
Frontiers in Bioscience-Landmark (FBL) is published by IMR Press from Volume 26 Issue 5 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.

1 Department of Biological Sciences, Middle East Technical University, Ankara, Turkey
2 Cancer System Biology Laboratory (CanSyL), Middle East Technical University, Ankara, Turkey
Abstract
17beta-estradiol (E2), the main circulating estrogen hormone, is involved in a wide variety of physiological functions ranging from the development to the maintenance of many tissues and organs. The effects of E2 on cells are primarily conveyed by the transcription factors, estrogen receptor (ER) alpha and beta. The regulation of responsive genes by the well-defined ER alpha in response to E2 relies on complex and highly organized processes that dynamically integrate functions of many transcription regulators to induce spatiotemporal alterations in chromatin state and structure. Changes in gene expressions result in cell-specific responses that include proliferation, differentiation and death. Deregulation of E2-ER alpha signaling contributes to the initiation and progression of target tissue malignancies. We aim here to provide a review of recent findings on dynamic transcriptional events mediated by E2-ER alpha with the anticipation that a better understanding of complex regulatory mechanisms underlying ER actions would be a critical basis for the development of effective prognostic tools for and therapeutic interventions against estrogen target tissue malignancies.
Keywords
- Estrogen
- Estrogen receptor
- Molecular mechanism
- Signaling
- Structure
- Review
Nuclear hormone receptors (NHRs) are members of a large nuclear receptor family that acts as transcription factors. NHRs include the androgen receptor (AR), estrogen receptor (ER) α and β, glucocorticoid receptor (GR), mineralocorticoid receptor (MR) and progesterone receptor (PR), all of which play diverse roles in cellular processes throughout the body (1, 2). The activity of NHRs is modulated by steroid hormones that diffuse across the plasma membrane enabling systemic extracellular signals to regulate tissue-specific intracellular events (1, 2).
Estrogens, including estrone (E1), estradiol (E2) and estriol (E3), as steroid hormones, are primarily synthesized and secreted by the gonads (3, 4). 17β-estradiol (E2), the most potent estrogen hormone in the circulation, is involved in a wide variety of vital physiological functions ranging from the development and maintenance of reproductive organs to the regulation of cardiovascular, musculoskeletal, immune, and central nervous system homeostasis (3, 4). E2 also contributes to the initiation and development of target tissue malignancies (3, 4). The effects of E2 in cells are primarily mediated by ERα (NR3A1) and ERβ (NR3A2). The dissection of ER-mediated E2 signaling in estrogen target tissues largely stems from knockout (KO) animal models (5-7). Although ERs display some species-specific differences in tissue distributions, ERα predominates, whereas ERβ plays a minor role, in the uterus, mammary glands, pituitary, skeletal muscle, adipose tissue and bone. ERβ, on the other hand, contributes to E2 signaling in the ovary, prostate, lung, cardiovascular system and central nervous system. Even within a single tissue, the expression pattern of each ER subtype is cell-type specific. In the ovary, for example, ERβ is expressed in the granulosa cells, but ERα is more abundantly expressed in the theca cells (5-7). Reflecting the different ER-subtype distribution patterns, ERα-KO and ERβ-KO mice show distinct phenotypes. ERα-KO female mice are, for example, infertile with a hypotrophic uterus as well as with anovulatory and hemorrhagic ovaries (5-7). In contrast, ERβ-KO female mice are subfertile and display reduced ovulation, likely as a result of a retardation in granulosa cell differentiation (5-7).
Although a significant progress has been made towards understanding the mechanism of ERβ signaling since its discovery in 1996 (8, 9), many aspects of ERβ actions and its role in physiology and pathophysiology of E2 signaling remain unclear (5, 10, 11). This is, due to, at least in part, the lack of established experimental cell models synthesizing ERβ endogenously and of receptor-specific antibodies (10, 12). Nevertheless, studies on ERα as a critical mediator in the initiation/development and as an important therapeutic target of the estrogen responsive tissue malignancies have broadened of our understanding how E2-ER signaling-mediated dynamic transcriptional events are manifested as cellular responses, for which this communication aims to give a brief account.
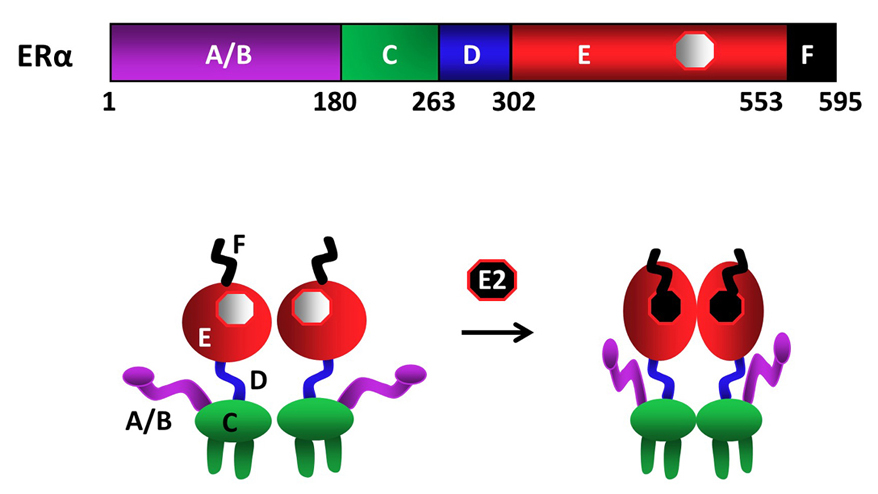
The human ERα gene (ESR1), spanning approximately 300 kb located at q24-q27 of chromosome 6, includes eight exons (13-15). Seven different promoters located upstream of the first coding exon are involved in the transcription of the ERα gene (13-15). The detection of distinct ERα transcripts in different tissues suggests the composition of regulatory promoter elements is critical for tissue-specific expression of the ERα gene. Although the different promoter usage gives rise to ERα transcript variants differing in their 5’-untranslated region, transcripts primarily encode a 595 amino-acid protein with an estimated molecular mass of 66 kDa (13-15).
As other members of the NHR family, ERα is a modular protein such that distinct structural regions of the receptor display unique functional features (fig1) (9-12). In the ERα gene, exon 1 encodes the A/B region. The C region, or the DNA Binding Domain (DBD), is encoded by the exons 2 and 3. Exon 4 encodes a part of the C region, all of the region D and part of the E region. The hormone (ligand)-binding domain (LBD or E/F domain) is encoded by exons 4 through 8 (13, 16).
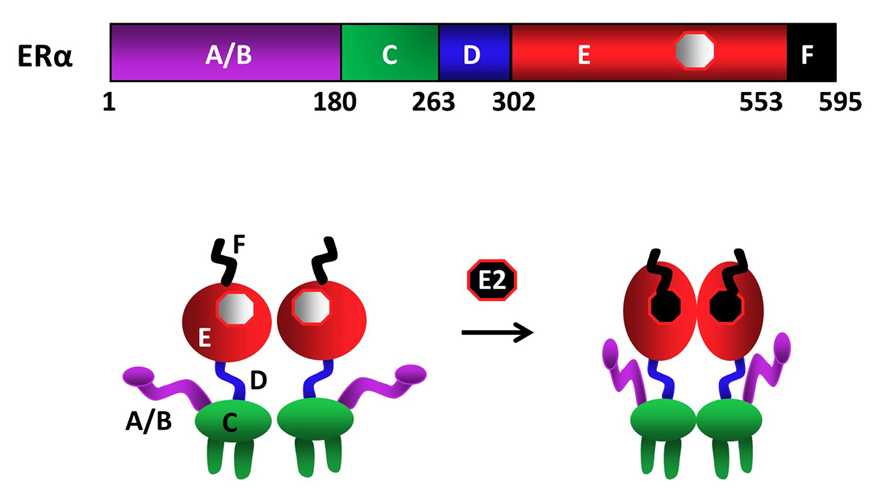 Figure 1
Figure 1Schematics of ERα structural regions. ERα is composed of 595 amino-acids. The amino terminal A/B domain, the central C region, or the DNA binding domain (DBD), the flexible hinge, or D, domain containing a nuclear localization signal, and the multi-functional carboxyl terminal (E/F) domain are indicated. ERα is a dimer with or without the endogenous ligand 17β-estradiol (E2), binding of which induces conformational changes in the receptor dimer. Figure is modified from (210).
Upon synthesis in cells of the estrogen target tissues, ERα dimerizes and is primarily translocated into the nucleus independent of E2. A fraction (about 5%) of the newly synthesized ERα also localizes to peri-membrane and mitochondria (17). The binding of E2 is the pivotal step in the cellular action of ERα. E2 binding induces a major structural re-organization in ERα. This structural change converts the inactive receptor to the functionally active form by generating functional surfaces that enhance the stability (18) and the interactions with co-regulatory proteins of the ERα dimer (19).
The LBDs of NHRs display a three-layered antiparallel α-helical fold (20, 21). This fold is universal within the receptor superfamily and is formed with 10-12 helices with the same numbering scheme used for all NHRs (20, 21). The ERα-LBD has 12 helices. The antiparallel α-helical fold comprising a central core layer of three helices (H5/6, H9 and H10) is packed between two additional layers of helices (H1-4 and H7, H8, H11) of the ERα-LBD (fig2c). This helical arrangement generates a scaffold that maintains a ligand-binding cavity. The remaining secondary structural elements, a small two-stranded antiparallel β-sheet and the dynamically mobile H12 (22, 23), flank the main three-layered motif (20, 21). The relatively unstructured carboxyl-terminus F domain contains an α-helical region and an extended β-strand separated by regions of random coil with a short extension near the carboxyl-terminus (24). The F domain appears to involve in the modulation gene transcription in a ligand-specific manner as the deletion of the entire F domain reduces E2-induced transcription (25, 26), likely through an altered structural state of the receptor to interact with co-regulatory proteins (26).
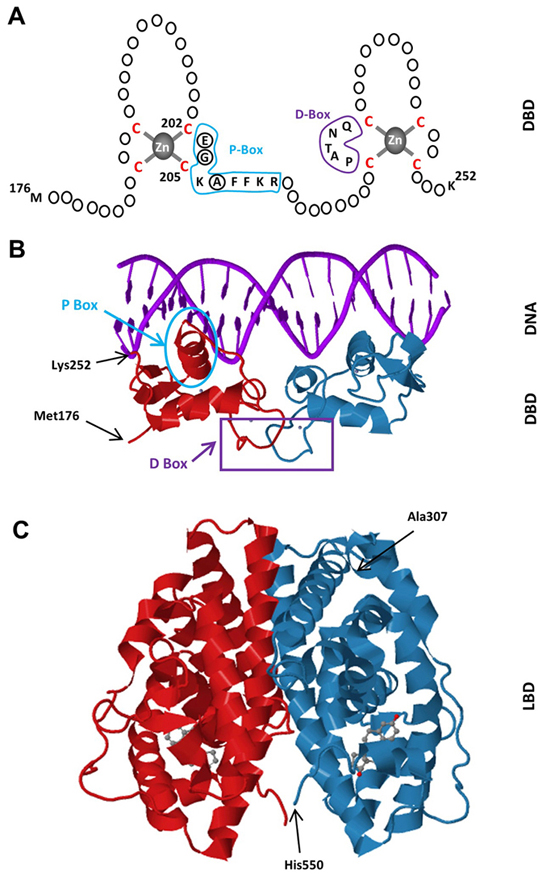 Figure 2
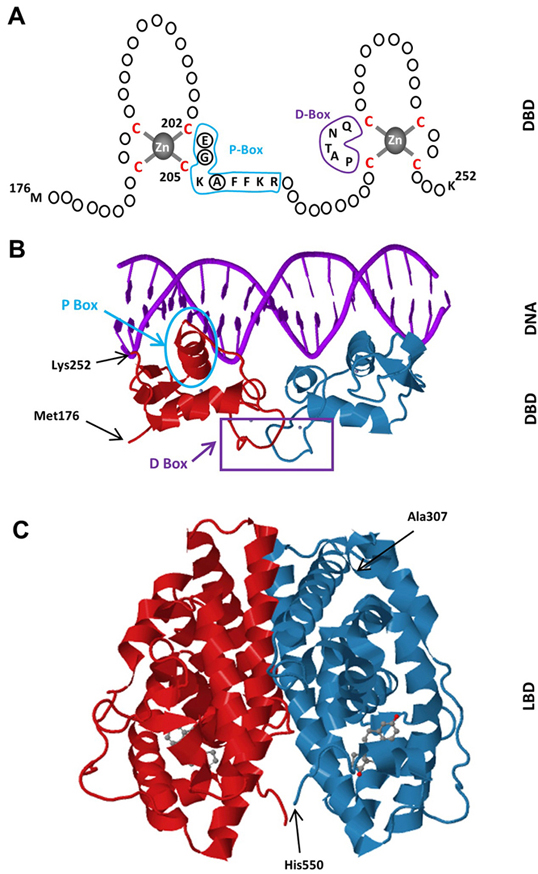
Figure 2Schematics of the DBD of ERα. The DBD of ERα monomer contains two zinc-binding motifs formed by a zinc ion (grey) coordinated by four cysteine residues (red). A region of the first zinc-finger module, the P-box (blue) which contains amino-acids particularly glutamic acid (E), glycine (G) and alanine (A) at positions 203, 204 and 207 respectively (circularized), determine the DNA binding specificity critical for sequence discrimination and binding to ERE. The residues (purple) in the second zinc finger module, the D-box, are involved in the discrimination of half-site spacing. B. Tertiary structure of the ERα-DBD (residues Met176-Lys252) as dimer bound to the consensus DNA sequence, GGTCAnnnTGACC, (Estrogen Responsive Element, ERE) (PBD ID: 1HCQ; (63)). The P (blue) and D (purple) boxes are indicated. C. The tertiary structure of the ERα-LBD (residues Ala307-His550) dimer bound to E2 (grey) (PDB ID: 1QKU; (255).
Dimer formation is essential for ERα function; as mutations that interfere with dimerization render the receptor transcriptionally inactive (20). Although the DBD of each monomer also contributes to dimerization of ERα, the predominant dimerization interface is formed with H11 of each ERα-LBD monomer (20, 21, 27). LBDs interact via a stretch of conserved hydrophobic residues at their amino-terminal ends with additional dimer interactions provided by the residues of H8 and the loop between H9 and H10 (20, 21). The E2 cavity buries the ligand in a highly hydrophobic environment composed of residues from helices 3, 6, 7, 8, 11, and 12. The ligand recognition is achieved through hydrogen bonds and the complementarity of the hydrophobic residues that line the cavity (20, 21, 28). It appears that E2 binding positions the dynamically mobile H12 over the ligand binding cavity forming a lid on the cavity by hydrophobic interactions as well as by hydrogen bonds (20-23). This positioning of H12 is a prerequisite for transcriptional activation, thereby a ligand-dependent activation function (AF2), as it generates a surface capable of interacting with co-activators (20, 21, 28). In this E2-mediated H12 agonist conformation, the E2 bound LBD can accept a short helical segment, the LXXLL motif (or nuclear receptor box, NRB, where L is leucine and X is any residue) from a variety of co-activator proteins, exemplified by the members of p160 Steroid Receptor Co-activator (SRC) family including SRC1-3 (19, 29, 30). Recent cryo-electron microscopy (cryo-EM) studies indicate that each of the ligand-bound ERα monomers in the dimer independently recruits one SRC-3 protein through LBD; the two SRC-3s in turn bind to different regions of one p300 protein through multiple contacts (31).
ERα also binds to various molecules with agonist, mixed agonist-antagonist or full antagonist properties (32, 33). Mixed agonist-antagonists, also called selective estrogen receptor modulators (SERMs), display distinct pharmacological effects depending on estrogen target tissues. Tamoxifen, for example, has been widely used for clinical treatment of breast cancers as an antagonist yet it acts as an agonist in most of estrogen target tissues (20, 29). Raloxifene, on the other hand, has protective effects on bone and displays anti-proliferative effects on breast cancer cells. Antagonists of ERα also known as the selective estrogen receptor down-regulator (SERDs), exemplified by fulvestrant, act as complete antagonists. While most of the key amino acids in the ligand binding cavity responsible for binding SERMs or SERDs are identical, structural differences among ligands, for example a large side chain emanating from the core of fulvestrant, prevents H12 of ERα from docking in agonist conformation (20, 29). This conformational shift in H12 prevents co-activator binding and transcription activation (20, 29). Independent of intracellular locations, AF2 of the ERα-LBD is indispensable in receptor actions. AF2 mutant knock-in mouse models bearing point mutations or deletions in the AF2 region to disrupt the AF2-mediated transactivation ability of ERα were shown to display female and male phenotypes indistinguishable from those of the ERα-KO mouse models (34, 35).
In most NHRs, antagonist binding locates H12 to a position outside the AF2 region leading to interactions with the corepressor-nuclear receptor (CoRNR) consensus motif (LXXXIXXXL; where L is leucine, I isoleucine and X is any residue) of corepressor proteins (36). However, the importance of NHR corepressors in ERα signaling remains unclear. Nevertheless, studies indicated that both agonist- and antagonist-bound ERα are able to recruit a variety of proteins that can repress receptor activity. It appears that a region in the carboxyl-terminus that encompasses AF2 of the unliganded (apo)-ERα interacts with corepressor proteins in vitro and in cellula (37-40). Importantly, the binding of E2 to ERα releases corepressors from the receptor (40). Studies indicate that retinoic acid and thyroid hormone receptors can act as ligand-independent repressors or ligand-dependent activators depending on the exchange of N-CoR (nuclear receptor corepressor), or its homolog SMRT (silencing mediator for retinoid and thyroid hormone receptors), containing corepressor complexes for coactivators in response to cognate ligands (41, 42). N-CoR/SMRT has been found as a component of holocorepressor complexes that also include histone and chromatin remodelers, TGFα-activated kinase 1 binding protein 2 (TAB2) as a sensory protein as well as transducin-β-like (TBL1) and TBL1-related protein (TBLR1), which act as adapter proteins for the recruitment of the ubiquitin/proteasome enzymes (43, 44). It appears that the molecular basis of N-CoR/SMRT recruitment is similar to that of coactivator recruitment. This involves cooperative binding of two helical interaction motifs within the N-CoR carboxyl terminus to nuclear receptors. The receptor interaction motifs exhibit a consensus sequence of LXXI/HIXXXI/L (L, I, H refer to leucine, isoleucine and histidine, respectively; while X denotes any amino-acid), or CoRNR box, representing an extended helix compared to the coactivator LXXLL motif (36, 45). This CoRNR box interacts with specific residues in the same receptor pocket required for coactivator binding. The use of a common binding pocket by many coactivators and corepressors indicates that corepressor/coactivator exchange mechanisms are critical for the responsive gene expression. A previously unrecognized internal CoRNR motif within H12 of ERα was discovered to be critical for the interaction with co-repressor proteins (46). This motif is able to compete with corepressors for binding to the AF2 surface, thereby reducing the ability of ERα to directly interact with corepressors (46). Although E2 binding to ERα appears to be sufficient to dissociate N-CoR from the receptor, in vitro and in cellula studies suggest that an active co-regulator exchange mechanism is involved. It was shown that apoERα bound to ERE-bearing promoters is associated with complexes containing N-CoR/SMRT (37). Moreover, an evolutionarily conserved amino-terminal L/HX7LL motif with an α-helical structure in the A domain of ERα appears to be required for the interaction of the receptor with TAB2 as a component of the N-CoR corepressor complex (43). This interaction could also be critical for basal transcriptional levels of responsive genes, as suggested by the observations that the removal of the amino-terminal A domain increases transcriptional responses to apoERα (47, 48). Furthermore, the binding of E2 augments the interaction of TAB2 with the amino-terminus of ERα (43). Although the mechanism is unclear, it could involve a trans-conformational change in the amino-terminus of the receptor induced by the E2 binding to LBD or an E2-induced functional integration of the carboxyl- and amino-termini. It appears that TAB2 allows TBL1 and TBLR1 to recruit the ubiquitin/proteasome enzymes to the holocorepressor for dismissal and subsequent degradation (43). This TBL1/TBLR1-mediated N-CoR removal is seemingly required for the subsequent productive transcriptional cycle of E2-ERα mediated by the recruitment of coactivator containing complexes (44).
In addition, dynamic modelling of tamoxifen occupied ERα suggests that in the presence of tamoxifen, the ERα-LBD assumes flexible conformations fluctuating between agonist and antagonist confirmation that could underlie mixed agonist-antagonist property of the compound (49). Along with blocking ER-cofactor interactions (50), fulvestrant as an effective SERD prevents the binding of ERα to DNA by altering the stability, turnover, and intra-nuclear location of the receptor (51-54).
Linking the LBD of ERα to the central C domain, the D domain provides flexibility between the amino- and carboxyl-terminus of ERα, thereby modulating the functional interactions between the termini of ERα (55). The D domain also contains patches of amino acids that act cooperatively for the nuclear localization of the receptor (56).
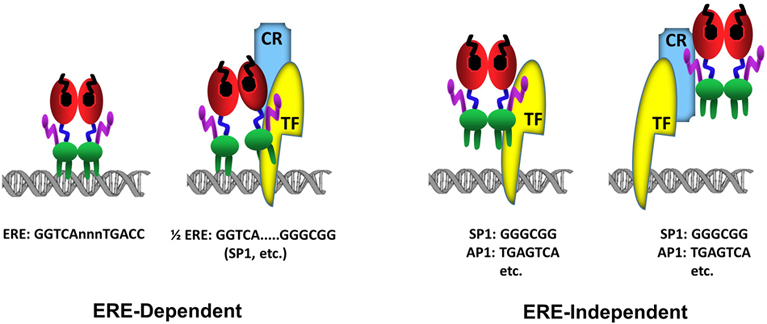
The C domain, or the DBD, mediates the DNA binding ability of ERα. Most high affinity binding sites for ERα (estrogen receptor binding sites, ERBSs) were initially characterized in the proximal regulatory sequences of target genes. However, chromatin/genome-wide ERBSs identified with chromatin-immunoprecipitation (ChIP) coupled with massively parallel sequencing (ChIP-Seq), pair end-tagging technology (ChIP-PET) and microarrays (ChIP-on-Chip) in cell models have indicated that genome contains remarkably large number of ERBSs found at large distances from transcriptional start sites (TSS) (57-60). The predominant motif enriched in these ERBSs is the estrogen response element (EREs) (fig3). EREs are permutations of the 5’-GGTCAnnnTGACC-3’ DNA palindrome wherein ‘n’ denotes a non-specific three nucleotide spacer (59, 61, 62). Being highly conserved among NHRs and shares the same three-dimensional structure (22), the ERα-DBD contains two zinc-binding motifs and each motif contains an α-helix nucleated at its amino-terminus through binding a zinc ion (63) (fig2a & b). Two helices are oriented perpendicularly to each other and cross at their mid-points (63). Each DBD of the ERα dimer makes analogous contacts with one of the inverted motifs, resulting in a rotationally symmetric structure (63). Two monomers of the DBD bind to adjacent major grooves from one side of the DNA double helix. Distinct residues in a region of the first zinc-finger module of DBD, the P-box, particularly Glu203, Gly204 and Ala207, determine the DNA binding specificity critical for sequence discrimination (13, 64, 65) and binding to ERE (66). The residues in the second zinc finger like module, the D-box, are involved in the discrimination of half-site spacing through a protein–protein interaction between two ER monomers (13, 64, 65). The regulation of gene expressions by binding of E2-ER to EREs is referred to as the ERE-dependent signaling pathway (67-72). ERBSs contain EREs that deviate from the consensus by one or more nucleotides (50, 61, 73). Although these EREs confer estrogen responsiveness mediated by ERα, they are less potent regulators of transcription than the consensus ERE (50,61,73-75). This appears to be due to the ERE-induced conformational change in the DBD of ERα: Single nucleotide changes in the consensus ERE, for example, require the formation of new interconnected hydrogen bonds between the response element and the DBD of ERα, thereby altering the conformation of the DBD (76).
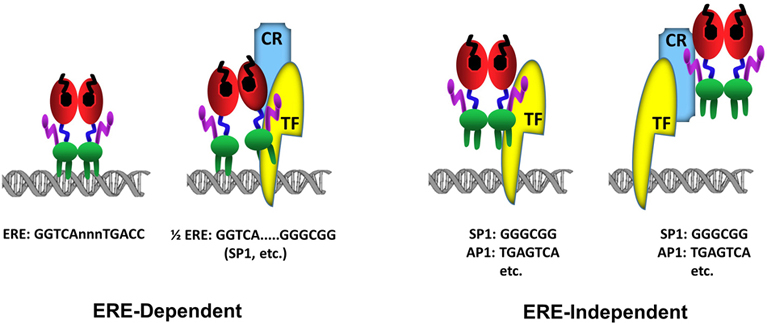 Figure 3
Figure 3Schematics of ERα-DNA interactions. In the ERE-dependent transcriptions, ERα directly interacts with an ERE sequence (GGTCAnnnTGACC). ERα can also interact with a composite element composed of a ½ERE sequence (GGTCA) and a response element for a transcription factor (TF). In this binding mode, the interaction of ERα with DNA is stabilized (anchored) by a direct, or indirect through coregulatory protein (CR), interaction with a Transcription Factor (TF) bound to its cognate response element, for example GC box (GGGCGG) for SP1 protein binding, juxtaposed to the ½ERE sequence. The ERE-independent binding refers to the indirect association of ERα with DNA through a direct, or through coregulatory protein (CR), interaction with a Transcription Factor (TF) bound to its cognate response element, for example GGGCGG motif for SP1 protein or TGAGTCA motif for AP1.
Genome-wide ERα binding studies also indicate that a large number of ERBSs contains half-site EREs or ½ERE: GGTCA (62, 77) (fig3). Although the mechanism by which ERα can bind to these ERBSs containing ERE half-sites in cellula remains unclear, some of ERBSs are enriched in transcription factor recognition motifs, for example Stimulatory protein 1 (SP1), that flank a ½ERE (59). While ERα does not bind to ½ERE in vitro, ERα can bind ½ERE when stabilized by direct or indirect protein interactions with transcription factors bound to their cognate response elements (61). ERα was, for example, shown to modulate the expression of progesterone receptor (78), transforming growth factor α (79), Vitellogenin A1 (80), heat shock protein 27 (81) as well as Cyclin G2 (82) genes through a composite binding motif that contains a ½ERE and a closely spaced Sp1 binding site. It is likely therefore that ERBSs enriched in ½EREs contain also binding motifs for, some of as yet undefined, TFs that anchor ERα to DNA.
E2-ERα has also the ability to regulate gene expression through direct or indirect, via co-regulatory proteins, interactions with transcription factors, exemplified with Stimulatory protein 1 (SP1), AP1, CCAAT/Enhancer Binding Protein Beta (CEBPB), Steroidogenic Factor 1 (SF1/ NR5A1), and Paired Like Homeodomain 1 (PITX1) bound to their cognate regulatory elements on DNA (68,69,71,81-87). In this route of transcription, which is referred to as the ERE-independent signaling (fig3), ERα establishes contact with TFs through regions that also encompass the ERα-DBD, while the integrated effects of the amino and carboxyl termini of the receptor are responsible for the modulation of transcription (67-72).
The highly divergent amino-terminal A/B domain of many members of the NHR family contains an activation function (AF1) (88). Observations that mutant ERα lacking the carboxyl-terminal AF2 domain does not alter transcription in many mammalian cells but activates transcription in a ligand and promoter-dependent manner in yeast and chicken cells (89-92) suggest that the function of the ERα amino-terminus is dependent upon the structural integrity of the LBD, nature of the ligand and the cellular-context. Structural studies further suggested that the A domain (the first 37 amino-acids of the amino-terminus) silences ligand-independent transactivation of ERα in cells that show AF2-independent AF1 activity. Moreover, the deletion of the A domain results in the manifestation of a repression activity of the receptor exhibited only in the presence of tamoxifen in mammalian cells (47). An α-helix in the A domain of the A/B region of ERα appears to be responsible for the silencing effect on ERα ligand-independent activities by interacting with the carboxyl-terminal E/F domain of the receptor in the absence of ligand (93).
Despite the important functions of the amino-terminus in ERα activity, biochemical and structural features for the underlying mechanism of AF1 action is incomplete. This is because the amino-termini of NHRs including ERα are intrinsically disordered (ID) (88,94-96). It has been proposed that ID leads to the formation of a large collection of rapidly inter-converting receptor conformations (88,94-96). ID structure allows the amino-terminus to rapidly and reversibly adopt various configurations. These conformational changes are controlled by allosteric cooperativity between different domains and interactions with proteins as well as posttranslational modifications, particularly phosphorylation (97). For example, the TATA box-binding protein (TBP) was shown to interact with and to induce a more ordered structure in the amino-terminus of ERα (96). The phosphorylation of serine 118 (S118) of the amino-terminus of ERα bound to E2 or SERM (tamoxifen) by growth factor signaling results in recruitment of the peptidyl prolyl cis/trans isomerase Pin1 that isomerizes the serine118-proline119 bond from a cis to trans isomer (97). This isomerization leads to a local conformational change that promotes ligand-independent as well as agonist- or SERM-inducible activity of ERα (97). These conformational changes seem to be critical for stable interactions with other coregulatory proteins to establish an effective transcription (88, 94, 95).
Previous studies also indicated that the B region of the amino-terminus contains two functional subdomains (referred to as Box-1 and Box-2, corresponding to aa 41-64 and 87-108, respectively). The Box-1 alone is responsible for the transcription activity of ERα (48, 98) as well as the partial agonistic activity of tamoxifen (98); while both the Box1 and Box-2 are required for the full transcription activity of the receptor in response to E2 (48, 98). It appears that the carboxyl-terminal region of the steroid receptor coactivator-2 (SRC-2) encompassing a glutamine-rich protein interaction domain, which is distinct from the LXXL containing NRB, interacts with the Box-1 region of ERα (48, 99, 100). Extending these observations, it was shown using cryo-EM that each of the A/B domain of the ERα monomers in the DNA-bound ERα dimer is located in proximity to the LBD and that the amino-terminus also participates in the SRC-3 recruitment together with LBD (31). This structural organization that brings together the amino- and the carboxyl-termini of ERα with co-regulatory proteins is the likely underlying mechanism for which the functional integration of both AF1 and AF2 is required for the full transcription activity of the receptor (34,35,89,101-103).
Plethora of factors including the amount and type of ER and/or complementary proteins required for receptor actions together with dynamic genome organization, which provides an architectural framework for the functional integration of enhancers and promoters, are ultimately responsible for the manifestation of E2-mediated cellular changes. However, the presence of ERα in various cellular locations also implies that the exertion of E2 effects on cellular phenotypes, which include cellular proliferation, differentiation, motility, and death dependent on estrogen target tissue, involves dynamically integrated and finely tuned ER-mediated signaling cascades. The extra-nuclear, referred to as the membrane-initiated E2 signaling, for example, mediates not only the second-to-minutes (or rapid) transcription-independent effects of ER but also post-translationally modulates the actions of nuclear ERs, transcription factors, coregulatory proteins and chromatin complexes contributing to protein-synthesis dependent signaling as well. It is therefore imperative that E2 signaling emanating from various intracellular locations, for which there are excellent reviews (10,32,104-112), is viewed as integrated rather than discrete, alternative events. Nevertheless, due to the critical importance of the nuclear ERα in the manifestation of cellular phenotype, we aim here to provide a brief view of dynamic transcriptional events mediated by E2 through the nuclear ERα.
The nucleus is enclosed by two layers of nuclear membranes that are separated by the perinuclear space (113, 114). The membranes are perforated with nuclear pores, which are multi-subunit nucleoporin complexes that mediate the selective transport of macromolecules. The inner nuclear membrane is lined with the nuclear lamina, which is a part of a scaffold involved in chromatin organization. Nuclear lamina is composed of lamins and lamin-associated proteins, including many integral proteins of the inner nuclear membrane, chromatin modifying proteins, transcriptional regulators and structural proteins (113, 114).
The dynamic structural organization of chromosomes within the nuclear space during the cell cycle provides regulatory separation for appropriate transcriptional control, facilitates ordered genome replication and contributes to genome integrity (115). Chromatin interaction mapping studies using various derivatives of chromosome conformation capture (3C) approaches (116) reveal that mammalian genomes are hierarchically organized into chromosome territories and are segmented/folded into functionally and epigenetically distinct Topologically Associating Domains (TADs) and subdomains. TADs show preferential contacts within and relative insulation from adjacent TADs and are generally conserved among cell types and across species (117-121). Within TADs, enhancer elements and active proximal promoters form chromatin loops, where looping is inferred from interaction frequencies between a point of interest and distal loci of the genome, that position enhancers and promoters in proximity (117-121).
Chromatin loop structures are mediated and/or stabilized by protein effectors, noncoding RNAs (ncRNAs), and histone post-translational modifications (PTMs) that are all important for defining cell specific physiological processes (122-124). Contacts between enhancer-promoter pairs within a TAD are sufficient for gene activation (125). Moreover, studies addressing long-range chromatin interactions associated with RNA polymerase II (Pol II) using genome-wide Chromatin Interaction Analysis with Paired-End-Tag sequencing (ChIA-PET) revealed a widespread promoter-centered intragenic, extragenic, and intergenic interactions (122). These interactions appear to further aggregate into higher-order clusters, wherein proximal and distal genes are engaged through promoter-promoter interactions leading to a cooperative and combinatorial control of transcription (122).
Genome wide ERα binding studies also indicate that the most of the distal ERBSs, which display an open chromatin architecture (DNase I hypersensitivity), correlate with gene expressions and with the presence of H3 lysine 4 monomethyl (H3K4me1), H3 lysine 4 dimethyl (H3K4me2), H3 lysine 27 acetyl (H3K27ac), histone variants (H2A.Z), coactivators (p300 and Mediator), chromatin loading of condensin I & II (126) and Pol II as well as the production of enhancer derived RNA (or enhancer RNA, eRNA), all of which marks the enhancers (127). This suggests that the most of the ERBSs act as enhancers; and that by bringing distant enhancers and proximal promoters, DNA looping is an essential aspect of ER-induced target gene expressions. Although the underlying mechanism(s) is yet unclear, an increased frequency of contacts between distal regions with proximal promoters upon binding of ERα to ERBSs suggests interactions of ERα with ERBSs facilitate chromatin looping. Indeed, a number of estrogen responsive genes including B-cell lymphoma 2, BCL2; (128), carbonic anhydrase XII, CA12; (129), Cathepsin D, CTSD; (130), Growth Regulation By Estrogen In Breast Cancer, GREB1 (131-133); Purinergic Receptor P2Y,P2RY2 (132); Progesterone Receptor, PGR (134); Erb-B2 Receptor Tyrosine Kinase 2, ERBB2 (135); Ret Proto-Oncogene, RET (136); Siah E3 Ubiquitin Protein Ligase 2, SIAH2 (137); and Trefoil Factor 1, TFF1 (138, 139) were shown to be regulated by distal ERBSs that communicate with the responsive promoters through chromatin looping.
Remarkably, several chromatin regions of estrogen-regulated genes contain multiple ERBSs, some of which with variant EREs (62), at varying distances from the TSS, exemplified by TFF1 (138, 140) and GREB1 (131). This implies cooperativity between widely separated enhancer units, or dynamic interactomes, for transcriptional regulation of target genes through the formation of multiple chromatin loops with the promoter and/or among enhancers (128, 135, 141). Indeed, a recent study using a CRISPR interference-based approach, referred to as the Enhancer-interference (Enhancer-i), to ablate the activity of ERBS alone and in combinations revealed that ERBSs are hierarchically organized such that one ERBS can act as the predominant site to coordinate the expression of TFF1 in cooperation with other ERBSs within the gene locus (142).
It appears that the cohesin complex is critical for the formation and/or maintenance of chromatin loops and TADs. TAD boundaries are established by the insulator protein CTCF, which is also critical for intra-chromosomal interactions within TADs, to regulate (activate/repress) gene expression in collaborations with various protein partners in a cell-type specific manner (143, 144). Accordingly, the deletion of a boundary between two TADs was shown to cause aberrant contacts among previously insulated loci (118) and can cause atypical gene expression through improper enhancer-promoter contacts (145, 146).
TADs associated with the nuclear lamina are referred to as lamina-associated domains, LADs. Primarily heterochromatin in nature, LADs are relatively gene-poor, show low levels of transcription and are depleted for active histone (117-121). Indeed, experimental relocation of specific chromosomes to the nuclear periphery through tethering to an engineered protein of the inner nuclear membrane results in the transcriptional repression of otherwise active genes (147, 148). Conversely, forced activation of a gene located inside a LAD can induce relocation of the gene towards the interior of the nucleus (149).
ERα binding events regulate gene expressions within mostly a spatially confined environment, which in many cases involves the folding of multiple regulatory regions into one transcriptional hub that does not require co-localization in the nucleus. Studies indicate that E2-ERα does not affect the relative nuclear position of the chromosome territories within which estrogen responsive genes, namely TFF1 and GRIP1, are located (chromosome 2 and 21 respectively) (150). There are also evidences that architectural proteins CTCF and cohesins are involved in the partitioning of the genome into ERα-regulatory blocks, which contain ERBSs and estrogen-regulated genes in a cell-specific manner. The binding of CTCF to enhancer regions was shown to result in the modulation of E2-induced gene transcription; as the depletion of CTCF affects transcriptions of estrogen target genes associated with cell division and increases the rate of proliferation of cell models derived from breast cancer (140, 151, 152). It appears that CTCF by providing boundaries for transcriptional blocks influences the binding of ERα to chromatin and also prevents additional enhancer-promoter ERα looping (140, 151, 152). Similarly, cohesins, which were also found at sites occupied by ERα in a CTCF-independent manner, appear to stabilize higher-order complexes including direct looping of distant regulatory regions (140, 153).
Pioneering factors are defined as a class of proteins capable of binding to their target sequences on nucleosomal DNA and initiating regulatory events that include pausing for induction, or repression, of transcription by creating accessible binding sites for transcription factors in a silent chromatin (154, 155). Genome-wide binding studies also indicated that ERBSs are enriched with DNA interaction sites for a number of pioneering factors which include Activator Protein 2γ, AP2γ (136); the Forkhead Box Protein 1, FOXA1 (60, 156, 157); GATA Binding Proteins, particularly GATA3 in mammary carcinomas (158) and GATA4 in osteoblasts (159); Pre-B-cell leukemia homeobox 1, PBX1 (160); Transducin-like Enhancer protein 1, TLE1 (161). Although redundancy, as well as collaborative effects, among these factors is yet unclear, the repression of synthesis by siRNA approaches of each pioneering factor has been shown to effectively impair the binding of ERα to DNA, long-range chromatin interactions and gene transcriptions (60,156-159,161). Moreover, it appears that the majority of ERBSs associated with long-range chromatin interactions co-localizes for the binding sites of these pioneering factors, at least for AP2γ and FOXA1 (136). It is therefore suggested that acting as primers and/or facilitators for transcription, pioneering factors interact with compact chromatin to modify chromatin structure as an early event and guides the recruitment of E2-ERα to, at least subset of, ERBSs (160).
It was also reported that the repression of ERα synthesis decreases FOXA1 or AP2γ binding in cell models of breast cancer (162). This together with the findings from single-molecule tracking approaches (157) suggest that reciprocal influences, or dynamic assisted loading, in the binding of ERα and pioneering factors to DNA are critical for defining the chromatin landscape of estrogen responsive cells.
An insightful study has further highlighted the importance of integrated actions of ERα, FOXA1, and GATA3 in the expression of responsive genes for the manifestation of hormone responsive cellular phenotype (57). While E2 is a proliferative agent in cell models derived from breast carcinoma expressing/synthesizing ERα (MCF7 and T47D cells), E2 effectively represses proliferation in ER-negative model cells that express the exogenously introduced ERα cDNA (MDA-MB-231 and BT-549 cells) (66,163-165). It was found that the co-expression of ERα, FOXA1 and GATA3 together, but not alone, are necessary to re-program ERα-negative MDA-MB-231 and BT-549 cells for the establishment of estrogen-responsive growth resembling to that of ERα-positive MCF7 cells (57).
The majority of newly synthesized and dimerized ERα is, as discussed, translocated to the nucleus through a multipartite NLS located in the D region. Although the mechanism by which ERα is translocated into the nucleus remains unclear, the import of nuclear hormone receptors to the nucleus is controlled by a multimeric chaperone machinery (166). The interaction of NLS with importins and microtubule-associated molecular motor proteins appears to mediate NHR transport to the nucleus (166). It was also reported that ERα contains a leucine-rich nuclear export sequence (NES) in the LBD (167, 168). The NES through binding to an exportin is suggested to modulate the nucleocytoplasmic shuttling of ERα (167, 168).
The nuclear ERα is a highly mobile protein dynamically partitioned between target sites on chromatin and nuclear matrix (52, 54). E2-mediated transcriptional events are rapid, extensive and transient (169). Detailed studies using Global Nuclear Run-On and Sequencing (GRO-seq) approach that maps the position, amount, and orientation of transcriptionally engaged all RNA polymerases genome-wide (170) to identify the immediate effects of estrogen signaling on the entire transcriptome in MCF7 cells suggest that early actions of E2 occurs at the level of transcriptions mediated by not only Pol II but also Pol I and Pol III with similar kinetics without affecting the stability or degradation rate of RNA (169). It appears that although long-range enhancer-promoter interactions play a pivotal role in actions of E2-ERα, immediate effects of E2 (within 10 minutes) is primarily observable at ERBSs that are located near TSS of responsive genes (169).
Although many features of active transcriptional enhancers have been characterized, the order of events leading to enhancer formation and activation as well as the dynamics of co-regulator interactions within the enhancer complex is yet unclear. Nevertheless, early kinetic studies using ChIP-PCR targeted at EREs in close proximity to the promoter of the estrogen responsive TFF1, cyclin D1 (CCND1), cathepsin D (CATD) and c-Myc (MYC) genes indicated that the association of ERα and Pol II with promoters shows a periodic engagement (171-174). Moreover, detailed studies utilizing the transcriptionally synchronized estrogen responsive TFF1 promoter as a model through the use of α-amanitin to reversibly inhibit Pol II activity indicate that the engagement of ERα with the proximal ERE of TFF1occurs cyclically with short periods (175). The upstream regulatory region of the TFF1 gene contains binding sites for different transcription factors that respond to growth factors and E2 (176, 177). The responsiveness of TFF1 to E2-ERα is mediated by a non-consensus ERE located at 405-393 bp upstream of the TSS, which is preceded by a TATA box element (176, 177). It appears that upon binding to this proximal ERE, E2-ERα initiates events that allow the relocation of nucleosomes associated with the TATA box to expose the previously occluded TATA box (175) through interactions with ATP-dependent chromatin re-modelling complex SWI/SNF together with some co-regulatory proteins with histone methyltransferases (HMT) and histone acteyltransferase (HAT) activities. Despite the recruitment by E2-ERα of the Mediator complex through the Med1 subunit which also interact with p300, a co-integrator with HAT activity, and Pol II (178), Pol II remains un(hypo)-phosphorylated. This consequently renders the initial cycle transcriptionally unproductive but generates a chromatin environment permissive for transcription (179). Further protein alterations, including ubiquitination, of ERα and associated co-regulators, disassemble the transcription complex (75, 76, 171, 180). This is followed by promoter remodeling through the association of chromatin modifiers with basal transcription factors. This oscillating promoter restructuring is suggested to provide a mechanism that enables a rapid adaptation of transcription to E2-ERα (75, 76, 171, 180). Kinetic studies further indicate that the interaction of E2-ERα with ERE initiates a series of interdependent events that result in an extended periodicity of cyclic engagement (75, 76, 171, 180). An interconnected ensemble of multi-subunit transcription factor complexes governs transcriptional activation. The binding of E2-ERα to ERE is immediately followed by the recruitment of the SWI-SNF chromatin re-modelling complex that locks the nucleosomes into a stable orientation. This is followed by the recruitment of HMTs and HATs to modify histones. E2-ERα recruits members of the p160 co-activator family that includes SRCs. The SRC family co-activators are also HATs that acetylate the chromosomal histone proteins. This results in destabilization of histone-DNA contacts and chromatin decompaction to allow the positional phasing of nucleosomes. These ERα-associated SRCs subsequently serve as a platform for the recruitment of p300, a sequential recruitment that coincides with an increased level of histone acetylation. A subsequent exchange of one molecule of SRCs, specifically SRC3, with coactivator-associated arginine methyltransferase 1 (CARM1) induces conformational changes in p300 to enhance p300-mediated histone acetylation (181). Increased histone acetylation further promotes CARM1-mediated histone methylation (181). These histone modifications subsequently lead to the recruitment of Pol II, which is then phosphorylated on serine 5 at the carboxyl-terminal domain (CTD) by the basal transcription complex, converting the polymerase to an elongation-competent form (75, 76, 171, 180). Following these events, the dissociation of p300 from and the subsequent binding of CREB-binding protein (CBP) and p300/CBP-associated factor (pCAF) to the complex take place. CBP alone or together with pCAF further modifies chromatin through histone acetylation and methylation. CBP also appears to be involved in the termination of transactivation by acetylating the acetyltransferases. The acetylation of the p160 proteins by CBP leads to the disruption of the p160 coregulator-receptor complex (75, 76, 171, 180). At the end of the transcription cycle, ATP-dependent remodelling complex, NuRD, repositions the nucleosome associated with the TATA box. This results in the occlusion of TATA box and the exclusion of factors such as TBP, from it, leading to the termination of transcription and re-modelling of chromatin (182). These extensive alterations in the chromatin architecture provide the necessary scaffold for ERα to enhance transcription (37, 175, 183, 184).
Studies also suggest that DNA methylation, particularly of CpG dinucleotides, occurs during the initial phase of every productive cycle and is associated with the recruitment of methyl CpG binding protein 2 (MeCP2) and DNA (Cytosine-5-)-Methyltransferase 1 (DNMT1) to the promoter which coincides with recruitment of the NuRD (185-187). Moreover, E2-ERα mediated restructuring and transcriptional competence of the responsive gene promoter appear to require the generation of a DNA double-stranded DNA break (detailed in section 5) promoted by topoisomerase II and/or APOBEC3B to aid in relieving torsional stress generated by transcription-induced DNA supercoiling (185).
Thus, ERα-mediated transcriptional events are tightly associated with structural changes in chromatin and DNA. These changes encompass the positional phasing and post-translational modification of nucleosomes, the methylation status of CpG dinucleotides and the formation of DNA breaks.
The ability of E2-ERα to mediate gene expression through functional interactions with , transcription factors, for example activator protein 1 (AP1) and Specificity Protein 1 (SP1), bound to their cognate element on DNA constitutes the ERE independent signaling (fig3) (67-72). This pathway is dependent upon receptor-subtype, the nature of ER-ligand and cell-context (67-72).
The AP1 transcription factor consists of members of the Jun, Fos, ATF (activating transcription factor) and MAF (musculoaponeurotic fibrosarcoma) basic region leucine zipper (bZIP) motif protein families. The leucine zipper domain allows the dimerization of Jun-Jun and Jun-Fos to regulate gene expression. Once dimerized, basic regions of the dimer interact with the consensus TGAGTCA sequence, known as 12-O-Tetradecanoylphorbol-13-acetate, TPA, -response elements, TREs (188). SP1, on the other hand, belongs to the Sp/KLF zinc finger transcription factor family that binds to the consensus GGGCGG sequence, referred to as the GC box element (188, 189). Both AP1 and SP1 proteins play critical roles in cellular proliferation, differentiation and death (188, 189).
Studies indicated that AP1 activity can be induced by E2 treatment and reduced by antiestrogens without increasing in c-Fos and c-Jun expressions (190). Subsequent studies further showed that ERα (71, 191) does not bind directly to TREs, but the receptor is recruited by protein-protein interactions to c-Jun through regions encompassing also ERα-DBD. It appears that ERα-mediated transcription from a TRE site is dependent upon AF1 and AF2 functions as the receptor lacking AF1 or AF2 fails to modulate the transcription (71, 192, 193). Although ERα and AP1 use similar co-regulators, as exemplified with p160 proteins and CBP (71, 192, 193), in transcription at ERE and TRE sites, the different combinatorial assembly of co-regulatory proteins seems to be critical for ERα-mediated signaling events through TRE-dependent pathway. Indeed, the observations that SERMs and SERDs can activate rather than repress transcriptional responses mediated by ERα at a TRE (71, 192, 193) suggest that the altered pharmacology of ER ligands could be explained by differences in the amount and/or type of the co-regulatory proteins, which show variations in cells from different tissue of origin (194).
Moreover, ERα in response to E2 cross-talks with the SP1 transcription factor to modulate the transcription of a variety of estrogen responsive genes (69, 84, 195). This interaction is mediated by tethering of ERα to GC-box response element bound SP1 protein (69, 84, 195). Moreover, it appears that the amino-terminal of region ERα is critical for responses from GC box element bearing promoters (69, 84, 195).
Various in cellula and in vivo studies have addressed the importance of the ERE-independent pathway in E2-ER signaling by dissecting nuclear ER signaling pathways. Findings established that Glu203, Gly204 and Ala207 residues of the P-box in the DNA-binding helix of human ERα (fig2A) determine the DNA binding specificity critical for sequence discrimination (64, 65, 196) and binding to ERE (66). Changing Glu203 and Gly204 residues to Ala in the DNA-binding helix of the human (197), and the corresponding residues of the mouse (191), ERα generates a mutant receptor capable of mediating E2 signaling only at the ERE-independent pathway. Analogous mutations in the DBD of the human ERβ (70, 198) also render the receptor functional only in the ERE-independent signaling pathway. Studies with a knock-in mouse model of a mutant P-box in the DNA-binding helix of mouse ERα (ERαAA) provide compelling support for the importance of the ERE-independent pathway in the regulation of various tissue functions, albeit in a tissue-specific manner (199-201). On the other hand, in an attempt to correlate genomic responses from the ERE-independent signaling pathway to alterations in cellular phenotypes, we found that changing Glu203Ala and Gly204Ala human ERα reduces but does not prevent functional features of ERα in the ERE-dependent signaling pathway (66). Moreover, Glu203Ala and Gly204Ala mutations could alter the response element specificity of ERα, as indicated by studies using ERαAA mouse uteri, which showed that the ERαAA mutant binds to hormone responsive motifs normally occupied by progesterone receptor, leading to E2 regulation of uterine transcripts that are normally progesterone responsive (202).
Previous studies indicated that a network of protein-DNA hydrogen bonds confers the binding specificity and stability of the human ERα to DNA (63, 203). For the consensus ERE, the network involves residues Glu203, Lys207, Lys210, and Arg211 of the DBD (203). Although the recognition of a non-consensus ERE is achieved by a rearrangement of side chains of various residues of the ERα-DBD, particularly Lys207 and Lys210, the interactions of Glu203 and Arg211 with DNA remain unaltered (203). Based on these observations, we replaced positively charged Arg211, which is a conserved residue among NHRs, with the negatively charged Glu residue in the ERα203/204 mutant. This generated an ERE binding defective ERα mutant (or ERαEBD) that abolished in vitro and in cellula ability of the receptor to interact with and to modulate transcription from an ERE while retaining the functionality at simulated ERE-independent signaling pathways in various cell lines (66). Furthermore, ERαEBD in response to E2 mediated a subset of estrogen responsive genes in a manner similar to E2-ERα, but it was insufficient to alter phenotypic features of cell models in contrast to E2-ERα (66). Identical results were also observed with an ERE binding mutant of ERβ (198). These findings suggest that genomic responses from the ERE-independent signaling pathway can be dissociated from the induction of phenotypic alterations. These findings also imply that the ERE-dependent pathway is the required signaling route for E2-ERs to induce cellular responses. This conclusion is supported by the observations derived from a knock-in mutant mouse (ERαEAAE) model bearing mutations at the DBD that synthesize an ERE-binding defective ERα mutant incapable of modulating transcription from the ERE-dependent signaling pathway but effectively regulating gene expressions at the ERE-independent signaling route (204). Displaying hypoplastic uteri, hemorrhagic ovaries, impaired mammary gland development and liver functions, the phenotypic features of the ERαEAAE mouse (204) resemble the general loss-of function phenotype of ERα-KO mouse models.
The critical importance of the ERE-dependent signaling pathway in inducing cellular alterations is also supported by experimental studies utilizing oligonucleotide decoys, ER-specific electrophilic agents or designer transcription factors. Short sequences of DNA containing a response element for a transcription factor have been used as “decoys” to bind the cognate transcription factor in cellula or in vivo. Binding of transcription factor to decoy DNA sequesters the transcription factor away from endogenous binding sites. This renders the transcription factor ineffective to regulate target gene expressions in a variety of systems. The use of a synthetic consensus ERE as the decoy in transfected ER-positive breast cancer cell models was shown to prevent the growth of cells in response to E2 (205). Similarly, the prevention of ER-ERE interactions by ER-specific electrophilic agents that preferentially disrupt zinc fingers of ERα effectively suppresses E2-mediated growth of ERα-positive breast cancer cell models in cellula and in vivo (206, 207). Moreover, we previously showed that the intrinsic specificity of the DNA binding domain of ERα to interact with ERE sequences can be exploited to engineer a monomeric ERE-binding module by co-joining two DNA binding domains with the hinge domain (208). Integration of strong transcription activation domains from other transcription factors into the ERE binding module generated monomeric transcription factors, or monotransregulators, with constitutive activity at ERE-driven gene promoters (163, 209, 210). These monotransregulators, but not the ERE-binding defective counterparts, altered cellular phenotypes by mimicking the effects of E2-ERα on gene transcriptions that require ERE interactions.
Studies showed that, in addition to Pol II mediated gene expressions, E2-ERα signaling also induces direct, rapid and transient transcriptions mediated by Pol I and Pol III with kinetics similar to those observed with Pol II (169). These findings indicate that E2-ERα signaling-induced cellular responses require coordinated transcriptional events mediated by all three polymerases (169). While Pol I produces ribosomal RNAs (rRNAs); Pol III generates structured RNAs, most prominently tRNAs and 5S rRNA (211). On the other hand, Pol II transcribes all protein-coding genes and some noncoding RNA (ncRNA) genes including long ncRNAs (lncRNAs), many small nuclear RNAs (snRNAs) and miRNAs (211). Enhancer RNAs (eRNAs) are considered to be lncRNAs (212). Transcribed from the DNA sequence of enhancer regions, eRNAs are relatively short non-coding RNA molecules of 50-2000 nucleotide long. eRNAs appear to play critical roles in the post-transcriptional regulation of mRNA stability and epigenetic control of chromatin activity (212). Due to the correlation between target gene expression and eRNA transcriptions, enhancers are also suggested to be transcription units (213) and that eRNA transcription marks active ERBSs (127).
Although the sequence and kinetics of events of eRNA transcription at enhancer is yet emerging, based on a large number of studies on signal-induced enhancer formation, recent models suggest that collaborative binding of pioneer proteins, or lineage-determining transcription factors, to enhancer regions leads to nucleosome remodeling, increased chromatin accessibility, histone modifications and assembly of basal transcription machinery (214-217). In response to a stimulation, signal-dependent transcription factors bind to response elements at enhancers and recruit coactivator complexes. This leads to further epigenetic modifications including histone acetylation and the recruitment of the transcriptional initiation complex including phosphorylated Pol II at serine 5 together with hypo-phosphorylated serine 2, a characteristic of the enhancer region for eRNA transcription (214-217). It appears that the most active enhancers of ERα-mediated transcriptions contain a large (2 MDa) complex, MegaTrans complex, of diverse DNA-binding transcription factors that serves as a mark that distinguishes the most active enhancers of the estrogen-regulated gene expressions (218).
Exhibiting shorter half-lives compared to mRNAs and lncRNAs, transcribed eRNAs are 5’-capped but are generally not spliced or polyadenylated. Polyadenylated eRNAs are usually unidirectionally transcribed from enhancers; while non-polyadenylated transcripts are commonly produced from bidirectional enhancers (214-217). The roles and functional importance of eRNAs are yet unclear and may be dependent on enhancer in a cell-type; eRNAs, nevertheless, appear to play significant regulatory roles in various aspects of the transcription process. eRNAs transcribed from neuronal enhancers were shown to facilitate the release of paused RNA polymerase II by directly interacting with the Negative Elongation Factor Complex-E subunit (NELF-E) of the NELF complex, which causes transcriptional pausing (219). Knockdown of eRNAs impairs the release of NELF from target promoters during transcriptional activation, coinciding with a decrease in target mRNA induction (219). eRNAs produced from enhancers also appear to be involved in the mediation of chromatin looping between enhancers and promoters of target genes by facilitating the recruitment of specific factors known to stabilize long-range chromatin interactions, including cohesin and the mediator complex (214-217). Reduction of eRNAs produced from the estrogen responsive TFF1, FOXC1, CA12, GREB1 and P2RY2 loci by siRNA approaches in the presence of E2 diminishes corresponding mRNA productions through the loss of the chromatin loops between the enhancers and the promoters of target genes (220). This appears to result from the interruption of eRNA interactions with cohesin subunits, as the depletion of RAD21, a component of the cohesin complex, also causes loss of enhancer–promoter interactions (220). Similarly, it was reported that eRNA association with the mediator complex is involved in long-range transcriptional activation, as the reduction of mediator subunits or eRNA decreases chromatin looping between the promoter and enhancer (213). A recent study further suggests that eRNAs directly interact with a highly basic region within the core HAT domain of CBP (221). This leads to enhanced histone acetylations, notably H3K27ac, at active enhancers, which, in turn, stimulate target gene expressions (221). eRNAs appear also to facilitate the loading of Pol II and the transcription initiation complex to promoters/enhancers (214-217).
It is therefore likely that ERα-mediated production of eRNA transcripts contributes to the stabilization of enhancer-promoter loops as well as to the generation of active histone marks to allow optimal gene expression.
Single (nick) or double stranded DNA breaks activate DNA-damage response (DDR) that leads to the recruitment of repair proteins to sites of DNA damage until repair is carried out (222-224). The earliest events that trigger the DDR cascade are the recognition of the lesion by the MRE11/NBS1/RAD50 complex (MRN), double-stranded DNA end sensing by KU, exposed single-stranded DNA recognition by Replication Protein A (RPA) and Poly (ADP-ribose) Polymerase 1 (PARP1), the latter which also senses DSBs (222). MRE11–RAD50–NBS1 (MRN) complex recognizes DSBs and recruits the ataxia telangiectasia mutated (ATM) protein kinase which is responsible for the phosphorylation of the histone variant H2AX (γH2AX). The recruitment of additional DDR components together with spreading of γH2AX at DNA lesion sites generates γH2AX foci. DSBs are repaired by primarily homologous recombination (HR) and non-homologous end joining (NHEJ) pathways. Caused by oxidative stress, including the formation of reactive oxygen species (ROS) and ionizing radiation, SSBs trigger base excision repair (BER), nucleotide excision repair (NER) or mismatch repair (MER) pathways (222-224).
Accumulating findings clearly indicate that DDR is central to the promotion of gene expression by ERα. E2 through ERα induces DSBs, as determined by the formation of γH2AX foci (225-227). These DSBs appear to involve a number of interconnected networks of proteins with a variety of enzymatic and catalytic activities that are critical in DNA damage recognition and repair, including components of BER and NHEJ pathways (228). This E2-ERα interacting network of proteins includes T:G mismatch-specific thymine DNA glycosylase (TGD) (229); Flap endonuclease 1 (FEN-1); 3-Methyladenine DNA glycosylase (MPG); Apurinic endonuclease 1 (APE1) (229-233). Similarly, ERα interacts with Breast cancer type 1 susceptibility protein (BRCA1) (234) as well as the co-factor of BRCA1 (COBRA1), which is an integral subunit of the human negative elongation factor (NELF) (235), of the HR and NHEJ pathways. ERα was also shown to interact with PARP1 (236).
The movement and rotation of along DNA template during transcription can generate negative supercoils behind and positive supercoils in front of the advancing polymerase (237, 238). Positive supercoils can impede transcription elongation; whereas negative supercoils support DNA melting and favor initiation (237, 238). The accumulation of torsional stresses is released by a family of enzymes, DNA topoisomerases (TOPOs), which regulate DNA topology. DNA Topoisomerase I (TOPOI) relaxes supercoiled template by nicking a single strand of duplex DNA and allowing one end to rotate with respect to the other around the intact strand (TOPOIB) or by passing one strand through the break (TOPOIA). DNA Topoisomerase II (TOPOII), on the other hand, cleaves both strands of a DNA duplex with two base (TOPOIIA) or four-base pair (TOPOIIB) overhangs and passes a second intact duplex through the transient break. It appears that TOPOII relaxes positive torsional stress more quickly than negative torsional stresses compared to TOPOI, which slowly relaxes both torsional stresses (239). TOPOs rejoin cleaved DNA ends through intrinsic intramolecular ligation activity following resolution of super-helical strain (240).
DNA breaks induced by E2-ERα appear to be important in relieving torsional stresses that impede polymerase progression and also in chromatin modelling during transcriptional processes. The pioneering work on topoisomerase-dependent transcriptions came from the observations that ERα-mediated transcriptions in response to E2 are coupled the mutual recruitment of TOPOIIB and PARP1, as well as KU and DNA-PK components of the NHEJ pathway, at the promoter of TFF1 (241). TOPOIIB then generates transient and site-specific breaks at the promoter that facilitate dynamic changes in local chromatin to initiate transcription; as the inhibition of TOPOIIB enzymatic activity with a specific small molecule inhibitor or the reduction of TOPOIIB levels by siRNA results in the disappearance of the breaks and target gene transcription (241). These findings suggest that topoisomerase-mediated DNA breaks are critical to relieve torsional stresses as well as to induce strand break responses and subsequent repairs for transcriptions.
The binding of ERα to an ERE at both the promoter and enhancer of the estrogen responsive B-Cell CLL/Lymphoma 2 (BCL2) gene, was shown to activate Lys-specific demethylase 1 (LSD1/KDM1A) to promote local histone H3-Lys9 demethylation through an oxidative process that releases reactive oxygen species, which in turn modifies the surrounding DNA (241). This results in the recruitment of N-glycosylase/DNA lyase (OGG1) for the removal of the damaged DNA bases by the components of the BER pathway (241). Removal of the oxidized bases generates transient nicks. These nicks are suggested to function as entry points for TOPOIIB, which relaxes the DNA strands to initiate local chromatin re-modelling and the subsequent formation of chromatin looping between the promoter and enhancer, the recruitment of components of the NHEJ pathway and of the transcription initiation complex for transcription (241). Interestingly, it is also observed that ERα-induced DSBs mediated by TOPOIIB at the promoter of TFF1 can also persist longer times (6-24h) after exposure to E2 and the repair involves protein components (specifically Rad51) of the HR pathway (225). These findings also imply that although E2-ERα mediated transcriptions are transiently couple to DNA breaks, transcription coupled DNA breaks could also result in the formation of stable TOPOIIB–DNA cleavage complexes that may contribute to genomic instability mediated by aberrant estrogen signaling (225).
The role of TOPOI in ERα-mediated transcriptional regulation remains unexplored. Nevertheless, a recent elegant and detailed study using ligand-induced binding of androgen receptor (AR) to response elements in prostate cancer cell enhancers as models indicates that TOPOI is an important component of enhancer mediated eRNA transcriptions (242). It was shown that AR binding to DNA induces a rapid recruitment of TOPOI followed by additional components of DNA damage repair machinery to the AR-regulated enhancers (242). Furthermore, the DNA nicking activity of TOPOI appears to be a prerequisite for robust eRNA synthesis and enhancer activation, as the knockdown of TOPOI by siRNA prevents AR-regulated eRNA as well as coding target gene transcriptions (242). The effect of TOPOI on enhancer-mediated eRNA transcriptions may not be specific for AR but for all ligand-induced transcription factor including ERα.
Collectively, these findings indicate that TOPOII, possibly TOPOI, is a critical component of ERα-mediated transcriptions at both the promoters and enhancers. Moreover, an alternative repair pathway involving the cytosine deaminase Apolipoprotein B mRNA Editing Enzyme Catalytic Subunit 3B (APOBEC3B; A3B) was recently shown to assist the expression of ERα target genes (226). It appears that the recruitment of A3B to ERBSs at the regulatory regions of estrogen responsive genes, exemplified with TFF1, GERB and PDZK1, in an E2-ERα dependent manner in cell models derived from breast adenocarcinoma causes local and transient C-to-U transitions. Generation of U:G mismatches induces DNA nicks through the actions of UNG and AP endonuclease that result in DNA breaks and subsequent repair through NHEJ; as the knockdown of A3B or of UNG effectively blocks DNA breaks. This in turn promote chromatin modifications and remodeling, reflected in activating histone modifications and the recruitment of SWI/SNF, to drive expression of ERα target genes (226).
Thus, although DDRs are central to the promotion of gene expression by ERα, various mechanistic features of a specific repair pathway could be dictated by the nature of DNA lesion or break in a gene- and cell-specific manner to ensure the maintenance of genome integrity.
During transcription, the pairing of the nascent RNA with the template DNA generates a DNA:RNA hybrid which leaves the non-template DNA in a single-stranded conformation to form a three-stranded nucleic acid structure referred to as the R loop (237, 243). Strand asymmetry in the distribution of guanines and cytosines, or GC skew, appear to predispose DNA sequences toward R-loop formation upon transcription (237, 243). R-loops are associated with DNase I hyper-accessibility reflected as chromatin decondensation and lower nucleosome occupancy as well as with an increase in RNAP density suggesting that R-loop formation causes transient RNAP stalling (237, 243). It is suggested that R loops can cause genomic instability by generating ssDNA as a substrate for DNA lesions and nicks as well as by causing replication–transcription collisions, leading to DNA recombination and DSBs (237).
A recently developed bisulfite-DNA-RNA immunoprecipitation sequencing approach, bis-DRIP-seq, was used to genome-wide mapping of R-loops in MCF7 cells (244). Bis-DRIP-seq selectively converts cytosine into uracil residue, by the use of bisulfite, within genomic DNA regions that contain single-stranded DNA. This is followed by immunoprecipitation with an antibody specific to DNA:RNA hybrids and subsequent sequencing. It was found that most of R-loops are transcription dependent and are enriched at active promoter regions and TSSs (244). Extending these observations, it was recently shown that E2-ERα induces a rapid formation of R-loops at E2-responsive gene promoters in MCF7 cells and that R-loop formations correlates with gene transcription (245). Importantly, most of the DSBs induced by E2-ERα during transcription appear to be R-loop dependent (245). This suggests that co-transcriptionally formed R-loops are a significant source of E2-induced DNA damage that could contribute to genetic instability in estrogen target tissue malignancies in general and breast cancer in particular.
Cis and/or trans ERBSs, acting as nucleation sites for ERα to initiate/enhance transcription by recruiting many interdependent coregulatory proteins that cause spatiotemporal alterations in chromatin state and structure for transcriptions in a cell-specific manner (fig4), remains the central for E2 signaling. Nevertheless, the recently generated wealth of information has provided a better understanding of complex regulatory mechanisms underlying ER actions at ERBSs. This, in turn, holds considerable promise for the development of novel biomarkers and predictors as well as therapeutic approaches. However, E2 target cell functions are carried out within the context of remarkably diverse signal transduction pathways that crosstalk with E2-ER signaling. Although beyond the scope of this communication and many excellent reviews could be referred (246-252), one important cross-talk is of E2-ERα with growth factor signaling pathways (GFSPs). GFSPs modify and are modified by E2-ERα signaling (253, 254). Adding further complexity to E2-ERα signaling, these modifications include phosphorylation, glycosylation, ubiquitination and acetylation events that not only modulate un-liganded or liganded ERα functions at every level but also alter the ligand pharmacology (246-252). Deregulation of growth factor signaling appears to play a vital role in ERα-driven neoplastic processes and also in the development of endocrine resistance during the treatment of estrogen target tissue malignancies, exemplified by breast cancers (246-252). Nonetheless, recent novel discoveries with the use of many integrated, multidisciplinary approaches and applications have provided unprecedented opportunities to study cross-layered regulatory interplays and will certainly provide the critical insights into cellular events that could have a substantial impact on systemic management of estrogen target tissue malignancies.
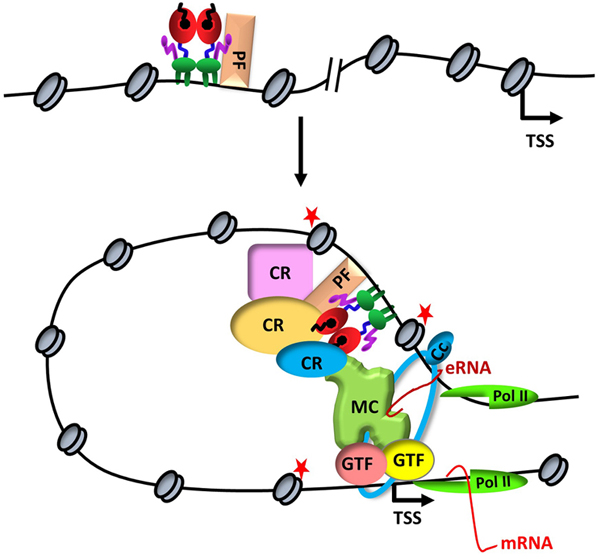 Figure 1
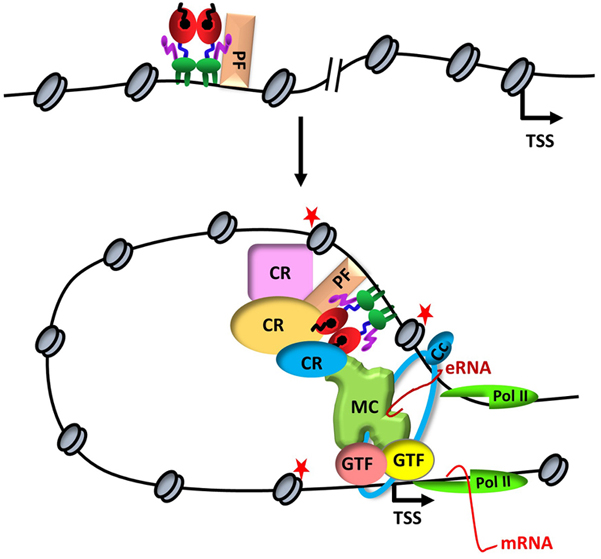
Figure 1ERα-mediated transcription. Upon binding to a distal estrogen receptor binding site (ERBS) in collaboration with a pioneer factors (PF), ERα leads to an interdependent sequence of events that involves the recruitment of coregulatory proteins (CR), nucleosome remodeling, increased chromatin accessibility, histone modifications (red stars), assembly of basal transcription machinery that includes mediator complex (MR) and Pol II, leading to enhancer RNA (eRNA) production and chromatin looping. eRNA then interacts with looping factors (cohesin complex, Cc) to facilitate/stabilize chromosomal looping between the enhancer and promoter and participates in the recruitment of Pol II to the promoter for transcription initiation of target gene (mRNA).
Gamze Ayaz and Pelin Yasar, contributed equally and should be considered as the first author. We thank Dr. A. Elif Erson-Bensan for critical reading of the manuscript. The work in the laboratory is supported by TUBITAK 114Z243, 315S045, 117Z213 and METU-BAP 2726 grants.