




Frontiers in Bioscience-Landmark (FBL) is published by IMR Press from Volume 26 Issue 5 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.

1 Department of Pathology, Immunology and Laboratory Medicine, College of Medicine, University of Florida, Gainesville, FL 32610, USA
2 Department of Orthopaedics, Ruijin Hospital North, School of Medicine, Shanghai Jiaotong University, Shanghai, China
3 Department of Pathology, Third Central Hospital of Nankai University, Tianjin, China
4 Department of Pathology and Laboratory Medicine, Kaiser Permanente Sacramento Medical Center, Sacramento, CA, USA
Abstract
Significant progress has been made in our understanding of the role of epigenetic modifiers in many types of human cancer. Here, we review currently available studies on the unique histone methyltransferase, SETD2, which is responsible for H3 lysine 36 tri-methylation (H3K36me3). SETD2 plays pivotal roles in RNA alternative splicing regulation, DNA damage repair, and cytoskeleton protein methylation; inactivation of SETD2 and resultant dysregulation of these functions may lead to tumorigenesis. Despite being a newly discovered tumor suppressor, SETD2 has been found to be mutated in multiple types of cancer, including gastrointestinal tumor. Some tumors can acquire a selective growth advantage after SETD2 inactivation, which could happen in different stages in tumor progression. Decreased level of H3K36me3 caused by SETD2 inactivation has been shown to associate with higher tumor grade, tumor stage, metastasis risk, and shorter survival. Some studies also suggest that SETD2 mutation is associated with therapy resistance, therefore these SETD2-deficient tumors may need different therapeutic strategies.
Keywords
- SETD2
- H3K36me3
- RNA Alternative Splicing
- Tubulin Methylation
- DNA Damage Repair
- Progression and Prognostic biomarker
- Review
As the basic unit of the chromosome, the bead-like nucleosome consists of 147 base pairs of DNA and eight histone molecules. Post-translational modifications (PTMs) of histone are crucial for genome regulation (1). Lysine methylation of histone is one of those modifications that can regulate the whole process of transcription. The lysine residues of histone can be methylated by adding 1, 2 or 3 methyls at K4, K9, K27, K36 or K79 of histone H3 and K20 of histone H4 (2). While most of these methylations distributed on promoter or intergenic regions, the enrichment pattern of histone H3 lysine 36 trimethylation (H3K36me3) seems to be quite different and is mainly observed at body regions of active genes (3, 4). However, the function of H3K36me3 remains largely unknown.
SETD2 is an H3K36-specific histone methyltransferase (HMT) that is evolutionarily conserved from yeast to human (5-7), and it is the sole HMT that required for catalyzing H3K36me3 in mammals (8, 9). Full length of SETD2 owns three conserved domains, the SET domain, the WW domain, and the Set2 RNA polymerase II large subunit Rpb1 interacting (SRI) domain (6). The SET domain is the catalytic portion of SETD2 that performs the H3K36me3 transfer. The WW domain, containing two conserved tryptophan residues, can bind to proline-rich proteins, is thus potentially required for some protein-protein interactions. The SRI domain can bind to the phosphorylated C-terminal domain of RNA polymerase II. All these three domains are conserved from yeast to humans. However, recent studies revealed that the yeast Set2 possessed an auto-inhibitory domain (AID) and histone interaction domain, and that these domains can regulate H3K36 methylation (10, 11); whether SETD2 also contains a region with similar function for H3K36me regulation is worth investigating in the future.
To understand the physiological function of SETD2 in normal development, a Setd2 conventional knockout mouse model was constructed by Hu et al., and they found that Setd2 deficient mouse is lethal at E10.5-E11.5 due to defects of embryo vascular remodeling (9). Furthermore, conditional knockout of Setd2 in mouse hematopoietic stem cells (HSCs), intestinal epithelium and mesenchymal stem cells (MSCs) revealed that the Setd2 deficiency severely impairs self-renewal of HSCs and intestinal stem/progenitor cells (12-14). In addition to affecting self-renewal, deletion of SETD2 also causes cell differentiation defects. For example, Setd2 absence leads to HSCs differentiation bias (12), impairs adipocytes differentiation from MSCs (12), inhibition of embryonic stem cell differentiation toward endoderm (15) and repression of myoblasts differentiation to skeletal muscle cells (16).
Interfering with the self-renewal and differentiation of stem/progenitor cells has been broadly regarded as the cellular basis of tumor initiation and progression. Indeed, SETD2 gene mutations have been frequently reported in clear cell renal cell carcinoma (ccRCC), acute leukemia, gliomas, and lung adenocarcinomas (17-20). SETD2 mutations also have been reported in some gastrointestinal tumors. As the critical role of SETD2 in the gastrointestinal (GI) tumor has not been reviewed yet, this review aims to examine the current literature about the critical function of SETD2 and to guide the understanding of SETD2 genetic variation in the initiation and progression of gastrointestinal tumors.
Gastrointestinal tumor (e.g., esophagus, stomach, liver, pancreas and colorectum tumors) incidence and mortality are rapidly growing world-wide (21), yet the reasons remain unclear. As most highly renewing tissues, the gradual accumulation of genetic and epigenetic errors may be a cause. Actually, genetic mutation of epigenetic genes has been broadly studied in tumor initiation, progression, relapse, and so on (22). SETD2 was reported to have tumor-suppressive function in various human cancers (20, 23, 24). Most frequently detected missense mutations of SETD2 are located at the SET domain (25). Dalgliesh et al. first identified inactivation mutations of SETD2 in ccRCC in 2010, in which 12 of 407 ccRCC samples had somatic truncating mutations of SETD2 (17). SETD2 mutations were also detected in 2 out of 10 primary ccRCC samples by their following investigation using targeted exome-sequencing (26). Another two cohort studies of ccRCC showed that the overall mutation frequency of SETD2 is around 11% (27, 28). In addition, Zhu et al. identified 19 somatic SETD2 mutations from totally 241 leukemia patients (20). Here, we will focus on the role of SETD2 in gastrointestinal tumors.
In a comparison of 168 colorectal cancer (CRC) specimens and 48 normal biopsies, Yuan et al. demonstrated that SETD2 mRNA and protein levels were downregulated, suggesting SETD2 as a tumor suppressor in CRC (13). Similarly, Chen et al. reported that SETD2 mRNA and protein levels are remarkably lower in the gastric cancer samples than in cancer-adjacent tissues indicated by immunohistochemistry, RT-PCR and immunoblotting analyses. 79.7% of primary gastric cancer sample tissues showed a lower expression level of SETD2 mRNA (29). Recently, non-coding RNA mediated transcriptional and translational regulation have been reported to control SETD2 expression. For example, since Tang et al. found that the long non-coding HOX transcript antisense RNA (HOTAIR) is an oncogenic long noncoding RNA in several tumors (30), a study by Li et al. proved that HOTAIR can compete with CREB-P300-RNA Pol II complex and prevents its loading to the SETD2 promoter, thereby inhibiting SETD2 at transcriptional level and reducing the level of H3K36me3 in the liver cancer sample (31), which implies that the oncogenic function of HOTAIR is partially mediated by down-regulating SETD2 and decreasing H3K36me3. At the translational level, Xiang et al. discovered that the microRNA miR-106b-5p can bind to the 3’-UTR of the SETD2 mRNA and suppresses SETD2 expression. Thus, SETD2 protein level was found to be increased by inhibition of miR-106b-5p, resulting in ccRCC cell cycle arrested at G0/G1 phase (31). Furthermore, SETD2 protein stability can also be regulated. Yeast two-hybrid screen and co-immunoprecipitation assays identified SPOP, a key subunit of the CUL3 ubiquitin E3 ligase complex, as a SETD2-interacting protein, which was responsible for SETD2 stability control through polyubiquitination (32). Hence, these transcriptional, translational and post-translational mechanisms for controlling SETD2 expression level and stability are worthy of attention in GI tumors.
Huang et al. propose that SETD2 is a tumor suppressor for gastrointestinal stromal tumor (GIST), as inactivation of SETD2 is related to the tumor progression. They found that, in patients with high-risk GIST, SETD2 contains loss-of-function somatic mutations. These mutations can only be detected in patients with high-risk GISTs and metastatic GISTs with a prevalent rate of these mutations around 11%, while they cannot be detected in low or intermediate risk patients. In univariate analysis, the patients with SETD2 loss-of-function were associated hypomethylated heterochromatin and had remarkably shorter relapse-free survival (33).
In a whole exome sequencing (WES) analyses of 20 patients with a single nonsyndromic, nonfunctional but with distant metastases pancreatic neuroendocrine tumor (PanNET), Roy et al. identified mutations in chromatin-remodeling genes, including SETD2, ARID1A, CHD8, and DNMT1 (34). In a separate analysis of 347 primary PanNETs, disruption of SETD2 function or mutations/deletions of ARID1A, DAXX, ATRX, and CDKN2A were found in 81% of primary PanNET patients with distant metastases. In the patients without any of those alterations, 98% of patients had disease-free survival times for 5 years, and 95% of patients had 10 years of disease-specific survival. Among patients with loss functions of any one of these genes, 39% of patients had disease-free survival times for 5 years, and 44% of patients had 10 years of disease-specific survival. The evidence suggests that loss of function of SETD2, ARID1A, ATRX, DAXX, and CDKN2A in primary PanNETs is associated with shorter survival, and impaired function of these genes including SETD2 may result in metastasis of PanNETs (35).
Similarly, to identify molecular alterations between patients who are older than 65 and patients who are younger than 45 in left-sided colorectal cancers (LCRC), Puccini et al. examined the tumor tissue samples from splenic flexure to the rectum of those patients using immunohistochemical approaches and NGS. They found that younger patients more frequently harbor mutations in genes related to cancer-predisposing syndromes, such as MSH2, MSH6, POLE, NF1, SMAD4, and BRCA2. They also detected significantly frequent mutation of histone modifier gene: KMT2A, KMT2C, KDM5C, and SETD2. The mutation frequency of SETD2 is relatively higher in LCRC in young patients (3.2% vs. 0.9%, p = .039). In addition, high tumor mutational burden (TMB-H) and high microsatellite instability (MSI-H) were also more frequent in younger patients (36).
Multiregion sequencing revealed that independent SETD2 mutation occurred at several branching sites of ccRCC clonal evolution (37). Using immunohistochemical analysis, Ho et al. found that H3K36me3 levels increased in primary ccRCC in contrast to metastatic ccRCC (38). In a study including 185 ccRCC patients, Hakimi et al. observed that SETD2 mutations were associated with advanced tumor stage (39). The results showed that patients bearing SETD2 mutations had a higher relapse rate and worse cancer-specific survival, with the median overall survival of 62.7 months in SETD2 mutated patients comparing 78.2 months in non-mutated patients (40). In addition, Sarakbi et al. reported that advanced tumor stages are associated with lower SETD2 expression levels in breast cancer (24). Fontebasso et al. proposed that SETD2 mutations are essential for the tumorigenesis of high-grade gliomas (19). In their cohort investigation, data of whole exome sequencing (WES) from 183 glioma samples revealed that SETD2 is mutated in 12.37% high-grade gliomas, while no SETD2 mutations have been detected in low-grade cases. Table 1 summarizes the currently available data on SETD2 alterations in a variety of human tumors.
| Tumor type | Number of cases | Major findings | References | |
|---|---|---|---|---|
| Breast cancer | 120 | Low expression of SETD2 is related to high tumor stage, metastasis, local recurrence and cancer-specific mortality | 24 | |
| Subgroups | Case tested | SETD2 mRNA (Mean ± SD) (CK19 normalisation) | ||
| • Normal tissue | 33 | 414434 ± 1106629 | ||
| • Tumor tissue | 120 | 20950 ± 37606 | ||
| • TNM stage 1 | 69 | 8551 ± 29766 | ||
| • TNM stage 2 | 40 | 8578 ± 24043 | ||
| • TNM stage 3 | 7 | 174 ± 414 | ||
| • TNM stage 4 | 4 | 679 ± 1081 | ||
| • Disease free | 81 | 12239 ± 43466 | ||
| • Alive with Metastasis | 7 | 1983 ± 5142 | ||
| • Local recurrence | 5 | 3513 ± 5683 | ||
| • Died of disease | 20 | 698 ± 1741 | ||
| Tumor type | Number of cases | Major findings | References | |
| Clear cell renal cell carcinoma | 146 | Decreased H3K36me3 in cancer cell and is associated with metastasis | 38 | |
| Kidney tissue type | Case tested | Percentage of nuclear staining of H3K36me3 | ||
| • Uninvoloved | 30 | ~ 90% | ||
| • Primary | 71 | ~ 70% | ||
| • Metastases | 45 | ~ 30% | ||
| Tumor type | Number of cases | Major findings | References | |
| Glioma | 183 | 12.37% high-grade gliomas have mutated SETD2. No low-grade gliomas harbor SETD2 mutation | 19 | |
| Tumor grade | Wild type | Mutated | Case tested | SETD2 mutation frequency(%) |
| • Grade 3-4 | 122 | 16 | 138 | 12.37 |
| • Pediatric | 62 | 11 | 73 | 15 |
| • Adult | 60 | 5 | 65 | 8.11 |
| • Grade 2 | 45 | 0 | 45 | 0 |
| • Pediatric | 23 | 0 | 23 | 0 |
| • Adult | 22 | 0 | 22 | 0 |
| • Overall | 167 | 16 | 183 | 8.7 |
| Tumor type | Number of cases | Major findings | References | |
| Clear cell renal cell carcinoma | 185 | SETD2 mutation is noted in 7.6% of ccRcc, is associated with higher stage | 39 | |
| Tumor grade | Wild type | Mutated | Case tested | SETD2 mutation frequency(%) |
| • Stage 3-4 | 93 | 11 | 104 | 11.8 |
| • Stage 1-2 | 78 | 3 | 81 | 3.9 |
| • Overall | 171 | 14 | 185 | 7.6 |
Tumor progression is regulated by many different mechanisms, such as genome instability (41), tumor microenvironment (42, 43), immune deregulation (44), metabolic reprogramming (45), chemoresistance (46). A specific morphological transition from the epithelial to the mesenchymal morphology, a process named EMT (epithelial-mesenchymal transition), is a characteristic change during tumor progression (47). Despite a shortage of mechanistic studies of SETD2 that were performed exactly with GI tumors, lesson from other models may also help us to understand how SETD2 may control GI tumor progression through various mechanisms.
Existing bodies of evidence suggest that H3K36me3 catalyzed by SETD2 plays an important role in transcriptional regulation. In one of the earliest studies, the artificial binding of yeast set2 to promoter indicated that set2 repressed transcription (5). Tandem affinity purification revealed that set2 associated with elongating form of RNA polymerase II, suggesting that set2 couples with transcriptional elongation (48). Setd2 has been reported to regulate Fgfr3 transcription initiation via H3K36me3 at distal promotor region to influence mouse endoderm differentiation(15). Interestingly, SETD2 deficiency leads to DNA replication stress (RS) by a similar manner to regulate RRM2 transcription initiation in ccRCC cell lines(49).
In a mouse model of CRC, Yuan et al. reported that Setd2 can suppress Wnt signaling pathway, and ablation of SETD2 could promote tumorigenesis in mouse intestine (13). Setd2 can modulate the alternative splicing of the genes implicated in tumorigenesis. In their multivariate analysis of transcript splicing study, totally 711 genes showed changes of alternative splicing in the SETD2-deficient sample, 198 genes were found to have exon skipping and 279 with intron retention. These alterations of alternative splicing increased disheveled segment polarity protein 2 (DVL2) mRNA, enhanced Wnt/β-catenin signaling and affected intestinal cell differentiation and consequently resulted in tumorigenesis.
SETD2 can also regulate alternative splicing of Human mutL homolog 1 (hMLH1). Zhao et al. found that the abnormal transcripts of hMLH1 are associated with the low H3K36me3 status around hMLH1 exon 10-11 (50). In human gastric carcinoma BGC-823 cells, the alternative splicing pattern of hMLH1 transcripts changed after SETD2 inactivation. The researchers proposed that some reader proteins could recognize H3K36me3 and the splicing factors such as SRSF2 was then recruited to the alternative splice sites, resulting in the right splicing pattern which contains hMLH1 Exon10 and Exon11. Once H3K36me3 was impaired at those alternative splice sites via SETD2 inactivation, aberrant splicing occurred and caused inactivation of hMLH1, demonstrated by expressional and translational diversity of hMLH1 protein in gastric cancer, as illustrated in Figure 1. And this explains why some types of gastric cancer carry high-frequency microsatellite instability (MSI).
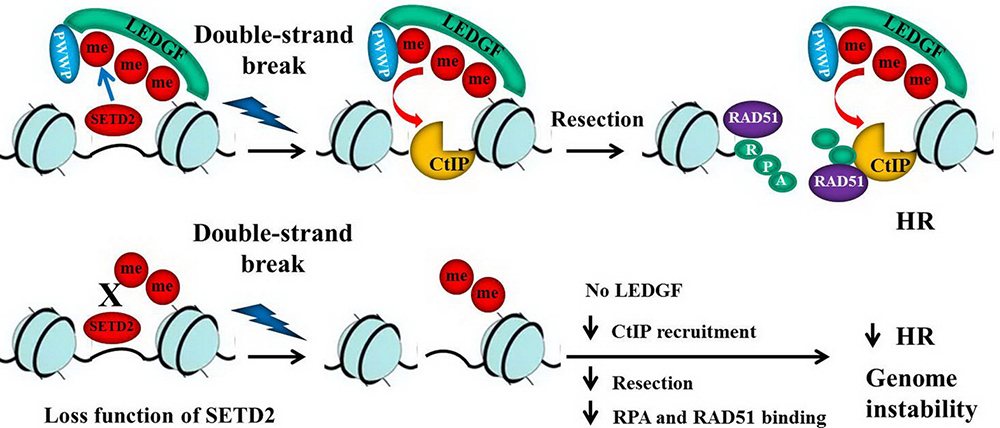 Figure 1
Figure 1With normal H3K36me3 level surrounding hMLH1 exon 10–11, medicated by SETD2, SRSF2 can be recruited to the functional splicing sites to enhance normal splicing. With SETD2 inactivation, aberrant splicing happens and this leads to inactivation of hMLH1 and high-frequency microsatellite instability.
DNA replication requires high fidelity to ensure the transmission of genetic information from one generation to the next, however, numerous obstacles of both intracellular and extracellular (e.g. limiting nucleotides, DNA lesions, repetitive DNA elements, oncogene-induced stress) cause DNA replication stress, uneffectively repaired RS will lead to aging (51) and genome instability for tumor formation and progression (41). Indeed, loss of function of SETD2 triggers RS to promote renal cancer evolution (52). Recently, Zhang et al. showed that conditional knockout of Setd2 in hematopoietic stem cell results in RS, leading to subsequent genome instability and malignant transformation (12).
The critical roles of SETD2/H3K36me3 are mediated by recruiting some chromatin-associated proteins which have a PWWP domain. The PWWP domain is a protein-protein interaction domain, it has a central core ‘Pro-Trp-Trp-Pro’ motif and is about 70 amino acids length (53). These chromatin-associated proteins include DNA methyltransferase DNMT3b (54), Lens epithelium-derived growth factor (LEDGF) (55) and MSH6 (56). LEDGF and MSH6 have been broadly reported to participate in DNA repair, indicating that beside RS, SETD2 also guard genome stability through DNA repair.
Served as the basic element for HR repair, the PWWP domain of LEDGF can recognize H3K36me3, and facilitate binding of LEDGF to the DNA DSBs of the chromatin. Thereafter, LEDGF recruits C-terminal binding protein interacting protein (CtIP), coupling replication protein A (RPA) and RAD51, and promotes resection, the essential step in HR repair. As illustrated in Figure 2, the impairing of the resection may occur after compromised H3K36me3. However, cells will repair DNA damage via other pathways, such as nonhomologous end-joining and/or microhomology-mediated end-joining. Those alternative mechanisms are fallible and could lead to genomic instability and subsequent tumorigenesis (57).
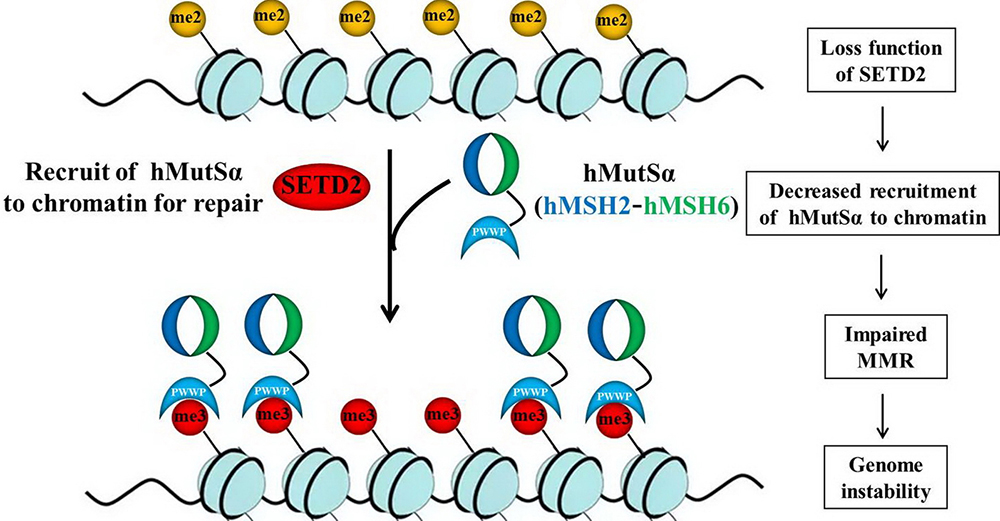 Figure 2
Figure 2PWWP domain of LEDGF can recognize H3K36me3, through which LEDGF can be recruited to DSBs to initiate DNA repair. LEDGF recruits CtIP, RPA, and RAD51, which promotes resection, an essential step in HR repair. Without H3K36 trimethylation, chromatin LEDGF association could be compromised, CtIP, RPA, and RAD51 cannot be recruited, which will impair resection and HR, leading to deletions and subsequent genome instability and tumorigenesis.
DNA mismatch repair (MMR) ensures replication fidelity by correcting insertion/deletion loops and base-base mismatches generated during DNA replication. hMutS-alpha (MSH2-MSH6) complex plays a pivotal role in MMR. Li et al. reported that the hMSH6 PWWP domain as a histone modification reader and can interact directly with H3K36me3. Thus, hMutS-alpha complex is recruited to the chromatin. Once SETD2 is inactivated, the recruitment of hMutS-alpha to the damaged DNA sites will be decreased (58). As illustrated in Figure 3, this finding explained why some MSI-positive cancer cells do not bear mutations of the well-studied MMR genes.
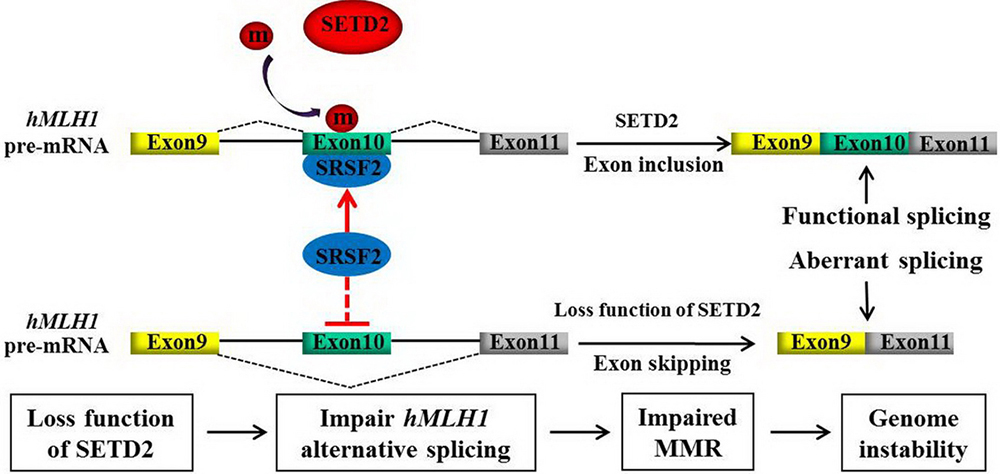 Figure 3
Figure 3SETD2 mediated H3K36me3 is essential to attract the hMutS-alpha (hMSH2-hMSH6) complex to the damaged DNA sites through recognizing between H3K36me3 and PWWP domain of hMSH6. Depletion of SETD2 will compromise this interaction and the subsequent localization, leading to a DNA MMR deficiency.
A recent discovery by Park et al. showed that SETD2 can maintain genomic stability by keeping the proper function of α-tubulin during mitosis and cytokinesis. SETD2 can tri-methylate α-tubulin on lysine 40 (α-TubK40me3), and ablation of SETD2 causes defects of mitotic spindle and cytokinesis, micronuclei, and polyploidy. This study suggested that the genomic unstable oncogenic phenotypes of SETD2-mutated cells may be caused by tubulin defects during cytokinesis and mitosis, as illustrated in Figure 4 (58). Nevertheless, this mechanism leading to chromosomal instability in colorectal cancer remains to be determined.
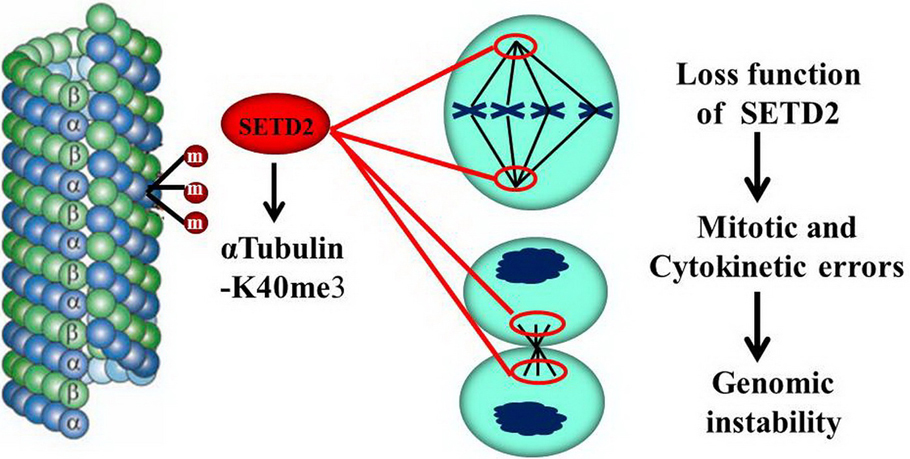 Figure 4
Figure 4During mitosis and cytokinesis, SETD2 can trimethylate alpha-tubulin on lysine 40 (alpha-TubK40me3), and ablation of SETD2 causes mitotic spindle and cytokinesis defects, micronuclei, and polyploidy, which causes genomic instability and tumorigenesis.
Tumor cells often form an immunosuppressive niche that can promote tumor progression, a non-biased high-throughput RNAi screening found that SETD2 as a critical effector molecular to promote IFNα-mediated cellular response against HBV replication via directly methylated STAT1 like tubulin methylation (59). Whether the progressive stage of GI tumor with SETD2 mutation also exists IFNα pathway inhibition is a worthy research direction in the future.
Several studies have reported a high frequency of SETD2 mutation in the relapse of acute leukemia (18) and hepatosplenic T-cell lymphoma (60), suggesting that SETD2 mutation promotes chemoresistance. Using CRISPR/Cas9 to knockout SETD2 in leukemia cell lines, Mar et al. found SETD2 impaired DNA damage recognition to induce resistance to DNA-damaging chemotherapy (61). To further clarify the mechanism of chemoresistance in leukemia, Mll-Af9 knock-in leukemia mouse model in Setd2 mutant was constructed, the authors demonstrate that SETD2 loss of function mutations confer chemoresistance on AL to DNA-damaging treatment by S and G2/M checkpoint defects (62). In other solid tumors, SETD2 also confer chemoresistance, Kim et al. found that SETD2 mediated H3K36me3 is critical for cellular sensitivity to cisplatin, and SETD2 inactivation may lead to cisplatin resistance. They reported that knockdown of SETD2 and ectopic expression of mutated SETD2 both can confer cisplatin resistance by inhibiting H3K36me3-ERK pathway in non-small cell lung cancer cells (63). Jiang et al. found that low expression of SETD2 was observed in osteosarcoma tissue, osteosarcoma cell growth can be inhibited both in vitro and in vivo by ectopic SETD2. Additionally, through regulating Wnt/β-catenin signaling, SETD2 can increase cisplatin-mediated apoptosis in osteosarcoma cells, and this was mediated by enhancing GSK-3β expression through increasing H3K36me3 level at the GSK-3β promoter, which resulted in β-catenin degradation and downregulation of its downstream gene cyclin D1, c-myc, and CD133 (64).
This review summarizes the currently available studies on the alterations of SETD2, a histone methyltransferase, in the tumorigenesis of human malignancies, especially in the gastrointestinal tumors. Not only SETD2 mutation but also downregulation expression occurs in GI tumor. Notably, these changes appear almost exclusively in the progressive phase and associate with poor prognosis. We mainly discuss the potential mechanism of GI tumor progression caused by SETD2 mutation, with some lessons learned from other cancer types. SETD2 mediated H3K36me3 can be recognized by effector protein, which connects critical tumor suppressor function of SETD2 to several critical biological processes in tumorigenesis, including mRNA alternative splicing, methylation of α-tubulin, and recruitment of the LEDGF protein or the hMutS-alpha complex during DNA damage repair. Once mutations of SETD2 occurs with a resultant low level of H3K36me3, the effector proteins cannot be recruited to their right location to maintain the genomic stability and integrity. Although genomic instability caused by SETD2 mutation acts as a driver for tumor progression, several recent studies fortunately revealed that some inhibitors can selectively kill SETD2-/- cells. For example, Yang et al. have found four chemical compounds in the database of Genomics of Drug Sensitivity in Cancer (GDSC) which can selectively inhibit SETD2-/- cell growth, and two of them target P13Kβ (65). Feng et al. reported that AZD6482 can also selectively inhibit SETD2-/- ccRCC cell lines (66). In the study conducted by Pfister et al., the G2-M cell cycle checker WEE1 inhibitor AZD1775 can inhibit H3K36me3-deficient renal cancer cell growth (49). This study suggests that WEE1 inhibitors may be a promising drug for H3K36me3-deficient tumors treatment. Future research on elucidating the functional consequences of SETD2 mutations, including discovering more H3K36me3 readers, may help identify novel targets for therapeutic intervention in patients with SETD2 mutant-harboring gastrointestinal cancers.
Ming Hu and Mu Hu contributed equally to this article, Ming Hu, Mu Hu, Qin Zhang developed the original idea and wrote the manuscript, Jinping Lai and Xiuli Liu contributed to the critical revision of the manuscript for important intellectual content.