



Frontiers in Bioscience-Landmark (FBL) is published by IMR Press from Volume 26 Issue 5 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.

1 Cellular and Molecular Signaling, New York, New York 10022
Abstract
Metabolic disorders, such as diabetes mellitus (DM), are increasingly becoming significant risk factors for the health of the global population and consume substantial portions of the gross domestic product of all nations. Although conventional therapies that include early diagnosis, nutritional modification of diet, and pharmacological treatments may limit disease progression, tight serum glucose control cannot prevent the onset of future disease complications. With these concerns, novel strategies for the treatment of metabolic disorders that involve the vitamin nicotinamide, the mechanistic target of rapamycin (mTOR), mTOR Complex 1 (mTORC1), mTOR Complex 2 (mTORC2), AMP activated protein kinase (AMPK), and the cellular pathways of autophagy and apoptosis offer exceptional promise to provide new avenues of treatment. Oversight of these pathways can promote cellular energy homeostasis, maintain mitochondrial function, improve glucose utilization, and preserve pancreatic β-cell function. Yet, the interplay among mTOR, AMPK, and autophagy pathways can be complex and affect desired clinical outcomes, necessitating further investigations to provide efficacious treatment strategies for metabolic dysfunction and DM.
Keywords
- Alzheimer’s disease
- AMPK
- Apoptosis
- Autophagy
- Dementia
- Diabetes Mellitus
- Erythropoietin
- Nicotinamide
- mTOR
- Oxidative Stress
- PARP
- Rapamycin
- Sirtuin
- SIRT1
- Review
Metabolic disorders, such as diabetes mellitus (DM), form a significant component of non-communicable diseases (NCDs). Of concern, NCDs are increasing in incidence throughout the world. According to the World Health Organization, approximately seventy percent of deaths that are recorded each year are the result of NCDs (1, 2). Wealthy as well as low income countries are affected. More than ten percent of the population less than sixty years of age is affected in high-income countries (1). Interestingly, of the over forty million people that die each year from NCDs, fifteen million individuals are younger with ages between thirty and sixty-nine years old. Yet, NCDs affect a greater proportion of the population in low and middle-income countries with at least one-third of the population under the age of sixty suffering from NCDs.
This rise in NCDs parallels the observed increase in life expectancy of the world’s population (3). The age of the world’s population continues to increase with new estimates of life expectancy approaching eighty years of age (4). With life expectancy marked by a one percent decrease in the age-adjusted death rate from the years 2000 through 2011 (5), the number of individuals over the age of sixty-five has been noted to double during the previous 50 years (6). Furthermore, the number of older individuals in large developing countries such as India and China also will increase from five to ten percent over the next several decades (7, 8). Multiple factors may account for the observed increased in lifespan for the world’s population. These include improvements in treatments for multiple disorders that involve endocrine disease, vascular disease, acute neurodegenerative disorders, and nutrition as well as improved access to preventive care (9-13).
As an important NCD, DM is increasingly being recognized as a target to develop novel treatment strategies to reduce death and disability for the world’s population (14-16) (Table 1). Approximately eighty percent of adults with DM are living in low- and middle-income countries (17). More than $20,000 USD are required to care for each individual with DM per year. The care for patients with DM equals approximately $760 billion United States Dollars (USD) (17) and consumes more than seventeen percent of the Gross Domestic Product in the United States (US) as reported by the Centers for Medicare and Medicaid Services (CMS) (18). With the loss of function and disability that results from DM, an estimated sixty-nine billion USDs are consumed from reduced productivity linked to DM.
| 1. As important NCDs, metabolic disorders and DM are increasingly being recognized as critical targets to develop novel treatment strategies to limit death and disability for the world’s population. |
To add to these financial concerns for DM, the number of individuals with DM is expected to rise to seven hundred million individuals by the year 2045 according to the International Diabetes Federation (17). Currently, it is believed that close to five hundred million individuals have DM (7, 19-22). An additional four hundred million individuals also have some form of metabolic disease and are at risk for developing DM but remain undiagnosed at this time (17, 23-25).
Obesity in the general population is considered to be another risk factor for the development of DM. Obesity results in impaired glucose tolerance that leads to DM progression (26-28). As a result, impaired glucose tolerance and obesity increases the risk of developing DM in young individuals (29). Obesity with excess body fat also can affect stem cell proliferation, aging, inflammation, oxidative stress injury, and mitochondrial function (28, 30-35).
DM affects all systems of the body. For example, in the peripheral nervous system, at least seventy percent of individuals with DM can develop some degree of diabetic peripheral neuropathy. DM can lead to both autonomic neuropathy (36) and peripheral nerve disease (37, 38). Assessments of peripheral neuropathies can be challenging, since the disorder is chronic in nature, may be sub-clinical, and prior deficits may go undetected even after improved control over glucose homeostasis has been initiated. In the central nervous system, DM can cause insulin resistance and dementia in patients with Alzheimer’s disease (AD) (16, 39, 40). DM can affect multiple cellular pathways that lead to the progression of cognitive loss (7, 41-45). DM also has been linked to mental illness (46, 47), cerebral vascular injury (7, 24, 48-51), impairment of microglial activity (16, 39, 40), and can impact stem cell proliferation (7, 41-45). In addition, DM can result in endothelial dysfunction (3, 7, 52-54), cardiovascular disease (25, 26, 53, 55-61), retinal disease (62-64), and immune function disorders (65-70).
Given the significant death and disability that metabolic disease and DM can cause in the global population with a severe financial drain on world economies, it is clear that new avenues of therapeutic discovery are required. With conventional therapies, early diagnosis of DM and rapid treatment can offer some degree of protection and may inhibit the progression of DM (3, 21, 71-75). Yet, tight serum glucose control does not always resolve the complications from DM (29, 76). In addition, use of diet control treatments may be effective to prevent hyperglycemic events, but these strategies have potential risks that can decrease organ mass through processes that involve autophagy (77). As a result, new avenues for therapeutic strategies to address metabolic disorders are urgently needed. One novel strategy that offers exciting prospects involves the agent nicotinamide and the pathways associated with the mechanistic target of rapamycin (mTOR), mTOR Complex 1 (mTORC1), mTOR Complex 2 (mTORC2), AMP activated protein kinase (AMPK), and programmed cell death with autophagy and apoptosis (Table 1).
The vitamin nicotinamide is the amide form of vitamin B3 (niacin). It is obtained through synthesis in the body or as a dietary source and supplement, such as from animal sources or plants (78). Nicotinic acid is the other form of the water-soluble vitamin B3 (79) . The principal form of niacin in dietary plant sources is nicotinic acid that is rapidly absorbed through the gastrointestinal epithelium (80). Nicotinamide is generated through the conversion of nicotinic acid in the liver or through the hydrolysis of the coenzyme ß-nicotinamide adenine dinucleotide (NAD+) (Figure 1). Once nicotinamide is obtained in the body, it serves as the precursor for NAD+ (81, 82). It is also necessary for the synthesis of nicotinamide adenine dinucleotide phosphate (NADP+) (83). Initially, nicotinamide is changed to its mononucleotide form (NMN) with the enzyme nicotinic acid/nicotinamide adenylyltransferase yielding the dinucleotides NAAD+ and NAD+. NAAD+ converts to NAD+ through NAD+ synthase (84) or NAD+ can be synthesized through nicotinamide riboside kinase that phosphorylates nicotinamide riboside to NMN (85, 86).
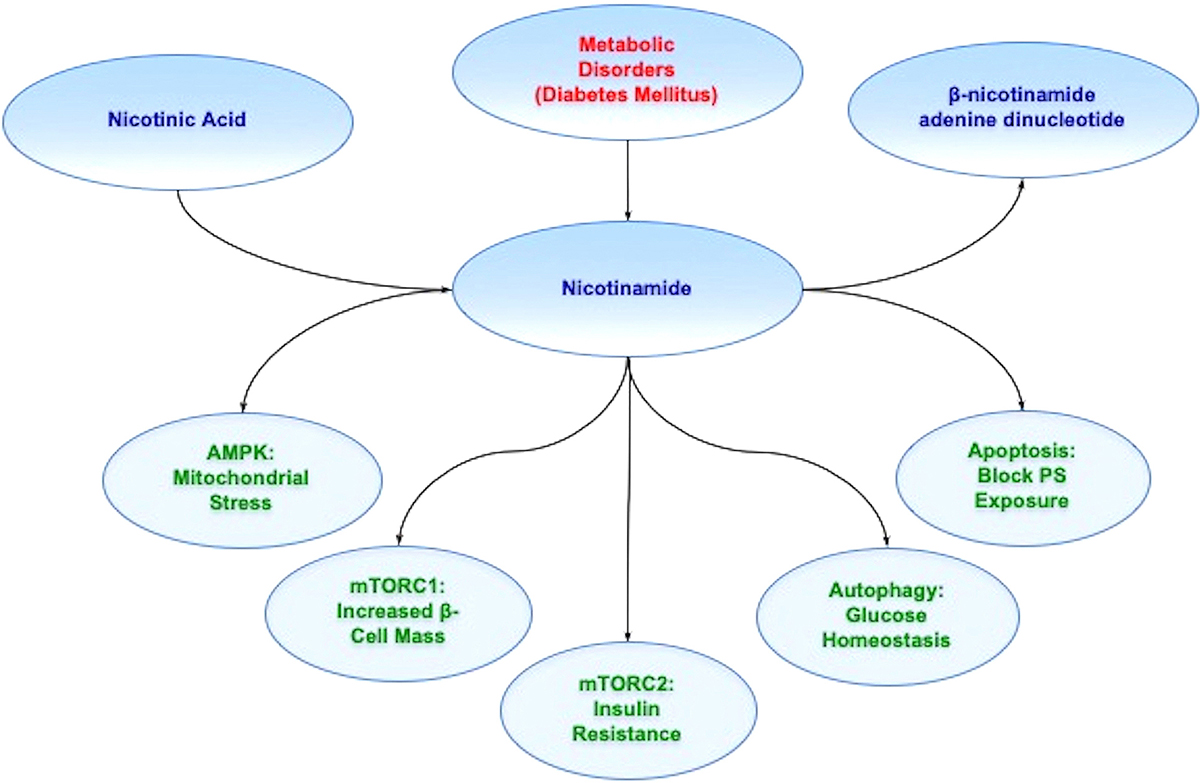 Figure 1
Figure 1Nicotinamide and metabolic disease. Nicotinamide is generated through the conversion of nicotinic acid in the liver as one source. Once nicotinamide is obtained in the body, it serves as the precursor for NAD+. Nicotinamide through the mechanistic target of rapamycin (mTOR), mTOR Complex 1 (mTORC1), mTOR Complex 2 (mTORC2), AMP activated protein kinase (AMPK), and pathways of autophagy and apoptosis offer innovative strategies to treat metabolic disorders such as diabetes mellitus (DM). For example, during DM, AMP activated protein kinase (AMPK) can limit mitochondrial stress, mTOR Complex 1 (mTORC1),can increase pancreatic ß-cell mass, mTOR Complex 2 (mTORC2) can improve insulin resistance, autophagy can foster glucose homeostasis, and blockade of apoptosis can prevent cellular membrane phosphatidylserine (PS) externalization to promote cell longevity. Further appreciation and understanding of the complexities of these pathways can foster new strategies for translation into innovative treatments for metabolic disorders, such as DM.
Nicotinamide through NAD+ has a critical physiological role in cellular metabolism and can be directly utilized by cells to synthesize NAD+ (82, 87, 88). Nicotinamide also participates in energy metabolism through the tricarboxylic acid cycle by utilizing NAD+ in the mitochondrial respiratory electron transport chain for the production of ATP, DNA synthesis, and DNA repair (89-91). Under some circumstances, nicotinamide can preserve mitochondrial function as a mechanism to enhance cellular survival (82, 92). Some studies suggest that the specific levels of NAD+ can be the critical factor for cell survival (88, 93, 94). Increased administration of nicotinamide may be useful against tumorigenesis (95) and lead to apoptotic cell death in cancer cells (96, 97).
If nicotinamide is depleted in the body, fatigue, loss of appetite, pigmented rashes of the skin, and oral ulcerations can result. More severe states of deficiency lead to pellagra that is characterized by cutaneous rashes, oral ulcerations, gastrointestinal difficulties, and cognitive loss (98). Pellagra can occur during conditions of low nicotinamide or the inability to absorb nicotinamide (99). As a result, the cellular pathways of nicotinamide are essential for energy metabolism and are directly tied to normal physiological processes as well as disease states that include inflammatory pathways (100), energy metabolism (72), vascular disease (101, 102), alcohol toxicity (103), and oxidative stress (88, 104, 105).
During periods of cellular injury that can involve oxidative stress (21, 106), reactive oxygen species can be scavenged by endogenous antioxidant systems that include nicotinamide, superoxide dismutase, glutathione peroxidase, catalase, and small molecule substances such as vitamins C, D, E, and K (107-113). However, nicotinamide can affect cellular survival during metabolic dysfunction and impact multiple systems of the body that are particularly affected by aging. Nicotinamide can foster protection during aging and mitochondrial dysfunction (79, 106, 114, 115), neuronal cell injury (103, 104, 116-119), vascular aging processes (29, 73, 101, 120), vascular demise and associated angiogenesis (101, 104, 117, 121-123), and neurodegenerative disorders such as AD (7, 87, 98, 124). Nicotinamide offers protection usually in a specific concentration range (81). Administration of nicotinamide in a range of 5.0 - 25.0 mmol/L can significantly protect neurons during oxidative stress injuries. This concentration range is similar to other injury paradigms in both animal models (125) and in cell culture models (82, 121, 126). It is important to note that elevated concentrations in some experimental models may not offer protection and can be detrimental (108, 127).
Nicotinamide plays a significant role during metabolic dysfunction and DM (29, 72, 73) (Table 1). Nicotinamide may lower insulin resistance and glucose release in combination with other factors to prevent the onset and progression of DM (128-130). Nicotinamide may protect against skeletal muscle atrophy during DM (131) and may reduce inflammation of the brain during DM with the administration of niacin (132). Prior work also has shown that nicotinamide can maintain normal fasting blood glucose with streptozotocin-induced DM in animal models (133, 134) and inhibit oxidative stress pathways that lead to cell death and apoptosis (121, 135-138). Nicotinamide can significantly improve glucose utilization, prevent excessive lactate production, and improve electrophysiologic capacity in ischemic animal models (139). Oral nicotinamide administration (1200mg/m2/day) protects pancreatic β-cell function and prevents clinical disease in islet-cell antibody-positive first-degree relatives of type-1 DM (140). Nicotinamide administration (25mg/kg) in patients with recent onset type-1 DM combined with intensive insulin therapy for up to two years after diagnosis can significantly reduce HbA1c levels (141). Yet, the duration of nicotinamide administration may influence the efficacy of this agent since long-term administration has been reported to support glucose intolerance in some animal models (93). Prolonged exposure of nicotinamide in other studies was reported to result in impaired pancreatic β-cell function and cell growth (142, 143). Nicotinamide also may inhibit cytochromes P450 and hepatic metabolism (144).
As previously noted during metabolic disease, nicotinamide is vital for maintaining important cellular energy homeostasis and mitochondrial function (78, 79, 82, 120). Nicotinamide can control mitochondrial function at the level of mitochondrial membrane pore formation to prevent the release of cytochrome c (82, 121, 136). Pretreatment of cells with either nicotinamide alone or in combination with the mitochondrial permeability transition pore inhibitor cyclosporin A prior to an injury paradigm can inhibit mitochondrial membrane depolarization (145, 146). Nicotinamide can block the chemical induction of mitochondrial membrane depolarization during exposure to either tert-butylhydroperoxide or atractyloside (104). There are other pathways that nicotinamide may use to maintain cellular metabolic homeostasis through the maintenance of mitochondrial membrane potential (121, 126). Nicotinamide can lead to the phosphorylation of Bad to prevent mitochondrial membrane depolarization and subsequent cytochrome c release (82, 121, 136), block the assembly of the mitochondrial permeability transition pore complex similar to the action of cyclosporin A (147), or stabilize cellular energy metabolism since the maintenance of mitochondrial membrane potential is an ATP facilitated process (148).
As a vital modulator of cell survival, nicotinamide oversees pathways of programmed cell death that involves apoptosis (149-151). The apoptotic pathway consists of two distinct phases. The early phase involves the loss of plasma membrane phosphatidylserine (PS) asymmetry (152-158). A later phase results in genomic DNA degradation (156, 159-163). Apoptosis is initiated through a cascade activation of nucleases and proteases that involve caspases (105, 106, 164-167). These processes can influence both the early phase of apoptosis with the loss of plasma membrane PS asymmetry and the later phase that leads to genomic DNA degradation. Loss of membrane PS asymmetry activates inflammatory cells to target, engulf, and remove injured cells (152, 157, 168, 169). If the engulfment by inflammatory cells can be prevented and cells that have membrane PS residues exposed are not removed, then functional cells expressing membrane PS residues can be rescued (62, 160, 170, 171). Yet, once the destruction of cellular DNA occurs, the process is usually not considered to be completely reversible (172).
Nicotinamide can address both phases of apoptotic cell injury. During different cellular injury paradigms, nicotinamide can prevent exposure of membrane PS residues to block inflammatory cell activation (81, 121, 126, 136). In particular, nicotinamide may reduce cardiovascular injury by blocking membrane PS exposure in vascular cells (82, 121), since membrane PS residue externalization in vascular cells can lead to hypercoagulation states (173) and cellular inflammation (174, 175). Nicotinamide may reverse a previously sustained insult (82, 104, 121, 126, 136, 176). Post-treatment strategies with nicotinamide that can follow apoptotic injury in “real-time” show that cellular injury can be reversed. Nicotinamide can reverse an initial progression of membrane PS inversion and prevent PS exposure (82, 126, 176, 177). These results support the hypothesis that if a cellular injury does not progress to DNA degradation, the injury can be reversible (82, 126, 176, 177).
In relation to apoptotic DNA degradation, nicotinamide prevents apoptotic DNA injury in vascular cells (104, 121), neurons (116, 124, 136, 178), keratinocytes (179), corneal endothelial cells (180, and photoreceptor cells {Kiuchi, 2002 #3733, 181). Nicotinamide also can inhibit DNA replication in some cell systems (182). Dependent upon the cellular conditions, nicotinamide may not be able to prevent DNA degradation (87). During periods of acidosis-induced cellular toxicity that involve decreased pH, nicotinamide cannot prevent cellular injury during intracellular acidification (126).
Autophagy is a process that recycles components of the cytoplasm in cells for tissue remodeling (183-185) (Table 1). As a result, it eliminates non-functional organelles (24, 149, 151, 186, 187). Macroautophagy recycles organelles and sequesters cytoplasmic proteins and organelles into autophagosomes. Autophagosomes subsequently combine with lysosomes for degradation and recycling (188, 189). Microautophagy consists of lysosomal membranes invagination for the sequestration and digestion of cytoplasmic components (8). Chaperone-mediated autophagy requires cytosolic chaperones to transport cytoplasmic components across lysosomal membranes (67, 190).
Similar to nicotinamide, autophagy is involved with clinical aging pathways (12, 105, 184). Studies with Drosophila show that neural aggregate accumulation observed with aging is linked to a reduction in the autophagy pathway. These neural aggregates lead to behavior impairments that can be resolved with the maintenance of autophagy pathways in neurons (191). Autophagy also is involved in a number of other disorders that may be tied to aging such as dementia (40, 192-196), AD (7, 12, 39, 40, 193, 197-201), Huntington’s disease (HD) (172, 202-204), and DM (21, 27, 39, 40, 62, 193, 205).
Nicotinamide has been tied to autophagic pathways, especially as an inhibitor of sirtuin pathways, such as those involved with silent mating type information regulation 2 homolog 1 (Saccharomyces cerevisiae) (SIRT1) (7, 16). Nicotinamide can lead to the induction of delayed autophagy and decreased survival in cancer cells (206). Mitochondrial autophagy can result in an increased NAD+/NADH ratio during nicotinamide administration (93, 105, 207) and in some cases induction of autophagy can affect mitochondrial mass (196, 208, 209). Chronic administration of nicotinamide can result in skeletal muscle autophagy (93). Nicotinamide has been shown to protect against palmitate-induced hepatotoxicity via SIRT1-dependent induction of autophagy (210).
Through autophagy, nicotinamide has been shown to rely upon the pathways of the mechanistic target of rapamycin (mTOR) (Table 1). For example, nicotinamide can protect hypoxic myocardial cells through the induction of autophagy and the modulation of mTOR pathways (211). mTOR, a 289-kDa serine/threonine protein kinase, is increasingly being recognized as a critical pathway for nicotinamide given the ability of this vitamin to control cellular metabolism and the programmed death pathways of autophagy and apoptosis. mTOR also is known as the mammalian target of rapamycin and the FK506-binding protein 12-rapamycin complex-associated protein 1 (203, 212) and is encoded by a single gene FRAP1 (213-215). The target of rapamycin (TOR) was initially discovered in Saccharomyces cerevisiae with the genes TOR1 and TOR2 (216). Using rapamycin-resistant TOR mutants, TOR1 and TOR2 are now known to encode the Tor1 and Tor2 isoforms in yeast (217). The compound rapamycin is a macrolide antibiotic in Streptomyces hygroscopicus that blocks TOR and mTOR activity (24).
mTOR serves as the principal component of the protein complexes mTOR Complex 1 (mTORC1) and mTOR Complex 2 (mTORC2) (218-220) (Figure 2). Rapamycin prevents mTORC1 activity by binding to immunophilin FK-506-binding protein 12 (FKBP12) that attaches to the FKBP12 -rapamycin-binding domain (FRB) at the carboxy (C) -terminal of mTOR to interfere with the FRB domain of mTORC1 (221). The mechanism of how rapamycin blocks mTORC1 activity with the interaction of the domain of FRB is not entirely clear. One pathway may involve allosteric changes on the catalytic domain as well as the inhibition of phosphorylation of protein kinase B (Akt) and p70 ribosomal S6 kinase (p70S6K) (222). mTORC1 is more sensitive to inhibition by rapamycin than mTORC2, but chronic administration of rapamycin can inhibit mTORC2 activity as a result of the disruption of the assembly of mTORC2.
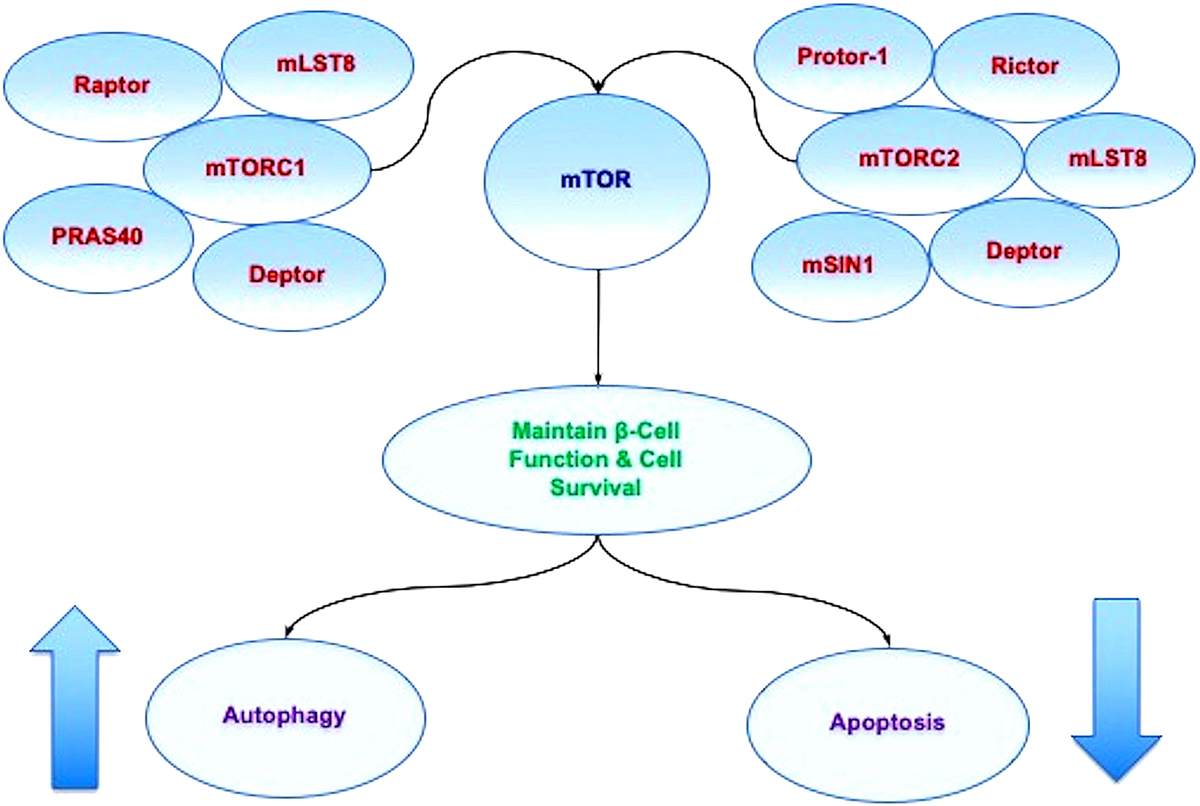 Figure 2
Figure 2mTOR oversight of autophagy and apoptosis. mTOR is the principal component of the protein complexes mTOR Complex 1 (mTORC1) and mTOR Complex 2 (mTORC2). mTORC1 is composed of Raptor, the proline rich Akt substrate 40 kDa (PRAS40), Deptor (DEP domain-containing mTOR interacting protein), and mammalian lethal with Sec13 protein 8, termed mLST8 (mLST8) (214). mTORC2 is composed of Rictor, mLST8, Deptor, the mammalian stress-activated protein kinase interacting protein (mSIN1), and the protein observed with Rictor-1 (Protor-1). Autophagy activity can be controlled through mTOR since activation of autophagy occurs during the inhibition of mTOR. As an example, mTOR inhibition also may be required for maintaining a balance between pancreatic -cell proliferation and cell size. Yet, mTOR activation can be beneficial at time s since mTOR activation protects pancreatic -cells against cholesterol-induced apoptosis, reduces glucolipotoxicity, and results in increased neuronal cell survival in cellular models of diabetes mellitus. These observations demonstrate that a fine balance in activity is required for mTOR, autophagic, and apoptotic pathways.
mTORC1 is composed of Raptor, the proline rich Akt substrate 40 kDa (PRAS40), Deptor (DEP domain-containing mTOR interacting protein), and mammalian lethal with Sec13 protein 8, termed mLST8 (mLST8) (216). mTORC1 binds to its constituents through the protein Ras homologue enriched in brain (Rheb) that phosphorylates the Raptor residue serine863 and other residues that include serine859, serine855, serine877, serine696, and threonine706 (223). The inability to phosphorylate serine863 limits mTORC1 activity, as shown using a site-direct mutation of serine863 (224). mTOR can control Raptor activity which can be blocked by rapamycin (224). Deptor, an inhibitor, blocks mTORC1 activity by binding to the FAT domain (FKBP12 -rapamycin-associated protein (FRAP), ataxia-telangiectasia (ATM), and the transactivation/transformation domain-associated protein) of mTOR. If the activity of Deptor is suppressed, Akt, mTORC1, and mTORC2 activities are increased (225). PRAS40 blocks mTORC1 activity by preventing the association of p70 ribosomal S6 kinase (p70S6K) and the eukaryotic initiation factor 4E (eIF4E)-binding protein 1 (4EBP1) with Raptor (188, 226). mTORC1 is active once PRAS40 is phosphorylated by Akt. This releases PRAS40 from Raptor to sequester PRAS40 in the cytoplasm of the cell with the docking protein 14-3-3 (227-231). mLST8, in contrast to Deptor and PRAS40, promotes mTOR kinase activity. This involves the binding of p70S6K and 4EBP1 to Raptor (232). In relation to cellular metabolism, mLST8 also controls insulin signaling through the mammalian forkhead transcription factor FoxO3 (67, 233). mLST8 is also necessary for Akt and protein kinase C-α (PKCα) phosphorylation and is required for Rictor to associate with mTOR (233).
mTORC2 is composed of Rictor, mLST8, Deptor, the mammalian stress-activated protein kinase interacting protein (mSIN1), and the protein observed with Rictor-1 (Protor-1) (188, 213). mTORC2 oversees cytoskeleton remodeling through PKCα and cell migration through the Rac guanine nucleotide exchange factors P-Rex1 and P-Rex2 and through Rho signaling (234). mTORC2 promotes activity of protein kinases that includes glucocorticoid induced protein kinase 1 (SGK1), a member of the protein kinase A/protein kinase G/protein kinase C (AGC) family of protein kinases. Protor-1, a Rictor-binding subunit of mTORC2, activates SGK1 (235, 236). The kinase domain of mTOR phosphorylates mSIN1 and blocks lysosomal degradation of this protein. Rictor and mSIN1 also phosphorylate Akt at serine473 and foster threonine308 phosphorylation by phosphoinositide-dependent kinase 1 (PDK1) to enhance cell survival.
Both mTORC1 and mTORC2 are tied to metabolic cellular regulation. These pathways are closely linked to the AMP activated protein kinase (AMPK) (16, 56, 237, 238) (Table 1). mTORC1 is associated with metabolic cellular pathways and can stimulate lipogenesis and fat storage (15, 239), may increase pancreatic ß-cell mass (240), and can improve glucose homeostasis (241). In the presence of other cellular pathways and systems such as with SIRT1, mTORC1 activity may require inhibition to preserve glucose homeostasis (242). mTORC2 also may be necessary to maintain glucose homeostasis (15), since loss of this pathway can promote severe hyperglycemia (243). Impairment in mTORC2 signaling can result in oxidative damage and insulin resistance (244). mTORC2 signaling also plays a significant role for the maintenance of pancreatic β-cell proliferation and mass (245).
In regards to AMPK, AMPK blocks mTORC1 activity through the hamartin (tuberous sclerosis 1)/tuberin (tuberous sclerosis 2) (TSC1/TSC2) complex that inhibits mTORC1 (246, 247) (Figure 3). Control of the TSC1/TSC2 complex also is controlled though phosphoinositide 3-kinase (PI 3-K), Akt, and its phosphorylation of TSC2. Extracellular signal-regulated kinases (ERKs), protein p90 ribosomal S6 kinase 1 (RSK1), and glycogen synthase kinase -3β (GSK-3β) can modulate the activity of the TSC1/TSC2 complex as well. TSC2 functions as a GTPase-activating protein (GAP) that converts G protein Rheb (Rheb-GTP) into the inactive GDP-bound form (Rheb-GDP). Once Rheb-GTP is active, Rheb-GTP associates with Raptor to control the binding of 4EBP1 to mTORC1 and increase mTORC1 activity (248). AMPK can phosphorylate TSC2 to increase GAP activity to change Rheb-GTP into the inactive Rheb-GDP and block mTORC1 activity (249).
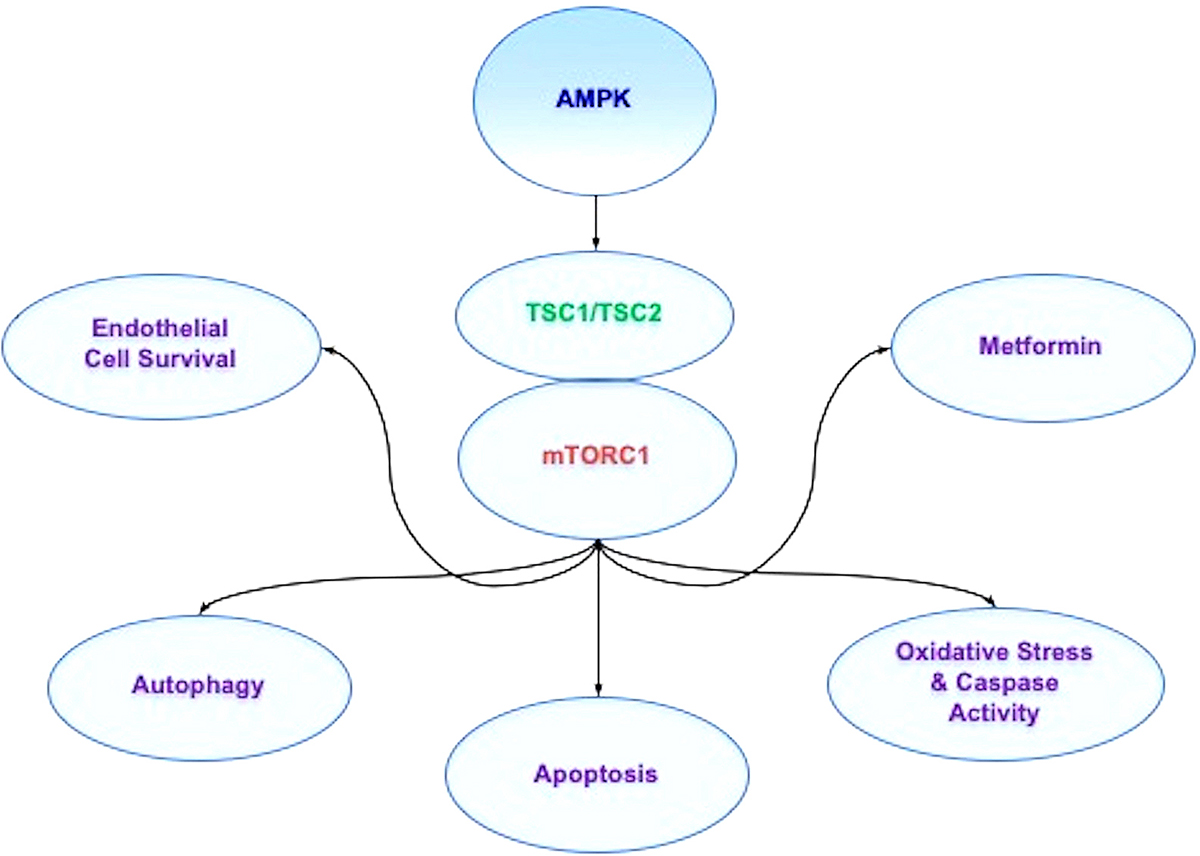 Figure 3
Figure 3AMPK affects multiple pathways through mTOR signaling. AMP activated protein kinase (AMPK) inhibits mTOR Complex 1 (mTORC1) activity through the hamartin (tuberous sclerosis 1)/tuberin (tuberous sclerosis 2) (TSC1/TSC2) complex. During periods of hyperglycemia, AMPK activity increases basal autophagy activity and maintains endothelial cell survival. In addition, AMPK can regulate apoptosis and autophagy during vascular disease and oxidative stress cell injury. Agents used to treat diabetes mellitus, such as biguanides and metformin, rely upon AMPK to restore cellular function. Through metformin, AMPK can be activated, leads to the induction of autophagy, and protects against cell apoptosis. Metformin also has been shown to limit lipid peroxidation in the brain and spinal cord and decrease caspase activity during toxic insults.
mTOR pathways are intimately tied to cellular metabolic function (24, 250). mTOR activation protects pancreatic β-cells against cholesterol-induced apoptosis (251), reduces glucolipotoxicity (252), and results in increased neuronal cell survival in cellular models of DM (253). mTOR activity controls insulin signaling in experimental models of AD and maintains astrocyte viability (254), promotes the differentiation of adipocytes (255), preserves endothelial cell function during hyperglycemia (54), and maintains glucose homeostasis (241). mTOR also may offer protection as a component of the Mediterranean diet as a focus on obesity in the population. The diet has been reported to reduce Aβ toxicity in astrocytes through enhanced Akt activity by consumption of polyphenol of olives and olive oil that ultimately could prevent the onset or progression of AD (254). In patients with metabolic syndrome, mTOR activation has been found to be diminished, suggesting that loss of mTOR may lead to insulin resistance and the increased risk of vascular thrombosis (256). In addition, growth factors that offer cellular protection against oxidative stress, such as insulin growth factor-1 (IGF-1) (257, 258) and erythropoietin (EPO) (259-270), may employ mTOR pathways as well (16, 227, 271-276). For example, EPO utilizes components of the mTOR pathway, such as PRAS40 and Akt, to enhance cell survival (227, 272, 273, 276) and limit toxic cellular environments (275, 277-279). EPO also plays a significant role as a potential cellular protectant during aging (280) and DM with the modulation of mTOR pathways, in part, to maintain cellular survival (24, 62, 73, 74, 175, 281-286).
Nicotinamide and mTOR downstream pathways also play a critical role during metabolic dysfunction (Table 1). For example, mTOR pathway activation with p70S6K and 4EBP1 can improve insulin secretion in pancreatic β-cells and increase resistance to β-cell streptozotocin toxicity and obesity in mice (240). Both p70S6K and 4EBP1 in the mTOR pathway are also utilized by nicotinamide to protect against radiation-induced apoptosis (179).
Interestingly, nicotinamide has a close relationship with poly (ADP-ribose) polymerase (PARP) (81, 87) that is also tied to mTOR and Akt (287, 288). PARP is a nuclear protein that binds to DNA strand breaks and cleaves NAD+ into nicotinamide and ADP-ribose. PARP catalyzes the synthesis of poly (ADP-ribose) from its substrate NAD+, which can foster the process of DNA repair (289). Nicotinamide can prevent PARP degradation and allow for DNA repair through the direct inhibition of caspase 3 (121, 126, 136). However, elevated concentrations of nicotinamide can lead to PARP degradation and apoptotic injury (290). Under some circumstances, a reduction in PARP activity may enhance neuronal cell survival, such as during cerebral ischemia (72). Prevention of NAD+ depletion during enhanced PARP activity also has been demonstrated to prevent cellular lysis during oxidative stress. In experimental studies of AD, PARP is present in the frontal and temporal cortex more frequently than in controls, suggesting that increased levels of functional PARP enzyme are present to potentially lead to the depletion of NAD+ stores (291). Reduction of PARP activity may be protective against oxidative stress and Aß (124). Furthermore, recent work illustrates that inhibition of the mTOR and PARP axis has protective effects on photoreceptors against visible-light-induced parthanatos (288), a form of programmed cell death that is distinct from other cell death processes such as necrosis and apoptosis and a PARP-1 dependent cell death.
Nicotinamide also can protect hypoxic cardiomyocytes and reduce intracellular mitochondrial stress through activation of the AMPK pathway (292). During periods of reduced dietary intake that may increase lifespan (116), AMPK activation can shift to beneficial oxidative metabolism (293) and has been shown to limit ischemic brain damage in diabetic animal models (294). AMPK activation also can strengthen memory retention in models of AD and DM (237), may assist with the elimination of ß-amyloid (Aß) in the brain (295), facilitate tau clearance (201), and limit chronic inflammation in the nervous system (12, 247, 296). AMPK can limit insulin resistance (297) and protect endothelial progenitor cells during periods of hyperglycemia (56) similar to the vascular protective properties of nicotinamide (104, 121, 298).
AMPK controls metabolic pathways through apoptosis and autophagy (Figure 3). During periods of hyperglycemia, AMPK activity can increase basal autophagy activity (149, 172) and maintain endothelial cell survival (54, 299). AMPK can regulate apoptosis and autophagy during coronary artery disease (300), endothelial dysfunction during hyperglycemia (54), and oxidative stress cell injury (301, 302). Anti-senescence activity also can be fostered through mTOR inhibition, AMPK activation, and the acceleration of autophagic flux (303). In addition to nicotinamide, other agents rely upon the ability of AMPK to modulate cellular metabolism. Agents used to treat DM, such as biguanides and metformin, use mTOR, AMPK, and autophagy to restore cellular function. Metformin inhibits mTOR activity, promotes autophagy, and may function at times in an AMPK-independent manner (304). Other reports note that through metformin, AMPK is activated, restores the induction of autophagy, and protects against diabetic cardiac cell apoptosis (305). Metformin also has been shown to limit lipid peroxidation in the brain and spinal cord and decrease caspase activity during toxic insults (306). Such results may be associated with the ability of autophagic pathways to limit oxidative stress under some circumstances (27, 307).
Interestingly, similar to nicotinamide having optimal dose concentrations for beneficial biological outcomes, autophagy also requires specific modulation of activity (Table 1). Under some circumstances, inhibition of autophagy may be required to achieve a benefit. In some experimental models of AD, autophagy activation can be one factor that leads to neuronal cell death (200). Increased activity of autophagy also can result in significant loss of cardiac and liver tissue in diabetic rats during attempts to achieve glycemic control through diet modification (77). Advanced glycation end products (AGEs), agents that can result in complications during DM, have also been shown to result in the activation of autophagy and vascular smooth muscle proliferation that can lead to atherosclerosis (308) and cardiomyopathy (309). Autophagy activation also can injure endothelial progenitor cells, lead to mitochondrial oxidative stress (310), and block angiogenesis (311) during periods of elevated glucose levels. Chronic inflammatory conditions, such as lichen planus, also have been linked to mTOR inhibition and autophagy activation (312). Interneuron progenitor growth in the brain relies upon mTOR activity with the inhibition of autophagy (313). During ischemic stroke in rodents, inhibition of autophagy may also limit infarct size and rescue cerebral neurons (314).
One mechanism for the modulation of autophagy activity may be through mTOR given that activation of autophagy occurs during the inhibition of mTOR (16, 315, 316). At times, inhibition of mTOR during DM can be beneficial and provide protection such as during cerebral ischemia (294). mTOR inhibition also may be necessary for maintaining a balance between pancreatic β-cell proliferation and cell size (245). Yet, other studies suggest that loss of mTOR activity can be detrimental. During mTOR inhibition with rapamycin, reduced β-cell function, insulin resistance, and decreased insulin secretion can promote the progression of DM (317). Decreased activity of mTOR also has been shown to increase mortality in a mouse model of DM (318). Translocation of glucose transporters to the plasma membrane in skeletal muscle can be blocked in the absence of mTOR activity (319). These studies may suggest that there may be a biological feedback for mTOR, such as through AMPK inhibition, to prevent excessive mTOR activity and allow for the activation of autophagy. For example, if mTOR activity is allowed to go unchecked with the down-regulation of AMPK activity during experimental studies, mTOR and p70S6K can lead to glucose intolerance by inhibiting the insulin receptor substrate 1 (IRS-1) (320).
Nicotinamide may require a close modulation of autophagy and mTOR to offer protection during disorders such as DM. Nicotinamide can offer cellular protection through autophagy (211) that requires inhibition of mTOR activity to allow for the activation of autophagy. Dysregulation of autophagy can potentially lead to the progression of cognitive loss with AD and the induction of DM (39). At least 33 autophagy-related genes (Atg) that have been identified in yeast with 40 autophagy-related genes involved in autophagosomes formation (185) that can affect multiple disorders including DM (15, 67, 321). Atg1, Atg13 (also known as Apg13), and Atg17 are associated with the PI 3-K, Akt, and TOR pathways (149). Autophagy haploinsufficiency with deletion of Atg7 gene in mouse models of obesity leads to increased insulin resistance with elevated lipids and inflammation (322). Loss of autophagic proteins Atg7, Atg5, and LC3 also can be responsible for diabetic nephropathy (323). Autophagy can be beneficial at the cellular level. Autophagy can remove misfolded proteins and eliminate non-functioning mitochondria to maintain β-cell function and prevent the onset of DM (324). Exercise in mice has been shown to promote the induction of autophagy and regulate glucose homeostasis (325). Autophagy also can improve insulin sensitivity during high fat diets in mice (297) and may be protective to microglia during acute glucose fluctuations (193).
It is important to recognize that in a number of circumstances, autophagy and apoptosis are closely linked to alter cellular survival. Inhibition of mTOR activity with activation of autophagy can reduce markers of senescence and aging in the skin of patients (326). Blockade of mTOR also can prevent aging and cell injury with extension of cell longevity through AMPK in endothelial cells (303). Additional work suggests that a balance is required during embryogenesis for autophagy and apoptosis that can affect body axis formation (327). Inhibition of mTOR pathways can trigger autophagy activation and affect tumor cell growth through the inhibition of apoptosis (328, 329). Yet, dependent upon the balance between autophagy and apoptotic pathways, other studies have shown that with nicotinamide one can block apoptosis in human colon cancer cells through the activation of autophagy (207). These studies support the premise that the balance between autophagy and apoptosis requires careful modulation to foster a desired biological outcome with nicotinamide.
Metabolic disorders, such as DM, pose a significant risk for the global population and contribute to the large percentage of deaths that are the result of NCDs (1, 2) (Table 1). Of even greater concern is the observation that of the over forty million people that die each year from NCDs, fifteen million individuals are between the ages of thirty and sixty-nine years old. In addition, NCDs affect a greater proportion of the population in low and middle-income countries. As the world’s population continues to age, life expectancy has been increasing that has occurred in parallel to the rise in NCDs (Figure 1).
As significant NCDs, metabolic disorders and DM are seen as critical targets to address and develop innovative strategies to limit death and disability for the global population afflicted by these disorders. Almost eighty percent of adults with DM reside in low- and middle-income countries and the care for patients with DM consumes more than seventeen percent of the Gross Domestic Product in the US (18). Furthermore, DM is believed to affect seven hundred million people by the year 2045 (17). DM affects all systems of the body that can involve the nervous system, cardiovascular system, and immune system. DM also alters new tissue generation and repair. Current conventional therapies for metabolic disorders and DM such as early recognition of disease onset, treatment with nutrition, and administration of pharmacological care can offer some degree of protection and may slow the progression of DM. However, treatments that lead to tight serum glucose control do not always resolve the eventual complications from DM and may pose potential risks that can decrease organ mass through autophagy. Given these concerns, innovative and novel strategies for metabolic disorders that involve DM are highly warranted for rapid development. Nicotinamide and the pathways associated with mTOR, mTORC1, mTORC2, AMPK, autophagy, and apoptosis fill this void to offer new avenues for the treatment of metabolic disorders.
Nicotinamide offers a partially unique perspective as a cellular protectant since it can foster cellular survival against oxidative stress not only during aging processes (72, 105, 114) and life span extension (29, 92, 116), but also during metabolic dysfunction (72, 88, 128, 131, 330). Nicotinamide can reverse a previously sustained insult by limiting the onset and progression of membrane PS exposure and apoptotic cell death. During DM, nicotinamide maintains cellular energy homeostasis and mitochondrial function, lowers insulin resistance and glucose release, protects against skeletal muscle atrophy, reduces inflammation of the brain, protects pancreatic β-cell function, improves glucose utilization, and prevents excessive lactate production. Nicotinamide employs mTOR pathways such that it can prevent myocardial cell death during hypoxia through the oversight of mTOR and autophagy. As a result, nicotinamide relies upon the inverse relationship between mTOR and autophagy pathways to control cellular survival. Similar to nicotinamide, mTOR activation can protect pancreatic β-cells and maintain glucose homeostasis. Through autophagy activation, the mTOR related pathway of AMPK can increase basal autophagy activity to prevent cellular apoptosis during DM. In addition, nicotinamide has been shown to protect hypoxic cardiomyocytes and reduce intracellular mitochondrial stress through activation of AMPK and autophagic pathways. Chronic administration of nicotinamide can activate skeletal muscle autophagy as a potential protective response to lipotoxicity.
However, there are a number of hurdles to consider for the targeting of nicotinamide, mTOR, and autophagy for the treatment of metabolic disorders. Nicotinamide appears to offer cellular protection during metabolic dysfunction in specific concentration ranges. Nicotinamide may protect against skeletal muscle atrophy during DM and can reduce inflammation of the brain during diabetes with the administration of niacin. Yet, the duration of nicotinamide administration may influence the efficacy of this agent since long-term administration has been reported to support glucose intolerance in some animal models and prolonged exposure of nicotinamide can result in impaired β-cell function and cell growth (142, 143). The acute and chronic levels of NAD+ that are generated may be vital to determine cellular survival. Similar to nicotinamide, autophagy may require critical modulation of activity. Nicotinamide can protect against palmitate-induced hepatotoxicity via SIRT1-dependnet induction of autophagy. In addition, autophagy can improve insulin sensitivity during high fat diets and may protect microglia during acute glucose fluctuations (193). Yet, autophagy activation also can injure endothelial progenitor cells, lead to mitochondrial oxidative stress, and block angiogenesis during periods of elevated glucose levels. mTOR, as a pathway that is inhibited during autophagy activation, may be a critical factor for nicotinamide to maintain cell viability during metabolic dysfunction. In some studies, loss of mTOR activity can be detrimental and result in reduced β-cell function, insulin resistance, and decreased insulin secretion that foster the progression of DM. In other scenarios, inhibition of mTOR during DM can be beneficial and provide protection such as during cerebral ischemia and may be necessary for maintaining a balance between pancreatic β-cell proliferation and cell size. Given these observations, it becomes clear that targeting nicotinamide as an effective agent to treat metabolic disorders may require careful scrutiny of the fine balance in activity required for mTOR and autophagic pathways. Additional investigations are warranted and would be highly advantageous to examine the intricate pathways of the vitamin nicotinamide and how they can be effectively utilized to provide innovative clinical strategies for metabolic dysfunction and DM.
This research was supported by the following grants to Kenneth Maiese: American Diabetes Association, American Heart Association, NIH NIEHS, NIH NIA, NIH NINDS, and NIH ARRA.
AD
Alzheimer's disease
AMP activated protein kinase
Autophagic related genes
DEP domain-containing mTOR interacting protein
Diabetes mellitus
Erythropoietin
Eukaryotic initiation factor 4E (eIF4E-binding protein 1
Extracellular signal-regulated kinases
Hamartin (tuberous sclerosis 1/tuberin (tuberous sclerosis 2
Insulin receptor substrate 1
Mammalian lethal with Sec13 protein 8
mLST8
Mechanistic target of rapamycin
mTOR Complex 1
mTOR Complex 2
Non-communicable diseases
p70 ribosomal S6 kinase
poly (ADP-ribose) polymerase (PARP)
Phosphatidylserine
Phosphoinositide-dependent kinase 1
Proline rich Akt substrate 40 kDa
Protein kinase B
Silent mating type information regulation 2 homolog 1 (Saccharomyces cerevisiae:
United States
United States dollars