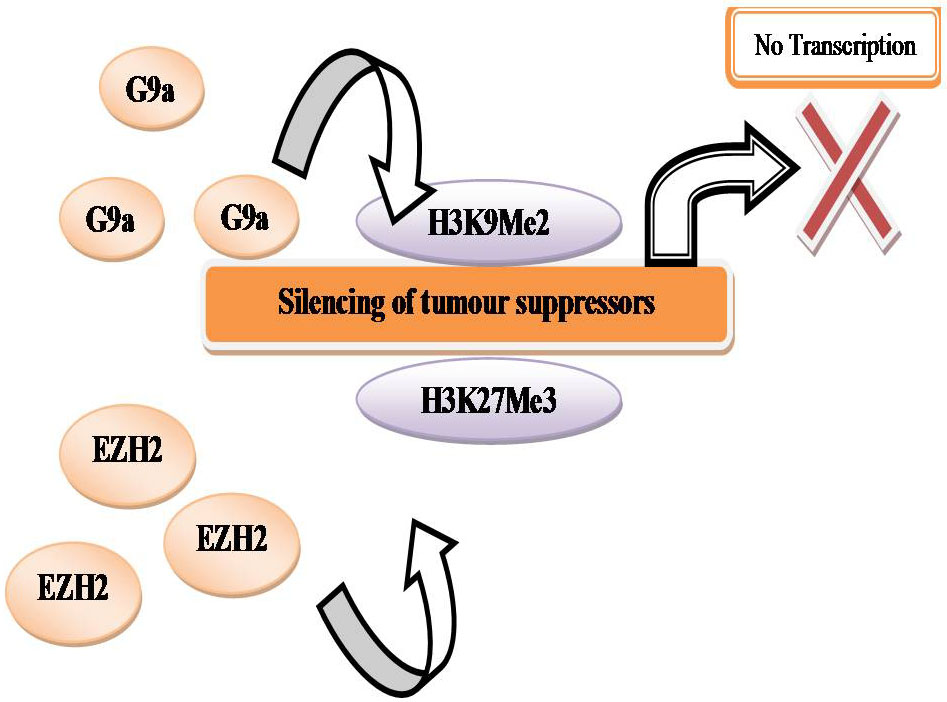 Figure 19
Figure 19Silencing of tumour suppressors.
Through fundamental research on how hypoxia-driven or related diseases such as cancer are initiated and progress and a functional link between hypoxia and epigenetics has been divulged. The drug resistant cancer cells can be driven by hypoxic tumor microenvironments (202), Disseminated Tumor Cells (DTCs) are detected in the peripheral blood, bone marrow or lymph nodes in cancer patients (203).
Metastasis can be emanated from DTCs and it can remain dormant in cancer patients with no sign of disease for several years before reactivation (204). The fate of DTCs can be influenced by a hypoxic microenvironment by up-regulating the key dormancy, NR2F1, DEC2, p27 genes (205). Among dormancy inducing genes, up regulation of NR2F1, an orphan nuclear receptor is epigenetically controlled. In dormant cells, NR2F1 is highly expressed but not in proliferative tumor cells. NR2F1 mRNA expression increases in proliferative tumor cells, when treated with 5-aza-deoxycytidine, an inhibitor of DNA methylation (206).
Additionally, in dormant tumor cells, transcriptional activation markers H3K4me3 and H3K27ac are enriched on NR2F1 transcription start site, whereas, in proliferative tumor cells the transcriptional repressive mark H3K27me3 is enriched in NR2F1 promoter (205).Even though NR2F1-dependent dormancy induced by hypoxic microenvironments, primary tumors under hypoxic microenvironments give rise to a subpopulation of dormant DTCs which elude chemotherapy (205). For the origin of cancer recurrence or metastasis, these post-hypoxic dormant DTCs may play an important role, which is resistant to therapeutics and this research suggests that hypoxic environment can give rise to various types of cancer heterogeneity. RRx-001 catalyzes the reduction of nitrite to nitric oxide, which accumulates in poorly oxygenated tumor. For the treatment of solid tumors, RRx-001 is currently under Phase II clinical trials, alone or in combination with other drug. Interestingly, RRx-001 automatically reduces expression and activity of DNMT1, DNMT3A, and DNMT3B and reduces global DNA methylation levels with apoptosis of cancer cells (207). RRx-001 can be a new hypoxia-selective epigenetic drug since RRx-001 has a different mechanism of action compared to conventional DNMT inhibitors. In some recent studies, various hypoxia-driven or related diseases showed that how epigenetic enzymes including histone methyltransferases and demethylases can dynamically affect and regulate. However, to confirm whether histone methylation-related enzymes are novel and potent targets of epigenetic drug and by clinical validation will be needed to confirm (208).
The most exciting recent advance for achieving durable management of advanced human cancers is immunotherapy, especially the concept of immune checkpoint blockade. However, with the exception of melanoma, most patients do not respond to immunotherapy alone. A growing body of work has shown that epigenetic drugs, specifically DNA methyltransferase inhibitors, can upregulate immune signaling in epithelial cancer cells through demethylation of endogenous retroviruses and cancer testis antigens. These demethylating agents may induce T-cell attraction and enhance immune checkpoint inhibitor efficacy in mouse models. Current clinical trials are testing this combination therapy as a potent new cancer management strategy (209).
The expression of immune-checkpoint proteins can be dysregulated by tumours as an important immune resistance mechanism. T cells have been the major focus of efforts to therapeutically manipulate endogenous antitumour immunity owing to: their capacity for the selective recognition of peptides derived from proteins in all cellular compartments; their capacity to directly recognize and kill antigen-expressing cells (by CD8+ effector T cells; also known as cytotoxic T lymphocytes (CTLs)); and their ability to orchestrate diverse immune responses (by CD4+ helper T cells), which integrates adaptive and innate effector mechanisms. Thus, agonists of co-stimulatory receptors or antagonists of inhibitory signals (the subject of this Review), both of which result in the amplification of antigen-specific T cell responses, are the primary agents in current clinical testing (Table 2). Indeed, the blockade of immune checkpoints seems to unleash the potential of the antitumour immune response in a fashion that is transforming human cancer therapeutics (210).
| Target | Cellular Role | Antibody or Ig fusion protein | State of clinical development1 |
|---|---|---|---|
| CTLA4 | Receptor for inhibition | Ipilimumab | FDA approved drug, used for melanoma, Phase II and III trials are under process for multiple cancers |
| Tremelimumab | Previously tested in a Phase III trial of patients with melanoma; not currently active | ||
| PD1 | Receptor for inhibition | MDX‑1106 (also known as BMS‑936558) | Phase I/II trials are done in patients having melanoma, renal and lung cancers |
| MK3475 | Phase I trial is done in multiple cancer conditions | ||
| CT‑011‡ | Phase I trial is done in multiple cancer conditions | ||
| AMP-2242 | Phase I trial is done in multiple cancer conditions | ||
| Receptor for apoptosis | Pembrolizumab | FDA approved for metastatic melanoma, first line treatment for metastatic non squamous non small cell lung cancer (NSCLC) | |
| Receptor for apoptosis | Nivolumab | FDA approved for metastatic melanoma, non squamous non small cell lung cancer (NSCLC) | |
| PDL1 | Ligand for PD1 | MDX‑1105 | Phase I trial in multiple cancers |
| Multiple mAbs | Phase I trials planned for 2012 | ||
| Ligand for apoptosis | Atezolizumab | FDA approved drug for urothelial carcinoma, non squamous non small cell lung cancer (NSCLC), Triple-Negative Breast Cancer (TNBC) | |
| Monoclonal antibody | FDA approved for the disease Metastatic Merkel cell Carcinoma (MCC) | ||
| Durvalumab | FDA approved for the disease metastatic urothelial carcinoma, non squamous non small cell lung cancer (NSCLC) | ||
| LAG3 | Inhibitory receptor | IMP321|| | Phase III trial in breast cancer |
| Multiple mAbs | Preclinical development | ||
| B7‑H3 | Receptor for inhibition | MGA271 | Phase I trial is done in multiple cancer conditions |
| B7‑H4 | Receptor for inhibition | Preclinical development | |
| TIM3 | Inhibitory receptor | Preclinical development |
Immune checkpoint inhibitors, cancer immunotherapy has shown encouraging clinical results. In solid tumors, the efficacy of immunotherapy is not as effectual as blood cancers (Figure 20). Consequently, in more types of cancer including various solid tumors, applying and expanding cancer immunotherapy is considered to be the main breakthrough in cancer treatment. Cancer immunotherapy may be resisted by the microenvironment of hypoxic solid tumor. Accordingly, the studies on the effect of solid tumor hypoxic microenvironment on immune suppression such as T cell exhaustion should be more actively conducted. In the well-oxygenated environment, the degree of T cell activation is stronger proposing that T cell activation is inhibited in the oxygen-poor tumor microenvironment (211). Additionally, immunosuppressive cells such as myeloid-derived suppressor cells (MDSCs) are attracted by tumor hypoxia (212). In the tumor microenvironment, hypoxia alters the function of MDSC and redirects their differentiation toward tumor-associated macrophage (213). As compared with splenic MDSCs, on tumor-infiltrating MDSCs the expression levels of PD-L1 immune checkpoint are known to be higher (214). As a HIF-1 direct target, in MDSCs by hypoxia PD-L1 is unregulated (216).Under hypoxia, MDSC-mediated T cell suppression decreased by the blockage of PD-L1, suggesting that in cancer patient’s combinatorial therapy targeting tumor hypoxia along with PD-L1 blockage might encourage the immune system. Recently, two remarkable studies have reported that in extensive chromatin changes, T cell exhaustion is highly associated (215, 216).
 Figure 20
Figure 20Dysregulation of epigenetic regulators or transcription factors (involved in hypoxia) are associated with hypoxia-driven or related diseases
In cancer cells and in immune cells, hypoxic stress causes epigenetic changes, to enable the application of a broad spectrum of immunotherapies, will expect more research in the relationship between hypoxia and epigenetics. Hypoxia, making cancer treatment difficult on tumor cells contributes to the therapeutic resistance and heterogeneity. The efficacy of cancer immunotherapy, as well as conventional therapy, was reduced by hypoxia (217). One of the reasons why cancer treatment is difficult, since of abnormal alteration of epigenetic modification by hypoxia. Epigenetic modifications reversibility makes epigenetic enzymes additional attractive therapeutic targets of cancer. Drugs targeting DNA methylation (DNMT inhibitors) and histone acetylation (HDAC inhibitor) are presently in the clinical trials or United States Food and Drug Administration (FDA) approved, but their adequacies are very limited in monotherapy. Hence, in order to achieve high effectiveness, it is necessary to study the effects of combinatorial treatment of epigenetic drugs and HIF-targeting therapy (217).
Due to the dynamic and reversible nature of epigenetic marks, these alterations represent attractive and therapeutically relevant targets in many diseases including cancer. Current epigenetic therapies are primarily directed towards two functional categories of epigenetic regulators: those that target the “writers”, enzymes that establish epigenetic marks, and those that target the “erasers”, enzymes that remove epigenetic marks (Table 3). Specifically, DNA methyltransferase inhibitors (DNMTi; writers) and histone deacetylase inhibitors (HDACis; erasers) are the main epigenetic therapy drug classes. DNMT and HDAC inhibitors exhibit anti-tumour functions by inducing differentiation, apoptosis, growth inhibition, cell cycle arrest, and cell death. DNMTis reactivate gene transcription by inhibiting the action of DNA methyltransferases (which add methyl groups to DNA) by directly incorporating into the DNA and trapping DNMTs for proteosomal degradation. The loss of DNMT is DNA replication dependent, and results in passive hypomethylation of DNA in daughter cells after cell division. Similarly, HDACis block the action of HDACs, which remove acetyl marks from tagged histones to increase global histone acetylation. These inhibitors might also work, at least in part, to re-activate gene expression by altering the global nuclear architecture. Loss of DNA methylation and/or increase in histone acetylation can result in a relaxed chromatin configuration, enabling access to transcriptional activators to restore gene transcription. Epigenetic drugs targeting these enzymes can restore, and in some cases overexpress, genes that have been epigenetically silenced in both immune and cancer cells (218, 219, and 220). Combining DNMT and HDAC inhibitors generally results in greater reexpression of epigenetically silenced tumour suppressor genes and cell cycle regulators (221).For the treatment of different malignancies, epigenetics therapy has been proven to be a successful approach. Two FDA approved cancer treatments are the inhibition of DNMTs and HDACs, while the fundamental mechanisms of DNMT and HDAC inhibition are not fully acknowledged (222).
| Category | Target enzyme | Tumor type | Compounds | Reference |
|---|---|---|---|---|
| Under Writers | DOT1L | Hematological cancers (under clinical trial phase) | EPZ-5676 | (225) |
| EZH2 | Advanced solid tumors, B-cell lymphoma (under clinical trial phase) | EPZ-6438 | (226) | |
| Cancer in organs such as breast, colon and prostate (under pre-clinical trial phase) | DZNep | (227) | ||
| Under Erasers | LSD1 | Relapsed or refractory acute leukemia (under clinical trial phase) | ORY-1001 | (228) |
| Small-cell carcinoma in lung (under clinical trial phase) | GSK2879552 | (229) | ||
| Under Readers | BET | Hematological malignancies, NUT midline carcinoma, solid tumors (under clinical trial phase) | I-BET762 | (230) |
| MLL-rearranged leukemia, multiple myeloma (under pre-clinical trial phase) | JQ1 | (231,232) |
In the past two decades, the FDA approval of various DNA methyltransferase inhibitors, collectively called DNA HMAs, and histone deacetylase inhibitors (HDACi) has brought epigenetic therapy to the forefront of cancer therapies. However, the benefits of epigenetic therapy are mainly restricted to the treatment of hematological malignancies. Other epigenetic modulators are currently under preclinical development, including second generation HMAs and small molecules targeting histone writers, readers, and erasers. A histone writer will deposit epigenetic marks on either DNA or histone tails, while a histone eraser removes these marks. Subsequently, epigenetic marks are recognized by the readers, and catalyze downstream cellular responses accordingly (223). A summary of FDA-approved epigenetic inhibitors and currently active clinical trials can be found in Table 4, (233).
| Epigenetic inhibitor | Target | Clinical status | Type of cancer |
|---|---|---|---|
| Vorinostat | pan-HDAC | FDA, 2006 | Refractory cutaneous T cell lymphoma |
| Romidepsin | pan-HDAC | FDA, 2009 | Cutaneous T cell lymphoma |
| Belinostat | pan-HDAC | FDA, 2014 | Peripheral T cell lymphoma |
| Chidamide | HDAC1 | Phase II | Peripheral T cell lymphoma |
| Givinostat | HDAC1 & 2 | Phase III | Refractory leukemia and myeloma |
| Panobinostat | pan-HDAC | FDA, 2015 | Multiple myeloma |
| Kevetrin | MDM2 p53 & Rb E2F pathways | Phase II | Ovarian cancer and spleen metastasis |
| KA2507 | HDAC6 | Phase I | Solid tumors |
| ACY-1215 | HDAC6 | Phase II | Lymphoid malignancies |
| Entinostat | HDAC1/HDAC3 | Phase I | Recurrent or refractory solid tumors |
| Vorinostat | pan-HDAC | Phase I/II | Colorectal and renal solid tumors |
| Azacitidine | FDA, 2006 | Acute myeloid leukaemia, chronic myelomonocytic leukaemia | |
| Vorinostat | FDA, 2006 | Cutaneous T cell lymphoma | |
| Romidepsin | FDA, 2009 | Cutaneous T cell lymphoma | |
| 5-Azacytidine | FDA, 2004 | Myelodysplastic syndromes | |
| Ruxolitinib | FDA, 2011 | Myelofibrosis | |
| Decitabine | FDA, 2006 | Acute myeloid leukaemia | |
| SGI-110 | Phase I/II | Higher risk myelodysplastic syndrome | |
| SGI-110 | Phase II | Gastrointestinal stromal tumors | |
| Azacitidine | Phase I | Recurrent and/or metastatic head and neck tumors | |
| CC-486 (oral azacitidine) | Phase II | Myelodysplastic syndromes | |
| CPI-1205 | EZH2 | Phase I | B cell lymphomas |
| Tazemetostat | EZH2 | Phase II | Solid tumor with an EZH2 mutation |
| Resminostat | EZH2 | Phase I | Colorectal carcinoma |
| Tazemetostat | EZH2 | Phase I/II | Advanced solid Tumors and B cell lymphomas |
| INCB059872 | LSD1 | Phase I/II | Advanced leukemia |
| IMG-7289 | LSD1 | Phase I | Acute leukemia |
| AZD5153 | BRD4 | Phase I | Advanced solid tumors and lymphomas |
| SF1126 | BRD4 | Phase I | Advanced hepatocellular carcinoma |
| EPZ-5676 | DOT1L | Phase I | Acute leukemia |
| Entinostat | PD-1/PD-L1 | Phase II | Breast and Non-small cell lung cancer |
| SB939 | FLT3-ITD | Phase II | Prostate cancer |
| Azacitidine plus Entinostat | HDAC/HMA | Phase II | Elderly patients with acute myeloid leukemia |
| Romidepsin plus oral Azacitidine | HDAC/HMA | Phase I/II | Relapsed/refractory lymphoid malignancies |
| Decitabine plus Vorinostat | HDAC/HMA | Phase I | Relapsed/refractory lymphoid malignancies |
| Triple: Entinostat, Nivolumab and Ipilimumab | HDAC/ICB | Phase I | Locally advanced or metastatic HER2-negative breast cancer |
| Entinostat plus Pembrolizumab | HDAC/ICB | Phase I | Advanced solid tumors |
| ACY-241 plus Nivolumab | HDAC6/ICB | Phase I | Unresectable non-small cell lung cancer |
| Azacitidine plus Durvalumab and Tremelimumab | HMA/ICB | Phase I/II | Metastatic head and neck cancer |
| Azacitidine plus Durvalumab | Phase I/II | Leukemia, myeloid, acute myelodysplastic syndromes | |
| Azacitidine plus Nivolumab and Ipilimumab | HMA/ICB | Phase II | Refractory/relapsed acute myeloid leukemia and newly diagnosed |
| CC-486 plus Durvalumab | HMA/ICB | Phase I/II | Relapsed and refractory peripheral T cell lymphoma |
| CC-486 plus Durvalumab | HMA/ICB | Phase II | Colorectal, ovarian, and breast tumors |
| CC-486 plus Pembrolizumab | HMA/ICB | Phase II | Metastatic melanoma |
| Guadecitabine plus Durvalumab | HMA/ICB | Phase I/II | Advanced kidney cancer |
| Guadecitabine plus Atezolizumab | HMA/ICB | Phase II | Refractory or resistant urothelial carcinoma |
| Decitabine plus Pembrolizumab | HMA/ICB | Phase I/II | Refractory or relapsed acute myeloid leukemia |
| SGI-110 plus Ipilimumab | HMA/ICB | Phase I | Unresectable or metastatic melanoma patients |
| Triple: Azacitidine, Entinostat and Nivolumab | HDAC/HMA/ICB | Phase II | Recurrent metastatic non-small cell lung cancer |
| CPI-1205 plus Ipilimumab | EZH2/ICB | Phase I/II | Advanced solid tumors |
Cancer genome sequencing has created an opportunity for precision medicine. Thus far, genetic alterations can only be used to guide treatment for small subsets of certain cancer types with these key alterations. Similar to mutations, epigenetic events are equally suitable for personalized medicine. DNA methylation alterations have been used to identify tumor-specific drug responsive markers. Methylation of MGMT sensitizes gliomas to alkylating agents is an example of epigenetic personalized medicine. Recent studies have revealed that 5-azacytidine and decitabine show activity in myelodysplasia, lung and other cancers. There are currently at least 20 kinds of histone deacetylase inhibitors in clinical testing. Inhibitors targeting other epigenetic regulators are being clinically tested, such as EZH2 inhibitor EPZ-6438. The most widely studied epigenetic change is DNA hypermethylation in the promoter region of tumor suppressor genes in human cancer. The identification of biomarkers that predict response to chemotherapy is also a part of personalized medicine. Methylation patterns can be useful to assess clinical outcomes or response to chemotherapeutic agents. DNA methylation profiling has identified tumor-specific drug responsive markers in different cancers. Many epigenetic chemosensitive markers have been found in different cancer types (Table 5). In the future, the combination of multiple epigenetic markers may effectively predict the chemosensitivity of various cancers (224).
| Gene name | Gene function | Tumor type application | Chemosensitivity prediction | Ref. |
|---|---|---|---|---|
| MGMT | DNA repair | Glioma, colon, lung, lymphoma | Sensitive to temozolomide, BCNU, ACNU, procarbazine | (234) |
| CHFR | Ubiquitin protein ligase | esophageal, gastric, cervical, lung, endometrial cancer and oral squamous cell carcinoma | Sensitive to paclitaxel and docetaxel | (235, 236) |
| FANCF | DNA damage response | Ovarian | Sensitivity to cisplatin | (237) |
| BRCA1 | DNA damage response | Breast, ovary | Sensitive to PARP inhibitors and alkylating agents | (238) |
| MLH1 | DNA repair | Colon, stomach, endometrium, ovary | Resistance to cisplatin | (239) |
| GSTP1 | Detoxification | Prostate, breast, kidney | Sensitivity to doxorubicin | (240) |
| PRKCDBP | Signal transduction | Colon | Resistance to TNF-α | (241) |
| SFN | Signal transduction | Lung | Sensitive to cisplatin and gemcitabine | (242) |
| TFAP2E | Transcriptional regulator | Colon | Sensitivity to fluorouracil | (243) |
| ABCB1 | Protein transport | Breast | Sensitivity to doxorubicin | (244) |
| APAF1 | Apoptotic activator | Melanoma | Resistance to Adriamycin | (245) |
| CDK10 | Cell cycle control | Breast | Resistance to anti-estrogens | (246) |
| IGFBP3 | Signal transduction | Lung | Resistance to cisplatin | (247) |
| MT1E | Antioxidant | Melanoma | Sensitivity to cisplatin | (248) |
| TGM2 | Apoptosis | Lung, breast, ovary | Resistance to doxorubicin and cisplatin | (249) |
| TP73 | Stress response | Renal, melanoma | Sensitivity to cisplatin | (250) |
By linking genomic sequencing and gene expression profiles, future studies may be able to analyze methods for recognizing response mechanisms. Additionally, histones may be phosphorylated, ubiquitinated, sumoylated, methylated and acetylated. But, in diseases, these modifications have been less studied and may also be able to divulge other therapeutic targets. A major challenge in epigenetic therapy is to know which genes are the driver and which genes are stimulated. New developments in Genome-wide sequencing, along with RNA data profiles, chromatin immunoprecipitation (ChIP), or bisulfate conversion have cause to an enormous amount of information that can be used to accurately identify epigenetic changes (Figure 21).
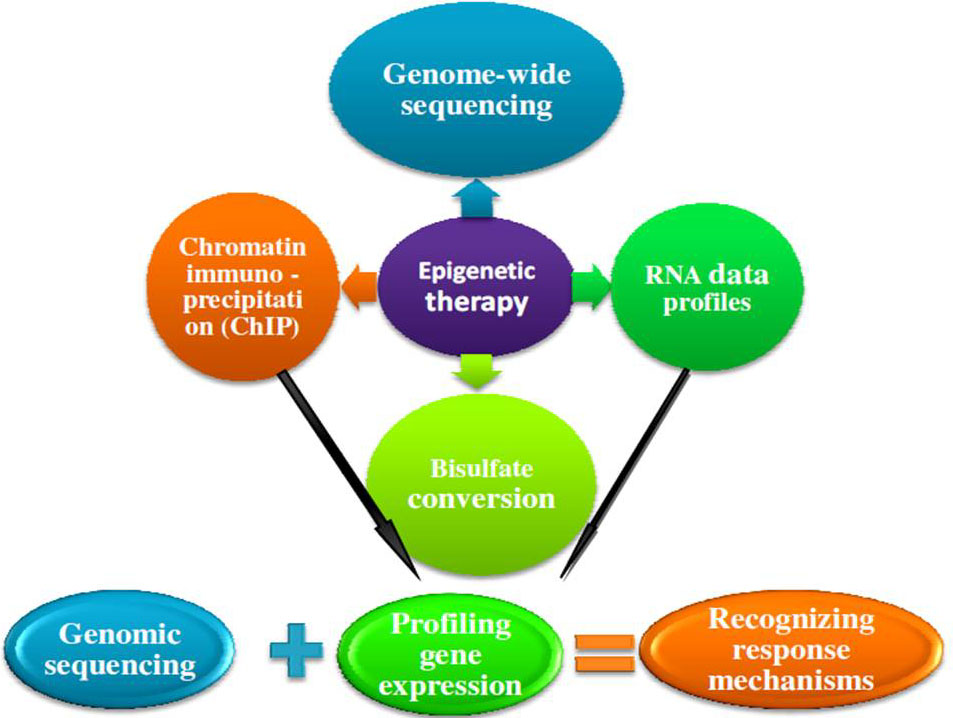 Figure 21
Figure 21Epigenetic therapy.
Analyzing and accommodating this enormous amount of information will help to recognize epigenetic alterations that appear as a source and consequence or completely dependent on each other. Appropriately in the future, patients may be screened using exact techniques or classified by genetic modifications in driver genes. In that method, it has made it feasible to achieve a personal and distinctive therapeutic approach to the treatment of each patient. At present, for the treatment of hematological malignancies, epigenetic therapy is profitably applied in clinics, but in the treatment of solid tumors, a little success has been achieved. The make use of epigenetics as a crucial contributing factor in the evolution of normal and abnormal cells will open new sights for the arrival of new therapeutic approaches. To provide certain treatments for reversal of the drug-resistant tumors traditional therapies may be combined with the epigenetic therapy. Also with this therapeutic approach, the drug dosages can be lessened to get rid of the side effects of treatment and, as a result, the patient’s healing problems and quality of life is increased (222).
BRP is profoundly thankful to the Science and Engineering Research Board, Department of Science and Technology, Govt. of India New Delhi, India (No. ECR/2016/001984) and Department of Science and Technology, Government of Odisha (Grant letter number 1188/ST, Bhubaneswar, dated 01.03.17, ST-(Bio)-02/2017) for providing funding.