



Frontiers in Bioscience-Landmark (FBL) is published by IMR Press from Volume 26 Issue 5 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.

1 Endocrinology Unit, Fondazione Policlinico Universitario A. Gemelli IRCCS, Roma – Università Cattolica del Sacro Cuore
2 Institute of Biochemistry and Clinical Biochemistry, Fondazione Policlinico Universitario A. Gemelli IRCCS, Roma
Abstract
Iodothyronine deiodinases are selenoproteins that regulate thyroid hormone metabolism. Of the three types of deiodinases, type 2 is the major regulator of intracellular triiodothyronine concentration in both the hypothalamus and pituitary, and therefore the major regulator of thyrotropin secretion. A defect in iodothyronine deiodinase activity can lead to a reduced sensitivity to thyroid hormones action and the most recent literature includes these defects in the so-called “syndromes of reduced sensitivity to thyroid hormones”. To date, the pathogenic variants of the selenocysteine insertion sequence-binding protein 2 (SECISBP2) gene are the first and only inherited disorder of iodothyronine metabolism described. Moreover, there is a growing interest in understanding the possible role of polymorphisms of DIO1 and DIO2 genes in some pathological conditions and in determining the requirement of levothyroxine replacement and the role of combined levothyroxine-liothyronine therapy in carrying subjects affected by hypothyroidism and who need replacement therapy. Results on this topic are still conflicting and more studies are needed to assess the efficacy of combined levothyroxine-liothyronine replacement therapy in this subset of patients.
Keywords
- Iodothyronine Deiodinases
- Thyroid Hormone
- Hypothalamus–Pituitary–Thyroid Axis
- SECISBP2
- DIO Polymorphism
- Reduced Sensitivity To Thyroid Hormone
- Review
Thyroid hormones (TH) activity is essential for many diverse processes in nearly all vertebrate tissues. The thyroid gland, in response to a cascade of signals including thyrotropin-releasing hormone (TRH) release from the hypothalamus and thyrotropin (TSH) release from the anterior pituitary (1), is responsible for the synthesis and release of triiodothyronine (T3) and thyroxine (T4), which exert their biological effects by binding to nuclear receptors (TH receptors, TRs).
TRs belong to the superfamily of nuclear receptors which includes the retinoid X receptor, vitamin D receptor, and oestrogen receptor (2). These receptors are ligand-dependent transcription factors that consist of a single polypeptide chain with three modular functional domains: (1) an amino-terminal domain that varies between the different isoforms and (2) a central DNA-binding domain and (3) a carboxy-terminal ligand-binding domain that are conserved between the different isoforms (3).
TRs are derived from two structurally similar human genes, thyroid hormone receptor alfa (THRA) and thyroid hormone receptor beta (THRB), located on chromosome 17 and chromosome 3, respectively. The alternately splicing of these two genes results in several homologous nuclear receptor isoforms that mainly differ in their amino-terminal domain and tissue distribution (4, 5). In humans, the alternative splicing of the TRbeta gene (THRB) gives rise to two T3-binding nuclear receptor isoforms (TRbeta1, TRbeta2), and to one truncated variant (TRbeta4) (6, 7). Similarly, the TRalfa gene (THRA) encodes one T3-binding variant (TRalfa1) and three splicing variants (TR alfa2, TR alfa3 and TR alfaDeltaΕ6), which have no T3-binding activity (8, 9).
The expression of TR isoforms is tissue-dependent: TRalfa1 is constitutively expressed since embryonic development, while TRbeta is expressed toward the later stages of development (10). Their distribution in human tissues is reported in Table 1.
| Isoform | Predominantly expression | Lower expression |
|---|---|---|
| TRbeta1 | kidneys, liver, brain, heart, thyroid | skeletal muscle, lungs, spleen |
| TRbeta2 | brain, hypothalamus, pituitary, retina, inner ears | lungs, heart |
| TRbeta3 | kidneys, liver, lungs | skeletal muscle, spleen, brain, heart |
| TRalfa1, TRalfa2 | brain | kidneys, skeletal muscle, lungs, heart, testes, liver |
Even though both T3 and T4 can exert biological effects, T3 is considered to be the biologically active thyroid hormone that binds to TRs, while T4 is considered a “prohormone” that must be converted to T3 in order to gain biological activity.
THs metabolism, including T4-to-T3 conversion, depends on iodothyronine deiodinase expression and activity, which can vary among different tissues and according to the age of development. In fact, the thyroid produces only about 20% of the active hormone T3, while 80% of the circulating levels of T3 in healthy human subjects is derived from the activity of iodothyronine deiodinase (11).
However, several studies have demonstrated that T4 has non-genomic roles that do not require conversion to T3 (12) Indeed, some observations indicate that TR alfa1 in brain can be activated by T4 also in absence of T3 (13).This phaenomenon is crucial in pregnancy, considering the direct evidences of the role of maternal T4 in neurogenesis (14).
There are three kinds of iodothyronine deiodinase but type 2 iodothyronine deiodinase (D2), expressed in the pituitary, brain, brown adipose tissue, placenta, intestinal wall and reproductive tract, is the major T4-activating iodothyronine deiodinase, producing T3 by the removal of an iodine residue from the outer phenolic ring of T4 (15-17). Furthermore, deiodinases are involved in the enterohepatic recycling of iodothyronines and a possible interference of microbiota on the peripheral TH metabolism has also been proposed (18).
D2 generates intracellular T3 from T4 in several human tissues, including the hypothalamus and pituitary. Importantly enough, the intracellular T3 concentration at both hypothalamic and pituitary levels is the major determinant of the control of TSH secretion. Therefore, D2 activity plays a crucial role in the regulation of the hypothalamus–pituitary–thyroid (HPT) axis, both in physiological and in pathological conditions. For example, the pharmacological effects of several drugs (e.g. amiodarone, propylthiouracil (PTU)) are secondary to their effects on iodothyronine deiodinase activities (19, 20), and the reduced TRH synthesis observed during the so-called “non-thyroidal illness syndrome” (NTIS) seems to be related to a marked increase in D2 mRNA expression both in tanycytes and in the hypothalamus (21). Furthermore, an impaired iodothyronine deiodinase activity can also result in different degrees of impaired sensitivity to TH action. To the best of our knowledge, the only known inherited defect of thyroid hormone metabolism involving iodothyronine deiodinase activity is the mutation of selenocysteine insertion sequence-binding protein 2 (SECISBP2, also known as SBP2), that is involved in iodothyronine deiodinase synthesis and degradation. This mutation interferes with the conversion of T4 to T3, resulting in a low T3 and a high T4 and reverse T3 (rT3). However, different polymorphism of DIO2 gene have been described in association with several diseases.
In this review, the role of iodothyronine deiodinases activity in the regulation of the HPT axis and the possible implications in conditions of altered sensitivity to TH action will be discussed.
T4 is the main form of TH produced by the thyroid gland, but it is only minimally active. T3 is the fully active TH responsible for biological actions, its affinity for TRs being about 10-fold higher than that of T4. In humans, approximately 80% of serum T3 is derived from T4. T3 has a shorter half-life than T4 (~12 h vs. 8 days, respectively) (22), and it is mostly produced outside of the thyroid gland through the removal of the outer ring (5′) iodine from T4 (23). Conversely, the removal of the inner T4 or T3 ring iodine produces reverse T3 and T2, respectively, which are biologically inactive molecules. These crucial steps are mediated by the iodothyronine deiodinases.
The deiodination cascade is also involved in the synthesis of funcional molecules, similar to TH. Iodothyronamine (3-T1AM) and thyronamine (T0AM) are endogenous signaling molecules with smilar stucture to TH. Their physiological mode of action is still controversial and sites of their biosynthesis are not fully elucidated. The biosynthesis of these compounds involves decarboxylation and more or less extensive deiodination (24). Monoiodothyronine (T1) and diiodothyronine (T2), the precursor of TH which undergo a coupling reaction to produce T3 and T4, seem to have a biological active role. In particular, T2 is considered a “biologically active iodothyronine”, which targets mostly mitochondria and is involved in bioenergetic metabolism regulation (25).
Iodothyronine deiodinases (encoded by DIO genes DIO1, DIO2, and DIO3) are selenoproteins that contribute to the intracellular regulation of TH action (Figure 1). They are homodimers anchored in cell membranes via a single transmembrane domain, while the catalytic globular domain faces the cytosol, and dimerization is required for their catalytic activity (26). Recently, some authors showed that the iodothyronine deiodinases share an evolutionary conserved TH-binding domain, which is similar to that of TH nuclear receptors and TH cell membrane transporters and which could represent the product of a common ancestor gene (27).
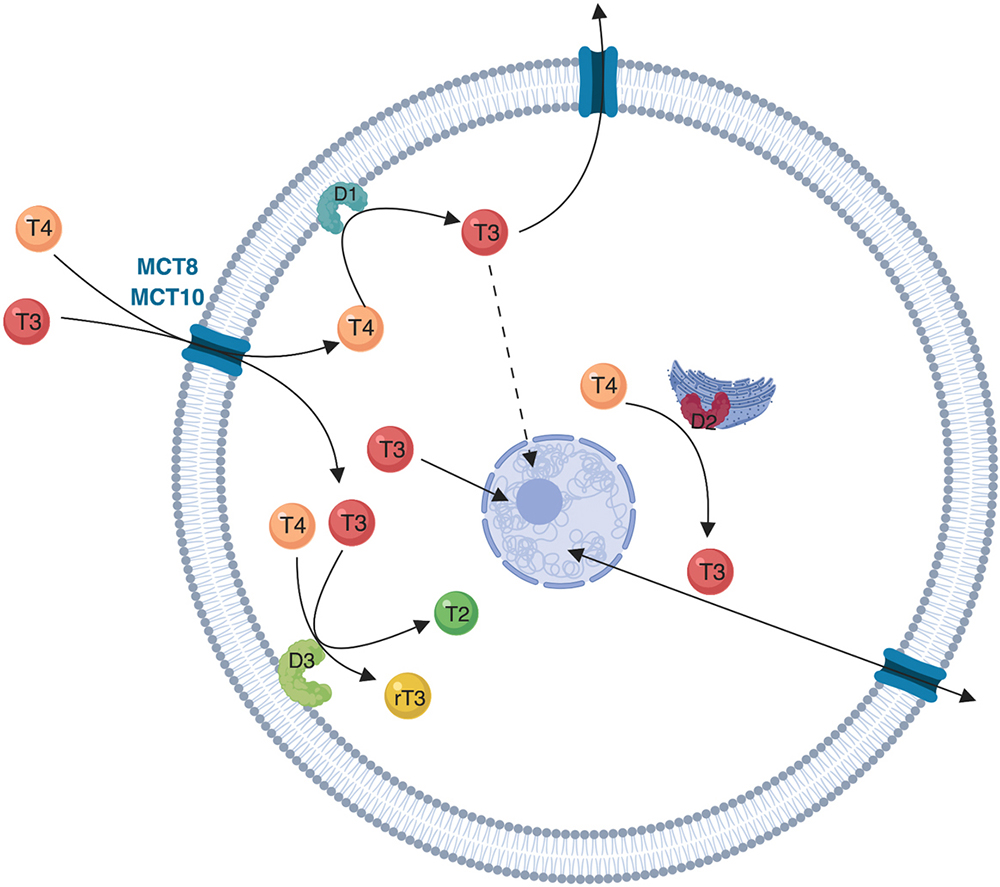 Figure 1
Figure 1Schematic representation of deiodinases localization and function. The cellular influx and efflux of thyroid hormones is facilitated by protein transporters (mainly MCT8 and MCT10). The intracellular metabolism of thyroid hormones depends on the activity of three iodothyronine deiodinases (D1, D2 and D3). D1 is a plasma membrane protein and is the only deiodinase that can function both as an outer ring deiodination, resulting in T4 to T3 conversion, and as an inner ring deiodination resulting in T4 to rT3 conversion (not shown). D2 is an endoplasmic reticulum protein that functions as an outer ring deiodinase converting T4 to T3. D2 is closer to the nucleus when compared to D1, and D2-originated T3 diffuses to the nucleus (solid line arrow) and remains in the target cell for a longer period of time when compared to D1-originated T3 (dotted line arrow). D3 is an inner ring deiodinase located in the plasma membrane that inactivates T4 to rT3 and T3 to T2.
DIO1 gene, maps on the short (p) arm of chromosome 1 at position 32.3 (1p32.3) (GRCh38/hg38: chr1:53,891,239-53,911,086; NC_000001.11) and encodes a protein (D1) of 294 amino acids. DIO2 and DIO3 genes, mapping on the long arm (q) of chromosome 14 in positions 31.1 (14q31.1) (GRCh38/hg38: chr14:80,197,525-80,387,757; NC_000014.9) and 32.31 (14q.32.31) (GRCh38/hg38: chr14:101,561,351-101,563,452; NC_000014.9) respectively, encode proteins of 273 (D2) and 304 (D3) amino acids, respectively. These selenoproteins contain the rare amino acid selenocysteine (Sec) at their active site. Sec is encoded by the UGA codon, which normally signals translation termination. The 3' UTRs of selenoprotein mRNAs contain a conserved stem-loop structure, designated the Sec insertion sequence (SECIS) element, that is necessary for the recognition of UGA as a Sec codon, rather than as a stop signal.
D1 and D2 activate TH, whereas D3, and to a lesser degree D1, inactivate both T4 and T3. The activity of iodothyronine deiodinase is related to the type of iodothyronine deiodinase expressed and to the degree of the enzyme activity in each tissue (23).
As mentioned previously, T3 binds to nuclear TRs to exert its biological effects. Since TR saturation depends on the intracellular concentration of T3, D2 and D3 play an opposite role in TH action by inversely affecting intracellular T3 levels. In fact, D2 produces T3 and thus activates TH signaling, whereas D3 inactivates T4 and T3, reducing TH signaling. Both pathways are relatively independent of circulating T4 or T3 levels (28).
D1 is predominantly expressed in the pituitary, thyroid, liver, and kidney. D1 is the only iodothyronine deiodinase that can function as either an outer- or an inner -ring iodothyronine iodothyronine deiodinase, and was the first to be cloned (29). D1 is located in the plasma membrane, and the amount of T3 produced by this pathway is thought to equilibrate with the plasma pool of T3. If in rodents D1 is responsible for about 50% of the daily extrathyroidal production of T3, in humans D1 plays a lesser role in maintaining circulating T3 (Figure 2) (30, 31).
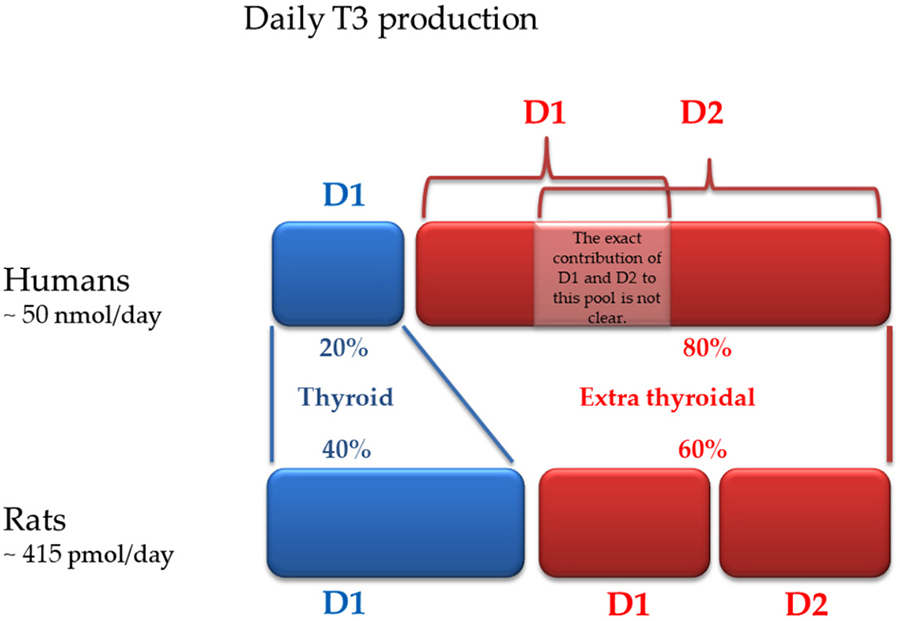 Figure 2
Figure 2Daily T3 production in humans and rodents: contribution of each deiodinase (modified from reference 31).
D2 is an endoplasmic reticulum resident protein. Its expression is low in most tissues during development, but it increases in the perinatal period (32). D2-mediated activity is an intracellular event: D2-originated T3, before exiting the cell and contributing to plasma T3 levels, diffuses to the nucleus and remains in the target cell for a longer period of time when compared to D1-originated T3. This is probably explained by the fact that D2-originated T3 exerts intracellular biological effects through the interaction with the local TRs (30). Tissues in which D2 plays a role are the cochlea, bone, brown adipose tissue, liver, and perhaps the muscle (22). D2 contributes to adaptive thermogenesis and metabolic control under norepinephrine control (33), and its expression is in turn regulated by food availability, which stimulates D2-mediated T3 production in mouse skeletal muscle (34). D2 also has a role in behavior and mood regulation, and animal models based on D2KO mouse exhibit a neurological phenotype characterized by diminished agility, altered global gait pattern, impairment of coordination and prehensile reflex, and reduced muscle strength (35). The human myocardium expresses D2 that is potentially capable of generating local T3. This is also the physiological basis contributing to the antiarrhythmic efficacy of amiodarone, which inhibits the deiodination of T4 to T3 (19). In fact, myocardial D2 increases fractional shortening, the velocity of circumferential fiber shortening, peak aortic outflow velocity, and aortic velocity acceleration. The noncompetitive D2 inhibitor amiodarone can potentially decrease thyroid hormone signaling in the heart. Furthermore, D2 expression has been detected in minimal amounts in both humans and murine muscle tissue, playing a role in muscle regeneration and exercise (36).
D3 is an inner-ring iodothyronine deiodinase with a half-life of approximately 12 h (22) that is expressed in numerous tissues in fetal life, but only in the central nervous system and placenta in adults. This enzyme is located in the plasma membrane and it is inserted into the membrane to easily inactivate TH (37). D3 activity predominates in developing tissues, preventing the exposure of cells to TH. D3-mediated T3 inactivation occurs in the brain, skin, and placenta: the latter could explain the need to increase TH replacement therapy during pregnancy (30, 38). On the other hand, in mature embryos, D3 expression is minimized and D2 increases, enhancing thyroid hormone signaling. However, D3 expression can be reactivated in multiple tissues in disease states, contributing to the low circulating levels of T3 observed in these conditions (39). D3 is expressed in both embryonic and adult pancreatic beta-cells in humans and mice, and D3 knock-out (KO) animals are glucose-intolerant, without changes in peripheral sensitivity to insulin (40). Severe heart diseases associated with ischemia or hypoxia increase the cardiac expression of D3 determining a local hypothyroidism (41). Increased D3 expression has been observed in animal models of heart remodeling due to myocardial infarction or chronic pulmonary hypertension with right ventricular hypertrophy (42). D3 overexpression has been found to be a common paraneoplastic syndrome in gastrointestinal stromal tumors (43), causing the so-called “consumptive hypothyroidism” in affected patients. Consumptive hypothyroidism is a systemic condition characterized by an accelerated degradation of circulating TH, at rates exceeding the synthetic capacity of the normal thyroid, due to an aberrant uncontrolled expression of D3 (44). This condition has previously been described in hemangiomas (45) and hemangioendotheliomas (46) and it is probably due to the great expression of D3 in these tumors (30). The diagnosis of consumptive hypothyroidism can be performed by the evidence of increased TH inactivation (elevated levels of serum reverse triiodothyronine or abnormal TH replacement therapy requirement). Similarly, the antineoplastic use of tyrosine kinase inhibitors imatinib and sunitinib is associated with hypothyroidism, which seems to result from the paradoxical increase in D3 observed in some tumors after exposure to these drugs (43). However, the cause of hypothyroidism in these situations is not fully understood, and it could be multifactorial, including enhanced D3 expression in other tissues (47).
A debate regarding the most important source of plasma T3 in humans has been going on for many years. In fact, although it was initially thought that D1 provided the majority of plasma T3 in humans, further investigations suggest that D2 have a major contribution (48). In line with this, treatment with PTU (which inhibits D1 activity) only causes an approximately 30% decrease in serum T3 in patients with primary hypothyroidism receiving fixed doses of exogenous T4, suggesting an important potential role of D2 in the generation of plasma T3 (49). The most supported hypothesis is that the human D1- or D2-derived T3 production varies significantly according to the thyroid status. As DIO1 expression is positively regulated by T3, D1-catalyzed T4 conversion to T3 is decreased in the hypothyroid state (50). In contrast, D2 half-life and DIO2 transcription was increased by low TH levels. In the opposite scenario, D1-catalyzed T3 production, in particular from the thyroid gland, predominates in the hyperthyroid state (51). In this condition, the thyroidal contribution to serum T3 levels increases in parallel with the disease severity, reaching nearly two-thirds of the total T3 production in severe hyperthyroidism (52). A decreased D1 activity has been reported in the NTIS and in several human neoplasia of epithelial origin, except for the follicular thyroid tumors and breast cancer, in which D1 expression is increased (29).
In conclusion, the ratio of D2 and D3 expression is important both in physiological conditions and in thyroid disease states. In fact, D2 expression and activity are both increased in TH deficiency, whereas they are reduced in thyrotoxicosis. In contrast, D3 expression is regulated reciprocally. Thus, this balance determines the homeostatic control of T3 availability to the nuclear receptor and it is particularly important in specific tissues, such as the brain, with the aim of “protecting” from possible deleterious changes in TH levels.
Normal thyroid function results from the complex and well-regulated activity of the HPT axis, which in conjunction with external factors maintains normal concentrations of TH. Pituitary TSH secretion represents the main regulator of thyroid development, growth, and function. The set point of the normal concentration of TSH is determined by the balance between the stimulatory action of TRH and the negative feedback of THs, as well as by other regulators originating from the central nervous system, peripheral tissues, and local pituitary mechanisms. Both circulating T4 and T3 play a role in the negative feedback loop that inhibits both TRH and TSH secretion: as mentioned previously, T4 is only minimally active, while T3 represents the “active” hormone which negatively regulates TSH and TRH genes.
The negative feedback loop is regulated by the so-called “hypophysiotropic TRH-synthesizing neurons”, located in the paraventricular nucleus (PVN) (53). Interestingly, low levels of THs increase TRH expression only in the hypophysiotropic neurons (54), while TRH gene expression is not regulated by T3 in non-hypophysiotropic TRH neurons (54). In addition to the negative feedback exerted by TH on TRH, other factors also contribute to the modulation of TRH neurons (e.g., neuropeptide Y (NPY), leptin, alfa-melanocortin-stimulating hormone (alfa-MSH), and agouti-related peptide (AgRP)) (23).
The TR-beta2 isoform is the dominant isoform mediating both the negative regulation of the TSH-beta gene expression and the negative regulation of the TRH gene expression in the hypothalamus (55). Interestingly, mice lacking the beta isoforms of TR (TRbetaKO) have significantly higher TH and TSH levels compared with mice KO for both TR and TRH (double KO), which have reduced TH and TSH levels. In fact, hypothyroid double KO mice failed to mount a significant increase in serum TSH levels. This confirms that TRH is absolutely required for both TSH and TH synthesis, even if it is not necessary for thyrotroph cell development (56).
An increase in circulating T4 is expected to increase T3 concentration and to reduce TSH gene expression, while a drop in serum T4 has the opposite effects. Importantly enough, variations of circulating T3 concentration have a direct effect on the hypothalamic and pituitary expression of TRH and TSH genes, while variations of T4 concentration have effect on HPT axis only thanks to D2 activity. However, the vast majority of T3 present in the brain is generated locally by D2-mediated deiodination of T4 (57). Therefore, D2 can be considered a key regulator of the negative feedback loop mediated by local TH (Figure 3).
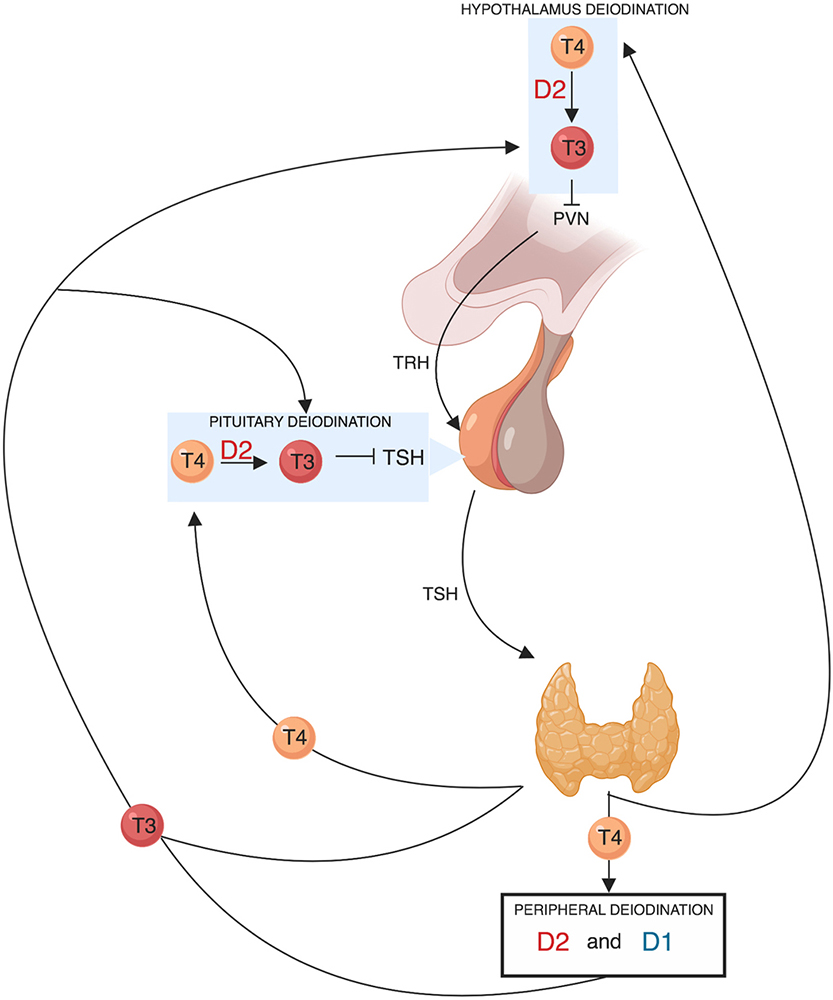 Figure 3
Figure 3Schematic representation of the role of deiodinases in the hypothalamic-pituitary-thyroid axis feedback regulation TRH derived from the parvocellular neurons of the hypothalamic paraventricular nucleus (PVN), stimulates TSH synthesis and secretion from thyrotroph cells located in the anterior pituitary gland. In turn, TSH regulates thyroid hormone production and release via binding to the TSH-receptor in thyroid follicular cells. The thyroid gland secretes T4 and about 20% of circulating T3. About 80% of circulating T3 is derived from the peripheral deiodination of T4 that is mediated by D2 (mainly) and D1. The circulating T3 signals a negative feedback mechanism both at the hypothalamic level by suppressing TRH secretion and at the pituitary level by suppressing TSH secretion. D2 is also expressed in the hypothalamus and in the anterior pituitary gland. The pituitary and hypothalamic deiodination of T4 to T3 and the local action exerted by the latter before exiting the cell explains how circulating T4 plays a key and probably even predominant role in regulating the negative feedback mechanism.
D2 has a relatively short half-life (approximately 40 minutes) related to its ubiquitination and destruction in the proteasomes. This process is accelerated by the interaction with T4, which is its natural substrate (51). This mechanism is at the basis of the rapid control of intracellular T3 production and of its interaction with TRs. Furthermore, D2 activity in HPT regulation is modulated by the pituitary adenylate cyclase-activating polypeptide (PACAP), which increases intracellular cAMP levels—a second messenger known to stimulate the DIO2 gene (58). D2 is co-expressed with TSH in pituitary thyrotrophs (59).
Tanycytes are specialized glial cells located in the ventrolateral walls and floor of the third ventricle, that seems to be crucial in regulating the negative feedback exerted by TH. Tanycytes are essential in guaranteeing strict TH levels in the hypothalamus. Tanycytes express TH transporters (particularly MCT8 and OATP1C1) and both D2 and D3. Therefore, while D2 is usually expressed by astrocytes in other brain regions, in the hypothalamus D2 is expressed in the tanycytes. D2 activity in tanycytes is crucial to permit the TH control of TRH expression. Tanycyte D2 has access to plasma or cerebral-spinal fluid-born T4, which is converted to T3. T3 in turn reaches the PVN or the pituitary gland via portal blood flow (60) and it enters TRH neurons via the MCT8 transporter where it regulates prepro-TRH expression and the processing of pro-TRH into TRH peptide (23). This process is rapid, occurring in about 5 hours (61).
Iodothyronine deiodinase activity has been considered to regulate the response of the HPT axis in different pathological conditions, including critical illness. However, data on this matter are still conflicting. The most accepted theory is that the extra-thyroidal conversion of T4 to T3 is reduced during critical illness due to the reduction in both hepatic/renal D1 activity and in skeletal muscle D2 activity (49). Furthermore, an increase in hepatic and skeletal muscle D3 activity may also be involved (62). Interestingly, the systemic administration of bacterial lipopolysaccharide (LPS) in normal mice increases D2 mRNA and its activity and reduces TRH expression. The T3 generated by the D2 pathway also directly affects the thyrotrophs, inhibiting TSH secretion (63). Nevertheless, recent evidences criticize these conclusions, arguing that the modifications in iodothyronine deiodinase expression in NTIS may be a consequence of changes occurring in T3 and T4, rather than the cause of these hormonal changes (64). In line with this, studies on both D3KO mice and D1/D2 KO mice subjected to treatment with LPS showed that the changes in T3 and T4 that occurred in response to LPS were essentially no different from those seen in wild-type (WT) animals (49). In addition, in WT mice treated with LPS, the decrease in plasma total T3 and total T4 has been found to precede the fall in hepatic D1 (65).
Iodothyronine deiodinase activity is the most important regulator of TH availability both in brain and in peripheral tissue. As a consequence, impaired D1 and D2 activity will virtually result in a reduced sensitivity to TH action. Until a few years ago, the mechanism of “resistance” to TH was considered as related almost exclusively to mutations in TRs, such that “Refetoff’s syndrome” was synonymous with “resistance to TH syndrome”. The recent literature has revised this concept, coining the so-called “syndrome of reduced sensitivity to thyroid hormone”, which includes other conditions that reduce the action or the availability of THs on target tissues. Defects that affect the synthesis of selenoproteins have also been included in this group. Furthermore, there is growing interest in the possible role of DIO1 and DIO2 polymorphisms in some pathological conditions and in determining the requirement of LT4 replacement therapy in carrying subjects.
In this section, the role of an impaired iodothyronine deiodinase activity as a model of reduced sensitivity to TH will be discussed, starting from the animal model of targeted disruption of DIO genes up to the possible consequences in clinical practice.
Studies aimed to evaluate the activity of iodothyronine deiodinase can be based on several approaches. One of these would be to specifically inhibit their activity by drugs. Unfortunately, there is no known pharmacological agent that specifically and completely inhibits the activity of each iodothyronine deiodinase individually. Furthermore, some of these compounds (e.g., PTU) also inhibit TH biosynthesis, and thus they are not ideal for in vivo studies where intact thyroid function is required. An alternative approach is to create animal models that lack the active form of an iodothyronine deiodinase enzyme. These elegant models might contribute to understanding the role of iodothyronine deiodinases not only in physiological, but also in pathological conditions. In particular, it is interesting to evaluate if animal models of impaired iodothyronine deiodinase activity can represent a model of reduced sensitivity to TH.
Schneider et al. produced a mouse strain lacking D2 activity using homologous recombination (66). Mice homologous for the targeted deletion (D2KO), in which no D2 activity was observed in any tissue (including astrocytes) in basal conditions, did not show abnormalities in their phenotype, and their development appeared normal except for a mild growth retardation in males. In the liver, thyroid, and pituitary, D1 levels were the same in WT and D2KO mice, while cerebral D3 activity in D2KO animals was twice that in WT. Interestingly, there were no differences in serum T3 levels between WT and D2KO animals, while serum T4 and TSH levels were significantly higher in the KO model. These findings suggest a pituitary resistance to the negative feedback exerted by plasma T4 in D2KO mice. Furthermore, the administration of both T4 and T3 was effective in hypothyroid WT mice in terms of TSH reduction, while only T3 was effective in D2KO models (66). In fact, in the presence of a targeted inactivation of the DIO2 gene, the exogenous administration of T3 overrode the absence of T4 activation in T3, restoring a “normal” feedback.
A more recent study performed in mice with inactivated pituitary D2, showed that basal TSH levels in neonatal D2KO mice were two-fold higher when compared with controls, but the TSH increase in response to hypothyroidism was reduced compared to controls, despite an identical level of pituitary TSHalfa and beta-subunit mRNAs. Therefore, a congenital severe reduction of thyrotroph D2 activity seems to cause a major impairment of the TSH response to hypothyroidism (67). Surprisingly, even if D2KO mice have elevated TSH levels, their TRH mRNA levels in the PVN were not increased, suggesting that the resistance to T4 suppression results mainly from DIO2 deficiency in the pituitary rather than in the hypothalamus (19).
It is well-known that analysis of the HPT axis in D2KO mice has demonstrated normal T3 levels, probably related to notably elevated T4 levels which are converted to T3 by the residual D1 activity (68). In contrast, other studies seem to demonstrate that iodothyronine deiodinase action is not critical for the maintenance of plasma T3 concentrations. In fact, D1 and D2 KO mice maintain normal plasma T3 concentrations (49). However, D2 KO animals have alterations in TSH regulations and thermogenesis, and the brain T3 concentration is reduced despite increased brain T4. Combined D1/D2 KO mice have plasma T4 levels almost twice those found in WT animals, and a plasma TSH 2.6 times the level found in WT or D1 KO (69). The most likely explanation for the maintenance of plasma T3 in D1/D2 KO animals is that the increased TSH concentration promotes an enhanced thyroidal production of T3. This also confirms that, despite the normal T3 plasma concentration, hypothalamic and pituitary T3 levels are decreased, causing an impaired negative feedback activity and an increase of TSH (60).
The role of D2 in regulating the sensitivity to THs is also related to its action in guaranteeing the normal development of TRs. Studies performed in zebrafish embryos and targeting D2 showed that, besides delays in phenotypic development, there is a reduction in THR-beta expression. Due to depletion of intracellular T3 levels, these effects were reversible by T3 administration (70).
In D1-deficient mice (D1KO), the general health and the reproductive capacity were unimpaired. The serum levels of T4 and rT3 were elevated, whereas those of TSH and T3, as mentioned previously, were unchanged. However, D1 deficiency caused marked changes in the metabolism and excretion of iodothyronines, including a significant increase in the fecal excretion of endogenous iodothyronines (71). This finding confirms the scavenging function for D1 that might be particularly important in the setting of iodine deficiency (29).
The intracellular levels of T3 define how much T3 is bound to TRs and the intensity of TH signaling, the so-called “thyroid status”. The “thyroid status” is the result of all T3-dependent signaling events and depends not only by the circulating T3 levels but also by the factors influencing the intracellular concentration of T3. In fact, the peripheral tissues can feel the “thyrotoxic” or “hypothyroid” status independently of serum TH thanks to cell specific factors which can alter the intracellular THs concentration and/or their activities. An example of a condition characterized by a mismatch between THs and TSH levels and tissue-specific “thyroid status”, is represented by the so-called “syndromes of reduced sensitivity to THs”, that include inherited disorders of the transport, metabolism, and action of THs, as recently stated by a new reclassification (72).
The first condition was described by Refetoff in 1967 (“resistance to thyroid hormone syndrome or “Refetoff’s syndrome”) (73). The most important clinical features of this condition are represented by elevated serum THs levels and non-suppressed TSH (“inappropriate TSH secretion”), often associated with goiter and no clear symptoms and signs of thyrotoxicosis, due to the possible coexistence of decreased and increased THs action in different tissues (74). This condition is predominantly due to a mutation in the THRB gene (75), but mutations in the THRA gene, which are related to a distinct phenotype, have recently been detected (76). Furthermore, a syndrome with reduced cellular access of T3 due to defects of THs cell membrane transport was identified (77, 78). Monocarboxylate transporter 8 (MCT8) and 10 (MCT10) are the most important TH transporters, which facilitate both TH transport into the cell and TH efflux from cells (79). Inactivating mutations in MCT8 cause a syndrome characterized by alterations in serum TH levels and neurological abnormalities (77, 78). Interestingly enough, some authors showed that over-expression of these transporter induces a transient elevation in intracellular T3 concentration but only a modest increase in nuclear T3 activity (79). Of note, mouse models with combined MCT8 and deiodinases deficiency have been generated to investigate the interplay of thyroid hormone metabolism and transport both in physiological and pathological conditions (80-83).
In this field, iodothyronine deiodinase activity plays a crucial role in assuring a normal intracellular “thyroid status”. In fact, D2 catalyzes the conversion of T4 to T3, increasing intracellular T3 levels and potentially leading to local thyrotoxicosis. The opposite is observed for D3 activity, which depletes the cell of T3 by deiodination to T2, causing local hypothyroidism. Therefore, due to the crucial role exerted in the regulation of TH metabolism, there is growing interest in how genetic variation and abnormalities in the iodothyronine deiodinases may influence thyroid function, not only by altering TH levels in blood, but also by altering their intracellular activity. Until just over 10 years ago, all of the known defects of TH metabolism observed in humans were acquired. For example, the most frequent defect is observed in “non-thyroidal illness syndrome” which produces the so-called “low T3 syndrome” (84).
In 2005, Dumitrescu et al. reported the first inherited disorder of iodothyronine metabolism, caused by a reduction in the synthesis of iodothyronine deiodinases (85). The involved gene, the selenocysteine insertion sequence-binding protein2 (SECISBP2), maps on the long (q) arm of chromosome 9 at position 22.2 (9q22.2) and spans a region of 46.644 bp (GRCh38/hg38: chr9:89,318,473-89,367,117; NC_000009.12) (86). The protein, composed of 854 amino acids, is one of the essential components of the machinery involved in co-translational insertion of selenocysteine (Sec) into selenoproteins. Sec is encoded by the UGA codon, which normally signals translation termination. The recoding of UGA as Sec codon requires a Sec insertion sequence (SECIS) element; present in the 3' untranslated regions of eukaryotic selenoprotein mRNAs. This protein specifically binds to the SECIS element, which is stimulated by a Sec-specific translation elongation factor.
Pathogenic variants in SECISBP2 gene have been associated with reduction in enzymatic activity of D2 and abnormal thyroid hormone metabolism. To date, 13 SECISBP2 variants have been reported as pathogenic. The most important clinical characteristics have been reported in Table 2. In particular, Dumitrescu described two unrelated families where affected children were either homozygous (p.Arg540Gln) or compound heterozygous (p.Lys438Ter/NM_024077.4:c.1212+29G>A) for SECISBP2 pathogenic variants (87). Significant clinical features were short stature, delayed bone age and delayed puberty. Accordingly with the impaired conversion of T4 to T3, laboratory findings are represented by high T4, low T3, high rT3, and slightly elevated serum TSH. When compared with their normal siblings, the affected children required higher doses and serum concentrations of T4 to reduce their TSH. In contrast, similar doses of T3 used in the affected and unaffected siblings had equal suppressive effects on TSH, suggesting a defect in T3 conversion.
| Family | SECISBP2 gene | Protein | N. affected | Defect | Main clinical characteristics | TFTs abnormalities | Overall phenotype | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | c.1619 G>A | p.Arg540Gln | 3 | homozygous | short stature, delayed bone age, | high T4, low T3, elevated TSH | mild | 85 |
| 2 | c.1312 A>T | p.Lys438Ter | 1 | compound heterozygous | transient growth retardation, | high T4, low T3, elevated TSH | mild | 85 |
| IVS8ds+29 G>A | fs | |||||||
| 3 | c.382 C>T | p.Arg128Ter | 1 | homozygous | growth retardation, | high T4, low T3, elevated TSH | mild | 91 |
| 4 | c.358 C>T | p.Arg120Ter | 1 | compound heterozygous | delayed bone maturation, impairement of mental and motor coordination, | high T4, low T3, elevated TSH | severe | 92 |
| c.2308 C>T | p.Arg770Ter | |||||||
| 5 | c.668delT | fs255Ter | 1 | compound heterozygous | delayed motor and speech milestones, hearing problems, infertility (azoospermia), axial muscular distrophy, cutaneous photosensitivity | high T4, normal T3 and TSH | mild | 94 |
| intron 6-155 delC | fs | |||||||
| 6 | c.2071 T>C | p.Cys691Arg | 1 | compound heterozygous | global developmental delay, short stature, hearing problems, axial muscular distrophy | high T4, low T3, normal TSH | mild | 94 |
| intronic SNP | fs | |||||||
| 7 | c.1529_1541dup CCAGCGCCCCACT | p.Met515 fs563Ter | 1 | compound heterozygous | short stature, delayed bone maturation | high T4, low T3, elevated TSH | mild | 95 |
| c.235 C>T | p.Gln79Ter | |||||||
| 8 | c.800_801insA | p.Lys267LysfsTer2 | 1 | homozygous | attention deficit disorder, muscle weakness | high T4, low T3, normal TSH | mild | 96 |
Studies performed on affected patients’ skin fibroblasts showed reduced baseline and cAMP-stimulated D2 enzymatic activity when compared to fibroblasts from unaffected individuals. However, the phenotype described by Dumitrescu is mild, because the deficiency is not complete, while a more severe deficiency might have more severe consequences (88).
There is no agreement about the best treatment in these patients. Supplementation with organic and inorganic selenium has been administered, but even if total serum selenium concentrations in SECISBP2-deficient subjects increased after selenomethionine supplementation, the effect was not indicative of improved selenoprotein synthesis (89). Treatment with LT3 showed an increase in height velocity and advancement in bone age with a narrowing of the gap between chronological and bone age (90).
Few years later, Di Cosmo reported a novel homozygous SECISBP2 variant, the p.Arg128Ter, producing an early arrest in the synthesis of the protein (91) while Azevedo (92) described a compound heterozygous 12-year-old girl carrying two new truncated variants (the p.Arg120Ter and the p.Arg770Ter). She was affected by a more severe phenotype due to impairment of the selenoprotein N-1 (SEPN-1)-related myopathy gene (93, 94).
Schoenmakers et al. detected compound heterozygous defects in two unrelated patients: proband 1 was heterozygous for a paternally inherited frameshift variant causing a premature stop codon at aminoacid 255 and a splicing defect causing misincorporation of additional intronic sequence between exon 6 and 7 on the other allele. Patient 2 was heterozygous for the maternally inherited missense variant p.Cys691Arg together with a paternally derived defect generating aberrantly spliced transcripts (Delta2-3-4 and Delta3-4) (94).
Finally, a pathogenic SECISBP2 genotype (p.M515SerfsTer48/p.Q79X) has been identified in Japanese boy treated with LT3 and recombinant growth hormone therapy to normalize thyroid function tests and improve bone maturation (95). The last reported case is a patient carrying the homozygous p.K267KfsTer2 variant (96).
The most impressive way to describe the impact of SECISBP2 pathogenic variants in clinical practice and the complexity of the underlying mechanisms which are mostly still unknown is represented by the refined words of Duntas who, referring to Greek mythology, compares the complexity of the SECISBP2 syndrome to Daedalean labyrinth: “the way out must be sought among the many mutations of SECISBP2 —several now known and others that remain to be identified—which express various defective selenoproteins that cause disease. In this crucial quest, better understanding of the thyroid hormone phenotype as well as of the Se concentrations could well prove to be Ariadne’s invaluable clue” (97).
Even though Gabriela Morreale de Escobar published two studies in 1995 and 1996 demonstrating that only the combined T4 + T3 treatment ensured euthyroidism in all tissues of thyroidectomized rats (98, 99), LT4 replacement therapy is considered the current standard of care for patients affected by hypothyroidism, and no consistently strong evidence supports the superiority of alternative therapies. The regulated peripheral conversion of LT4 to T3 in humans has previously been demonstrated (100). Such conversion, dependent upon the activity of iodothyronine deiodinases, allows normal T3 levels to be achieved with traditional LT4 therapy alone in patients who underwent near-total or total thyroidectomy, suggesting that T3 administration is not necessary to maintain serum T3 values at their endogenous prethyroidectomy levels (11). The goal of LT4 treatment is to normalize serum TSH levels and improve the biological markers of hypothyroidism (101).
However, it is mandatory to consider all the situations that could make TSH evaluation not reailable. Different drugs (glucocorticoids, dopamine agonists, somatostatin analogues, rexinoids, anti-epileptic drugs) can affect thyroid function and reduce TSH, as well as secondary hypothyroidism related to a concomitant pituitary disease (102, 103).
On the other hand, in conditions of elevated TSH, the impact of gastrointestinal disorders which can affect LT4 absorption must be considered (104).
The adequacy of this strategy to replace physiological requirements and reverse patients’ symptoms remains controversial. Therefore, for the subset of patients treated with LT4 and complaining of persistent hypothyroid symptoms (i.e., memory loss, weight gain, fatigue, depression, and reduced quality of life) despite normal TSH levels, studies comparing clinical improvement between LT4 monotherapy and combination therapy with LT4 and LT3 have been performed. Some authors reported the superiority of combination therapy for mood, anxiety, and depression (105), whereas others observed no significant superiority or only partial improvement (106). However, in consideration of the insufficient evidence of substantial benefit and the lack of long-term LT3 safety, combination therapy is not currently recommended for the routine treatment of hypothyroidism.
Aside from the lack of a feeling of well-being, which is a subjective parameter, another problem is represented by the difficulty in reaching adequate TSH levels in some subjects on TH replacement. In these cases, a possible degree of reduced sensitivity to THs could be suspected. Nevertheless, twin and family studies are able to estimate that the heritability of TSH levels can be 65% and of FT4 up to 39%, suggesting the strong role of genetic predisposition (107). Furthermore, in approximately 20% of patients, LT4 did not ensure physiological T3 levels (108), even if results on this matter are controversial (11). In this setting, a different genetic background, which causes a reduced bioavailability of both endogenous and synthetic LT4, could be helpful in explaining these biochemical discrepancies. Single nucleotide polymorphisms (SNPs) in DIO genes represent a widely studied field in this context.
Polymorphisms in DIO genes are associated with variations in serum TSH and iodothyronine concentrations in healthy subjects (109). The C-allele of DIO1 SNP (rs2235544) is associated with increased D1 activity, which results in slightly higher FT3 and lower FT4 and, in LT4-treated hypothyroid patients, in higher FT3/FT4 as compared to WT (110). Minor genotypes of others DIO1 variants (rs11206244, rs2294512, and rs4926616) have been associated with reduced psychological well-being (111).
The C/C genotype of the rs225014T>C (NP_001311391.1:p.Thr92Ala) SNP was reported in 10.7% of the general population and in 11.3% of LT4 users (112). However, the effects induced by this polymorphism on D2 enzymatic activity and on thyroid function tests are unclear.
Torlontano (113) reported that athyreotic differentiated thyroid cancer (DTC) patients homozygous for rs225014C allele needed a higher LT4 dose compared with carriers of the rs225014T allele. However, this observation could be applied only to patients with serum TSH concentrations in the range of semi-suppression (0.1−0.5 mU/L), but not in the “suppressed group” (TSH <0.1 mU/L).
Castagna found an association between low FT3 values and the rs225014 polymorphism in thyroidectomized patients. In particular, the mean post-surgery FT3 levels were significantly lower in patients carrying the rs225014C allele than in patients with the rs225014T allele, in whom FT3 postsurgical levels were similar to pre-surgery levels. On the basis of these findings, thyroidectomized patients carrying the p.Thr92Ala variant are at increased risk of reduced intracellular and serum T3 concentrations, and could be inadequately compensated for by LT4 (114). The most recent study found that the homozygous genotype DIO2 rs225014 (T/T) and the homozygous genotype rs225015 (G/G) were associated with higher levels of TSH compared with their other types. Therefore, patients carrying these genotypes present higher LT4 requirement (115).
In contrast, other studies excluded the effects of the rs225014 polymorphism on serum TSH, FT4, or T3 (116, 117). A recent paper evaluating LT4 therapy in athyreotic patients with differentiated thyroid cancer showed no significant association with DIO2 rs225014 and rs12885300 polymorphisms in predicting the dose requirement for TSH suppression. In this study, neither DIO1 SNP seemed to influence HPT axis regulation (118).
Interestingly, the p.Thr92Ala variant has been associated with diabetes, psychological well-being, hypertension, and the risk of osteoarthritis (116, 119, 120). In African Americans, the presence of the p.Thr92Ala is associated with a higher risk, compared to European Americans, of developing Alzheimer’s disease, supporting the hypothesis that p.Thr92Ala might represent one factor contributing to racial discrepancies in incident AD.
Furthermore, some studies have reported the association between polymorphism in DIO1 and DIO2 and autoimmune thyroid disease, reporting a low frequency of the T/T genotype for D2 rs225014 polymorphism (which is associated with higher D2 activity) in autoimmune thyroid diseases, and especially in Hashimoto’s thyroiditis (121).
Since their first discovery, iodothyronine deiodinases have represented a very challenging topic in TH biology, both in physiological and pathological conditions. The superfine regulation of the HPT axis by iodothyronine deiodinases is the result of a harmonious concert of the three enzymes, which guarantees a physiological steady-state in thyroid function. In pathological conditions, iodothyronine deiodinase activity is regulated to “protect” each tissue from both over- and under-exposition of THs, and several drugs (e.g., amiodarone) exert their pharmacological activity via their effects on iodothyronine deiodinases.
Animal models created by the targeted disruption of DIO genes have unveiled some of the mechanisms at the basis of these complex events. However, it is difficult to fully apply certain paradigms deriving from these experiments due to the different contribution of each iodothyronine deiodinase to the circulating and local pools of T3 that exists between animals (in particular rodents) and humans.
The biggest challenge is to better characterize the genetic background and the clinical features of the syndromes related to defects of the SBP2 gene, which is the only known genetic defect related to iodothyronine deiodinase activity. Unfortunately, probably due to the relatively recent discovery of this syndrome, few cases have been reported in the literature. Nevertheless, this condition should be considered and eventually properly evaluated in the differential diagnosis of the reduced sensitivity to TH syndromes.
A more difficult trail to run across is represented by the role of iodothyronine deiodinase polymorphism both in the presence of persistent symptoms of hypothyroidism during “biochemically adequate” LT4 replacement therapy and in the presence of abnormal increases of LT4 replacement therapy to normalize TSH levels. Data are conflicting, on the one hand authors considers the genetic background an important element to guarantee a “personalized medicine” instead of empirical treatment, on the other hand different studies exclude any significant association between patients carrying the polymorphism and controls. In conclusion, further studies are needed to evaluate the efficacy of a combined LT4–LT3 replacement therapy in this subset of patients.
The authors contributed equally to this work.
alfa-MSH
alfa-melanocortin-stimulating hormone
Agouti-related peptide
Differentiated thyroid cancer
Hypothalamus-pituitary-thyroid
L-amino acid transporters
Lipopolysaccharide
Loss of heterozygosity
Monocarboxylate transporters
Neuropeptide Y
Non-thyroidal illness syndrome
Paraventricular nucleus
Peroxisome proliferator-activated receptor
Phosphatidylinositol 3-OH kinase
Pituitary adenylate cyclase-activating polypeptide
Pituitary transforming gene
Propylthiouracil
Resistance to thyroid hormone
Retinoid X receptors
Reverse T3
Selenocysteine insertion sequence-binding protein 2
Selenocysteine-specific elongation factor tRNA
Selenocysteine-specific elongation factor
Single nucleotide polymorphisms
Thyroid hormone receptor alfa gene
Thyroid hormone receptor beta gene
Thyroid hormone receptors
Thyroid hormones
Thyroid response elements
Thyrotropin
Thyrotropin-releasing hormone
Thyroxine
Transporters of organic anions
Triiodothyronine
Type 1 iodothyronine deiodinase
Type 2 iodothyronine deiodinase
Type 3 iodothyronine deiodinase
Wild-type