









Frontiers in Bioscience-Landmark (FBL) is published by IMR Press from Volume 26 Issue 5 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.

1 National Technical University of Athens, School of Electrical and Computer Engineering, Biomedical Engineering Laboratory, NTUA Campus, Heroon Polytechniou 9, 15780 Goudi, Athens, Greece
2 National and Kapodistrian University of Athens, First Department of Pediatrics, Choremeio Research Laboratory, Thivon and Levadias 8, 11527 Goudi, Athens, Greece
Abstract
First steps in brain research progress were made during the early 19th century, whose swift progress was accompanied by the discovery of monoamines and their localization in the brain. Since the discovery of polarography in 1924, several variations of electrochemical techniques for in vitro and in vivo determination of monoamines have been developed, with the most prevalent being microdialysis and voltammetry. Voltammetry takes advantage of the chemical property of certain species to oxidize, videlicet to produce a current that can be measured and subsequently interpreted to concentration gradient. Voltammetric techniques require a three-electrode system and operate under the application of a potential at the working electrode, responsible to evoke the oxidation processes. Methodological variations include, among others, amperometry, cyclic voltammetry, differential pulse voltammetry, etc. In the present work we attempted to review the available knowledge on voltammetry, its uses and future endeavors since voltammetry is a promising method towards the investigation of brain and central nervous system physiology and pathophysiology.
Keywords
- Voltammetry
- Monoamines
- Microdialysis
- Electrochemistry
- Review
Research on the Central Nervous System (CNS) found its origins in the early 19th century. The first endeavors concerned the discoveries on the morphological and anatomical characteristics of the CNS, yet discoveries on the electrical nature of the nervous system preceded by a century due to the work of Luigi Galvani. Further on, during the mid-20th century (1924), the discovery of polarography, by Jaroslav Heyrovský, opened a new window to the investigation of CNS physiology. In particular, Heyrovský’s discovery concerned the property of certain electrodes to oxidize molecules that have a redox potential, rendering possible to determine the presence of certain molecules or elements in a solution and in some cases their relative quantity. In 1924, Heyrovský discovered the precursor of the voltammetric method using a mercury electrode and deducing that the current measured by this electrode when applying a voltage, grants information about the concentration of the oxidizable species in the solution (1). Later on, the same conception applied in carbon-paste or other types of electrodes constituted voltammetry (2). These discoveries created new possibilities for the experimentation potential such as the in vivo and in vitro electrochemical techniques that would allow measuring biological substances or molecules in intact living brain regions of animals (3).
Further on, monoamines were unknown until the late 19th century as they were first discovered during the early 20th century. Yet, their presence in the brain was not discovered until the mid-20th century by the work of Vogt et al. (1954) (4, 5). Monoamine neurotransmitters are small molecules functioning as neuromodulators that contain one amino group connected to an aromatic ring by a two-carbon chain (-CH2-CH2-). All monoamines are derived from aromatic amino acids like phenylalanine, tyrosine, tryptophan, and the thyroid hormones by the action of aromatic amino acid decarboxylase enzymes. Examples of monoamines discovered in the mid-20th century were dopamine, isolated in 1957 and serotonin isolated in 1948. Both of these monoamines were later on detected in the brain and thus this signaled the study of their role in brain physiology and homeostasis. In continuation to the monoamine discovery, monoaminergic systems were further explored and in particular, the neuronal networks that utilize monoamine neurotransmitters, participated in the regulation of cognitive processes such as emotion, arousal, and certain types of memory. Neurotransmitters are found to have an important role in the secretion and production of neurotrophin-3 by astrocytes, a chemical which maintains neuron integrity and provides neurons with trophic support (6). Drugs used to increase (or reduce) the effect of monoamine are sometimes used to treat patients with psychiatric disorders, including depression, anxiety, and schizophrenia (7). Yet, the study and determination of monoamines proved to be a difficult task since the most useful and meaningful way of monoamine investigation would be the in vivo experimentation. Other methodologies developed for the determination of monoamines, lacked the advantage of real-time investigation, which was necessary for the investigation of the role of monoamines in CNS physiology.
During the early 70’s, an electrochemical technique, which would appear to surpass the boundaries of in vivo, real-time monoamine detection emerged. The introduced method was termed voltammetry (3, 8). Voltammetry was based on the electrochemical property of biological fluids of certain molecules such as neurotransmitters and their metabolites (phenols, thiols, etc.) to oxidize when they come infinitesimally close to a metal or semi-conducting surface1 (3, 9, 10). The voltammetric method appeared to have many advantages some of which include the sub-millisecond temporal resolution, adequate spatial resolution, chemical identification of the species of interest, low perturbation on the brain regions, and matching between the chemical results and the behavioral parameters etc. (10-12). Voltammetry has evolved and several variations have been developed such as cyclic voltammetry, differential pulse voltammetry (DPV), hydrodynamic voltammetry, stripping voltammetry, amperometry and others. As aforementioned, the main advantage of voltammetry was the factual suitability for in vivo measurements, yet the main disadvantage of voltammetry is the actual lack of sensitivity and specificity. Further on, a variety of other techniques were developed for the study of CNS physiology. Several of these techniques had the advantage of being able to measure monoamine levels in in vitro systems, as well as in vivo systems such as cortical cup and push-pull cannula micro-dialysis. These methods had the disadvantage of producing excess tissue damage and could perform only indirect measurements of the species of interest (2, 3, 13). Yet, they had a high specificity and sensitivity potential.
All the aforementioned methodologies found applications in a great variety of scientific investigations and endeavors such as the study of neurotransmitter physiology, which is crucial due to their relation with brain disorders (depression, anxiety, obesity, etc.) (14-17).
In the mammalian Central Nervous System the excitation transmission is most frequently performed through chemical synapses. Other signal transduction mechanisms (e.g. electrical synapses) have a subordinate role. A neurotransmitter synthesized in the pre-synaptic neuron is released from the axonal endings of the neuron through the synaptic vesicles. This reacts, further on, with the postsynaptic receptors of the recipient cell and triggers specific reactions, such as a conductivity change, for certain ions. Synapses can be divided into two groups. Neuronal systems can be divided based on the neurotransmitter they produce. These include the acetylcholinergic (ACh) system and the monoaminergic system, which features synapses releasing monoamines or biogenic amines. These are small molecules containing an amino group (-NH3) and demonstrating the ability to be further divided into smaller groups. The first group concerns of the catecholamines, which include dopamine (DA), adrenaline (A), also known as epinephrine (E), and noradrenaline (NA), also known as norepinephrine (NE). The second group consists of serotonin, also known as 5-hydroxytryptamine (5-HT) and histamine.
Based on the aforementioned neurotransmitters, neuronal cells include the adrenergic synapses, in which norepinephrine or adrenaline is released. The most commonly occurring neurotransmitter in the nervous system is norepinephrine, utilized in the ganglion cells and in the sympathetic nervous system. Biosynthesis of the catecholamines begins with the amino acid tyrosine (Tyr) catalyzed by the enzyme Tyrosine Hydroxylase (TH). Tyrosine is absorbed by nerve cells via the main blood stream, meaning that tyrosine can cross the so-called Blood Brain Barrier (BBB). Tyrosine is packaged in the synaptic vesicles and catalyzed to 5-dihydroxyphenylalanine (DOPA). DOPA is further on catalyzed by the enzyme DOPA decarboxylase to form dopamine (DA). DA is catalyzed to NA by the enzyme DOPA-b-decarboxylase. DA is degraded by the enzyme monoamine oxidase (MAO) to provide the starting material for the initiation of the metabolic path from the beginning. One of the significant steps in this metabolic pathway is the catalysis of DA to dihydroxy-phenyl-acidic-acid (DOPAC) through MAO. This catalytic step is important because DOPAC is an oxidizable molecule, which can be estimated voltammetrically and thus indirectly determines the levels of DA. In addition, another important molecule and neurotransmitter is serotonin. Serotonin is released in the serotonergic neurons and consists of one of the most important signal transduction pathways. Serotonin differs from the catecholamines because it possesses an alkyl-containing indole ring along with a catechol ring. The alkyl group contains an amino group, which makes serotonin monoaminergic. The serotonin monoaminergic neurons are located mainly in the brain stem and the cerebellum. Further on, dopamine-releasing neurons are mainly localized in the brain stem and the diencephalon (18). Highest concentrations of dopamine have been detected in the anatomical structures of Tubus Olfactorius, Nucleus Accumbens and the Striatum. A smaller group of dopaminergic neurons is found in the hypothalamus and belongs to the Tuberoinfundibular-Hypophyseal system.
The dopaminergic neurons are further located in the Ventral Mesencephalon and are divided into a group located in the Ventral Tegmentum and cells located in the Substantia Nigra Pars Compacta (SNC). The neurons of the Ventral Tegmentum innervate the Frontal Cortex through the mesolimbic system, especially the Nucleus Accumbens. The neurons of the Substantia Nigra Pars Compacta (SNC) innervate the Striatum, as well as the Tectum and the Thalamus, where more than 95% of the neurons belong to the dopaminergic pathway. In Figure 1 the neuronal function of the basal ganglia is presented diagrammatically.
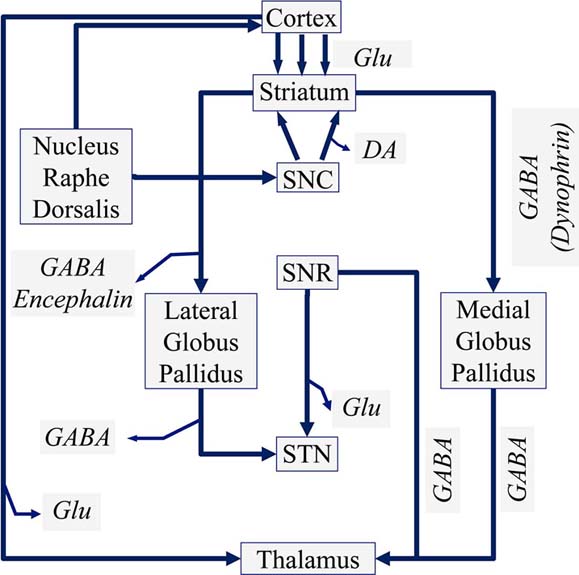 Figure 1
Figure 1Schematic diagram of a manual voltammeter (Adopted and modified from https://chem.libretexts.org/Textbook_Maps/Analytical_Chemistry_Textbook_Maps/Map%3A_Analytical_Chemistry_2.0._(Harvey)/11_Electrochemical_Methods/11.4.%3A_Voltammetric_Methods).
In voltammetry a time-dependent potential is applied to an electrochemical cell, measuring the resulting current as a function of that potential. The resulting plot of the current versus the applied potential is called a voltammogram, and it is the electrochemical equivalent of a spectrum in spectroscopy, providing quantitative and qualitative information about the species involved in the oxidation or reduction reaction.
Early voltammetric methods utilized two electrodes, while further voltammeters made use of a three-electrode potentiostat (Figure 2). In voltammetry a time-dependent potential excitation signal to the working electrode is applied, which changes the potential relative to the fixed potential of the reference electrode. This subsequently changes the current that flows between the working and auxiliary electrodes. The auxiliary electrode is usually a platinum wire while the reference electrode is usually a Sat’d Calomel (SCE) or an Ag/AgCl electrode.
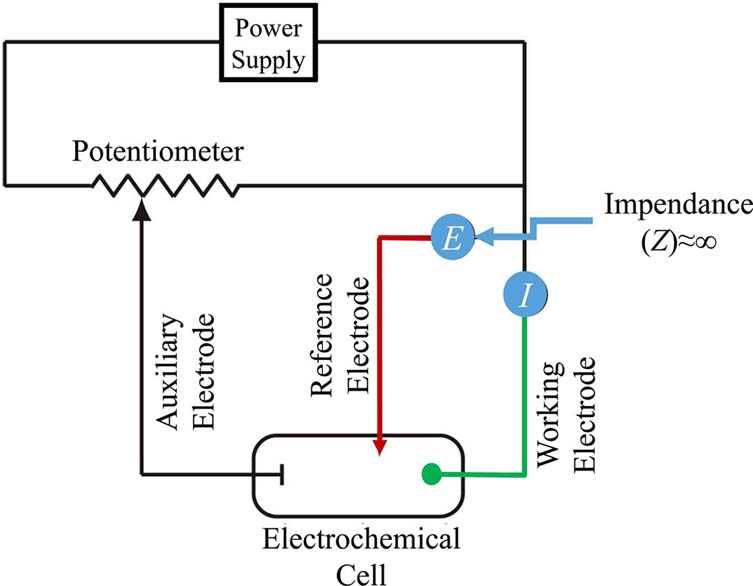 Figure 2
Figure 2Schematic diagram of a manual voltammeter (Adopted from Grote C. (1994) (18)) (Legend: SNC: Substantia Nigra Pars Compacta, SNR: Substantia Nigra Pars Reticularis, GABA: gamma aminobutyric acid, Glu: Glutamate).
The case of the working electrode is more complicated since it can be constructed by several different materials. These include mercury, platinum, gold, silver, and carbon. The earliest voltammetric techniques, including polarography, used a mercury working electrode. Due to the liquid nature of mercury, the working electrode is often a drop suspended from the end of a capillary tube. This is the case of the Hanging Mercury Drop Electrode (HMDE), where a drop of Hg is extruded by rotating a micrometer screw that pushes the mercury from a reservoir through a narrow capillary tube. Further on, in Dropping Mercury Electrode (DME) mercury drops form at the end of the capillary tube as a result of gravity. Unlike the HMDE, the mercury drop of a DME grows continuously-as mercury flows from the reservoir under the influence of gravity-and has a finite lifetime of several seconds. At the end of its lifetime the mercury drop is dislodged, either manually or on its own, and replaced by a new drop. Finally, the Static Mercury Drop Electrode (SMDE) uses a solenoid driven plunger to control the flow of mercury. Activation of the solenoid momentarily lifts the plunger, allowing mercury to flow through the capillary and forming a single, hanging Hg drop. Repeatedly activating the solenoid produces a series of Hg drops. In this way the SMDE may be used as either a HMDE or a DME.
Further on, voltammetry utilized working electrodes from carbon fibers, both single-thread as well as multi-thread (Figure 3).
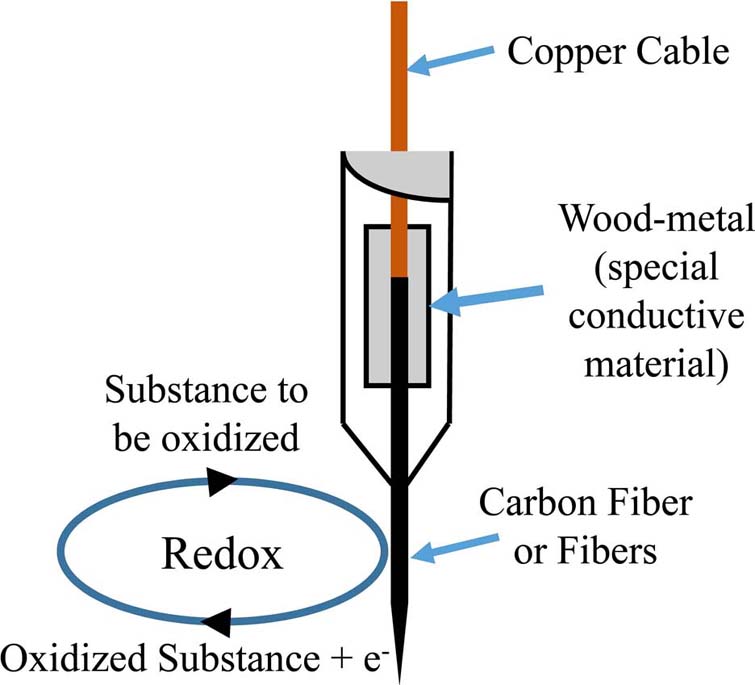 Figure 3
Figure 3Model of a voltammetry electrode and the respective Redox reaction on the surface of a carbon-fiber electrode.
Variations of the voltammetric methodology are based on the various alterations of the voltage applied on the working electrode (3). Each of these different voltammetric variations differ in their temporal resolution, sensitivity, and capability of chemical identification of the detected compounds (3). Several of these voltammetric methodological variations are presented below.
In amperometry, the potential applied to the working electrode is kept constant, sufficiently positive to oxidize the species of interest (2, 10, 13). The flowing current is measured with respect to time (8, 10). The applied, constant, potential imposes reduced capacitance at the electrode-solution surface and thus, lower noise (10), as well as sufficient faradaic/charging rate (2, 8). Amperometry also exhibits adequate temporal resolution (on the sub millisecond time scale (10, 13, 19)) for rapid measurements (changes in neurotransmitter concentrations, release, etc. (8, 13)). Adams et al. (2008) claim that, when amperometry is applied on a neurotransmitter release event, the shape of the graph provides several information about the circumstances of the event (20), yet it has been also reported that amperometry cannot provide specific chemical identification of the compound being detected (2, 3, 8, 10, 13, 19).
In cyclic voltammetry, an augmenting voltage ramp is applied (2), and the oxidation current occurs by the time the electrode potential matches with the sufficient oxidation potential of the species (2). Before this momentum, only the charging current is measured. Subsequently, applying a negative-going scan, reduction of the previously oxidized species takes place (8, 10). Kawagoe et al. (1993) reported that the oxidized product may undergo diffusion, so the reduced species during the cathodic scan could be underestimated (8). Critical capability of the cyclic voltammetry with respect to specificity and sensitivity, as well as the rest of the techniques described below (FSCV, DPV, etc.) is that it provides chemical identification of the oxidizable species of interest (2, 3, 10).
The applied potential is in a triangular waveform (10). FSCV is mainly used, in the study of histamine (15), serotonin (13, 16, 20, 21) and dopamine (13, 19, 22, 23), as well as it is utilized for the detection of the release and uptake of catecholamines in the chromaffin cells (24). FSCV displays the capacity of detecting monoamine concentrations on the nmol-umol range (13, 23).
DPV consists of a combination of amperometry and cyclic voltammetry (2, 3, 8, 12). Thus, DPV displays amperometry’s advantage of reduced charging current, along with higher selectivity and sensitivity as well as chemical identification (2, 8, 14, 25), although, it lacks temporal resolution (8) (slow scan rates that suppress charging current). The applied voltage consists of a series of a time-dependent gradually increasing voltage (Figure 4). Based on the applied voltage a significant characteristic is derived, which is that the current measured in DPV is the subtraction between the currents at the beginning and the end of each pulse (8, 12). Crespi F. (2011) applied DPV in the amygdala of rats in order to study activities of the neuropeptide Y (NPY) and its receptors (14). Further on, Clement et al. (1993) measured 5-HT and 5-HIAA in the nucleus raphe dorsalis of the rat using DPV (26, 27), and in addition Ozel et al. (2011) measured serotonin in vivo with DPV in zebrafish embryos (11). In addition, there is a variety of other similar techniques, such as Differential Normal Pulse Voltammetry (DNPV) (2, 13, 25), Differential Double Pulse Voltammetry (DDPV), Steady State Voltammetry (SSV) (2), Fast Differential Ramp Voltammetry (FDRV) (28), Differential Pulse Amperometry (DPA) (29, 30) and Square Wave Voltammetry (SWV) (31).
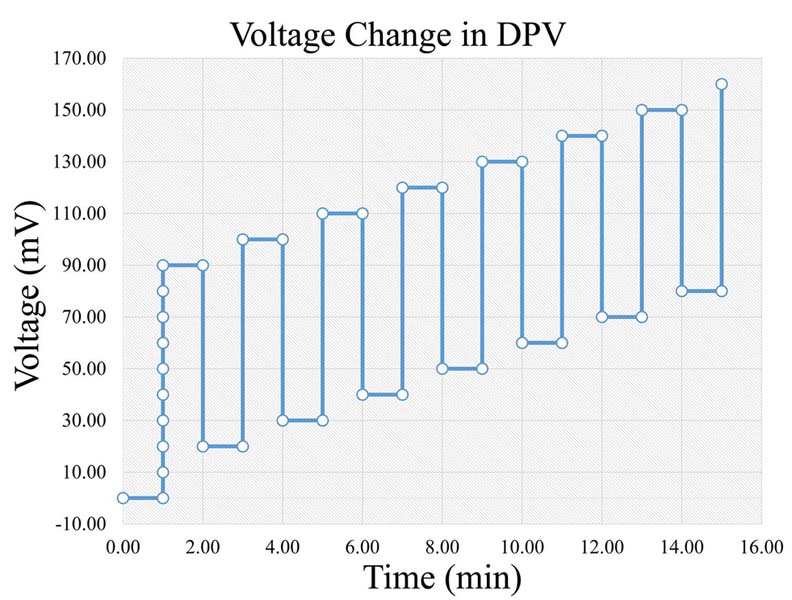 Figure 4
Figure 4Voltage change rate in DPV (Legend: DPV: Differential Pulse Voltammetry).
Besides the aforementioned voltammetric methodologies, a considerable discussion took place concerning the comparison between voltammetric and perfusion techniques. Perfusion techniques were applied during the second half of the 20th century, where some of them included the cortical cup, push pull cannula and microdialysis (2, 3). Of those perfusion methodologies the most sophisticated is microdialysis, mostly due to the dialysis membrane attached at the rim of a probe, averting extraneous diffusion and turbulence (2, 3). Electrodes/probes used in microdialysis have a diameter of about 200-300um, whereas, as mentioned above, voltammetric electrodes lie in a range of 10-35um (2, 3, 8, 13). As a result, microdialysis electrode induces whopping and undesirable perturbation on the region of the brain area being applied (2, 3, 8, 13). On the contrary, voltammetry electrodes cause minimal damage, and can measure electrochemical species accurately, in several cerebral regions (spatial resolution at the order of um) (2, 12, 13, 32).
Another characteristic of electrochemical techniques is time resolution. Voltammetric techniques are sufficient for measurements at intervals of about 100ms (3), or 250ms as reported by Stamford JA (1985) (2), and generally capture transient events of 10-500ms (22). This is crucial since detection of neurotransmitter release and uptake, occur in intervals less than 1sec (13, 22). Yet, voltammetric techniques manifest restricted potential on measuring non-transient, long timescale changes (12, 13). On the other hand, microdialysis measurements are confined at intervals 5-20min (3, 8) (minute to hour timescale (13)). Yet, microdialysis manifests high sensitivity and selectivity, able to detect any chemical species, regardless of its ability to oxidize (3, 8, 13).
Electrochemistry could be defined as the discipline that concerns the interaction between electrical energy and chemical charge. When a chemical reaction is caused by an externally supplied current, as in electrolysis, it is called an electrochemical reaction. A major category of electrochemical reactions are the reduction-oxidation (redox) reactions. Redox reactions refer to the electrochemical processes, where an electron transfer, to or from a molecule or ion, changes its oxidation state. These reactions occur spontaneously or after the application of an external voltage. In continuation, an electrochemical cell (or hereafter “cell”) is a device that produces an electric current from energy released by a redox reaction. Examples of electrochemical cells include the Galvanic cell, the Voltaic cell and the voltammetric cell.
Electrochemical cells include two conductive electrodes, an anode and a cathode, where the anode is the electrode causing oxidation and the cathode is the electrode causing reduction. Major component of the electrochemical cell is a solution that facilitated current flow called an electrolyte. Electrolytes are usually buffered solutions, which are also used as solvents for other species that are to be determined. In the case of voltammetric cells another electrode is added, which consists of the reference electrode.
Two principal equations govern the discipline of electrochemistry. The first is the Nernst Equation, which defines the effect of reactants concentration on the electrochemical cell potential, expressed as:
The second equation is the Gibbs Equation, which predicts whether a reaction can occur spontaneously based on free energy. Gibbs Equation is formed as:
In its simplest form, oxidation occurs when a metal is immersed in an aqueous or buffered solution. Oxidation takes place when ions of the electrolyte, react with the metal’s surface and donate electrons to it (10). An interesting example concerns the oxidation of Ascorbic Acid (AA) to Dehydroascorbate (DHA) (2). The generalized oxidation reaction can be described as follows: AA?DHA+ne- (Chemical Reaction 1), where ne- refers to the number of electrons donated (10, 30, 31). On the other hand, the reverse process, where electrons are transferred to the ions of the electrolyte is called reduction (31).
As aforementioned, voltammetry is an electrochemical analytical method in which the substances to be determined are oxidized. Thus, several physical measures determine the properties of the redox reactions participating in the electrochemical process. Hence, the current (i) flowing through the electrochemical cell responsible for the redox reactions is defined as:
Each pulse applied for time t1 to t2 that is dt=t2-t1, causes two phenomena. The first is a capacitive current of the form:
, Equation 4, where i is the capacitive current, KCapacitative is the capacitance constant and t is the time interval a voltage is applied. The capacitance current is very short but increases the background noise in the measurement (Figure 5). Further on, a second current forms, which is a Faraday current of the form:
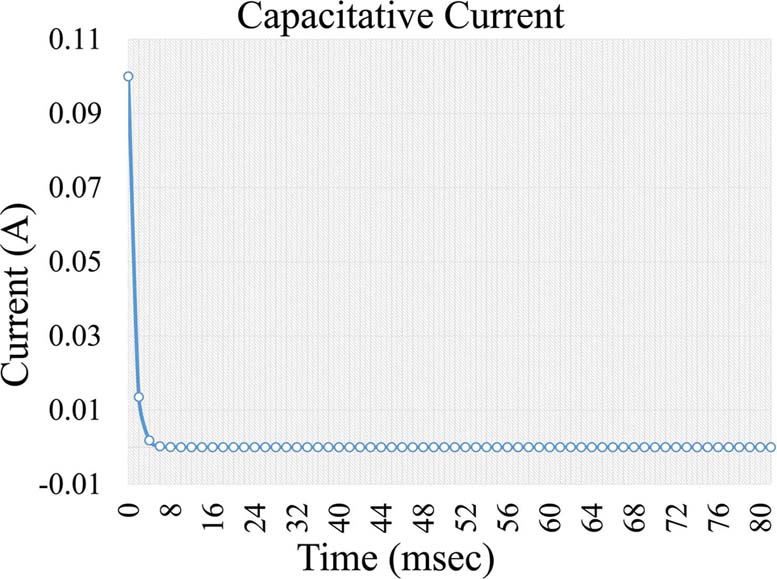 Figure 5
Figure 5Capacitive current formed during voltage application in an electrochemical cell.
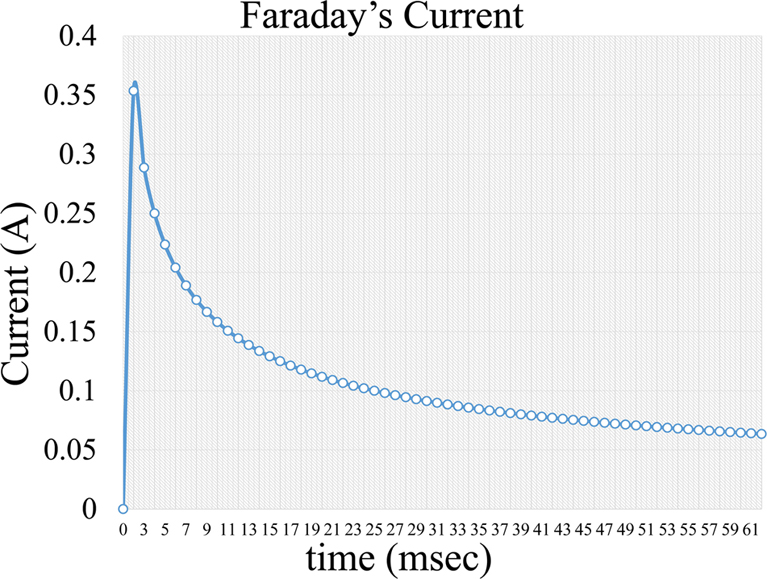 Figure 6
Figure 6Faradaic current formed during voltage application in an electrochemical cell.
Thus, the current measured in voltammetry consists of two components, the charging current which appears due to the capacitive nature of the layer between the electrode and the solution (10, 13) and the faradaic current due to the redox processes (2, 22). The theoretical optimal for the voltammetric measurements is the maximization of the Faradaic to Capacitive ratio (2). In several voltammetric techniques, a subtraction of the capacitive current (2, 8, 10, 13, 33) is pursued so that the Faradaic current can be isolated. The Faradaic current is considered to be smaller than the residual current (2, 10). Robinson et al. (2003) report that the Faradaic current may be about 3% of the charging current (13).
In particular, the Faradaic current measured by the polarograph is subtracted with respect to the different time points measured. This difference divided by the time lapse equals to the first derivative of the redox current. The quantity of the substances to be determined can be calculated from Equation 3.
Based on the aforementioned theory, during a voltammetric measurement a voltage is applied between the auxiliary and the working electrode. The voltage frequency and form depends on the type of voltammetry, as in the case of Differential Pulse Voltammetry (DPV) has the form presented inFigure 4). At the time point that the voltage reaches a certain level, the corresponding substances are being oxidized, creating the aforementioned measured current. The newly formed current is measured by the reference electrode.
As aforementioned, the reason that voltammetry could be used in a variety of applications comes from the fact that many biochemical species are oxidizable (12), videlicet they can produce a Faradaic current which is detectable and measurable. The detected current of an unknown species can be quantitatively determined by in vitro calibrations, where known concentrations are assigned to measured currents (13). In the case of brain research, numerous examples apply to the previous methodology, such as neurotransmitters. In a report by Crespi F. (2011), Differential Pulse Voltammetry (DPV) was used, where several oxidation voltages have been reported for certain neurotransmitters. For example, oxidation values included neuropeptide Y (NPY) oxidized at 600mV, at 595mV for the NPY 13-36 fragments and 580mV for hPP (14). In addition, measured currents recorded were 2.8.nA for NPY, 2.4.nA for hPP and 0.4.6 nA for NPY 13-36 (14). In another report by Cespuglio et al. (1981) oxidation potentials discovered, included -50mV for AA, 100mV for catecholamines, 300mV for 5-hydroxy compounds (5-HT, 5-HTP, 5-HIAA), 680mV for amino acids (tryptophan, tyrosine) and 680mV to 800mV for neuropeptides (in agreement to the results reported by Crespi F. (2011)) (12, 14). Further on, in a report by Gonon et al. (1980), reported oxidation potentials at -50mV for AA and 100mV for DOPAC in the striatum (34), while Clement et al. (1993) found a voltage peak at 280mV for 5-HIAA (27). Subsequently, detection of histamine exhibits a greater difficulty due to its electrochemical properties. Histamine is an oxidizable amine, but its oxidation process differs from that of DA or 5-HT due to the fact that it involves a charge transfer (15). Samaranayake et al. (2015) found an oxidation peak both in vitro and in vivo approximately at 300mV, whilst Pihel et al. (1995) were able to detect histamine only after appropriate electrode pre-treatments (35). Finally, in a very interesting report Kennedy et al. (1993) states an approach for the detection of insulin (36). In particular, this report showed that it was feasible to determine insulin levels of pancreatic b-cells, obtained from Langerhans’ islets. The investigators used an amperometric method utilizing carbon-fiber microelectrodes for measuring insulin excretion in single cells. Yet, although, this report supported the idea of detecting insulin with electrochemical methods, it did not provide proof, which was the case of later reports by other investigators (37, 38). Conclusively, several monoamines are detected by electrochemical techniques but a plethora of others have not been detected or at least not successfully detected. A reason for such a problematic issue has been attributed to the lack of electrode surface modifications (10).
The electrocatalytic effect appears in a multi-species solution, when the oxidation current of a specific species is enhanced, thus catalyzed, by the presence of another species, due to an iterative oxidation reaction (10). For instance, in the case of simultaneous presence of DA and AA in the same electrolyte an electrocatalytic effect takes place. Orthoquinone (DOQ), the oxidation product of dopamine, reacts with AA and to produce Dehydroascorbic acid (DHA), which is the oxidation product of ascorbic acid and DA (2, 10). This process is described by the following reaction (Chemical Reaction 2). Therefore, a fraction of DA concentration may be oxidized twice and thus measured twice, inducing erroneous results (2, 10). This electrocatalytic current is undesirable and may be reduced by applying high scan rates (i.e. in FSCV) or by the utilization of significant small electrodes (2, 10).
One of the rudimentary limitations of voltammetric techniques lies towards the fact that some species have similar oxidation potentials (2, 3, 11, 19, 25). Several neurotransmitters and their metabolites may oxidize within a narrow voltage range (e.g. Dopamine and DOPAC, Serotonin and 5-HIAA) (25), which if combined with the fact that metabolites display larger extracellular concentrations than their parent neurotransmitters (3, 25) leads to the induction that estimation of a chemical species cannot be adequate without ancillary methods (microdialysis, pharmacological identification) (3, 25, 39). Ozel et al. (2011), reported that ascorbic acid is the dominant interfering species that produces high concentrations in low voltage regions, as well as the difficulty to be distinguished from serotonin (11), whereas Kawagoe et al. (1993), indicate ascorbic acid and DOPAC as the major interfering compounds for dopamine (8), as well as uric acid along with 5-HIAA are the major interfering component for serotonin (8). In these reports, it is also demonstrated that norepinephrine and dopamine manifest similar voltammograms (8, 13).
In the complex extracellular environment, thus the case of in vivo experiments, sufficient selectivity is of paramount importance for the study of neurotransmitter physiology (11). In order to achieve high selectivity, several approaches can be used, which include i) electrode surface modifications (2, 3, 8, 13, 40), ii) selection of a region in the extracellular fluid in which the compound of interest maintains a high concentration whilst other interfering species retain with lower concentrations (3, 8, 13), iii) alterations of the scan rate (13), iv) pH alterations (9), v) pharmacological pretreatment (3, 8), v) utilization of suitable mathematical models (25) and vi) combination with other methods, such as microdialysis (8), or high pressure liquid chromatography (HPLC) (3).
In voltammetry a three electrode system is required (14). The three-electrode system includes a) a working electrode, where the measuring voltage is applied, b) a reference electrode, usually an Ag/AgCl (10-15 (although earlier the normal hydrogen electrode was considered as a standard but was substituted due to its difficulty in use (10)) as well as, a saturated calomel electrode has also been reported (41) and c) a counter/auxiliary electrode (commonly of platinum wire (9, 11, 12, 14)). The diameter of the working electrode ranges up to several micrometers (um), as described in plethora of reports. Electrodes utilized have usual diameters of 30um (14), 12um (25), 8um (12), 5-10um (20), 7um (15, 19), 10um (10), 10-20um (8) etc. Bath et al. (2000) mention two sorts of carbon fibers; a Thornel P55 (10um diameter) and a Thornel T650 (7um diameter) (22). Generally, the most commonly used working electrode diameter is about 1-35um in diameter (3, 8), while Hochstetler & Wightman (1998) claim that diameters of about 500nm-2um can be also constructed (10). Further on, Chen et al. (2003) fabricated carbon fiber electrodes with a diameter of 100-300nm (42). Finally, the length of the working electrode fluctuates at a range between 50-500um (10, 12, 25, 26). Working electrodes are mainly manufactures by carbon fibers or carbon-based fibers (2, 11, 12, 14, 15), because carbon is an electrochemically stable element, especially in the case of biochemical reactions (8, 10, 20), causing minimal tissue damage (20). Metal electrodes have been applied but reported too unstable for in vivo applications (8), yet Park et al. (2005) reported the use of diamond electrodes in electrochemistry (43), which provide increased stability, sensitivity and reusability, transcending the performance of carbon fiber microelectrodes (20). Ozel et al. (2011) reported that carbon fiber electrodes coated with chitosan (a polymer) produce a repulsive effect against ascorbic acid, uric acid and anions (5-HIAA, DOPAC), whilst at the same time serotonin is attracted and positively charged (11). Chitosan is biocompatible and exhibits broad mechanical strength and increased stability (11). Falat & Cheng (1982) contrived an adequate separation of AA and catechol with appropriate modifications of a carbon epoxy electrode (44). In addition, Nagy et al. (1982) reported a slightly different way by infusing ascorbate oxidase (AAOX) between the electrode surface and a surrounding membrane, which evoked the oxidation of AA before it contacted the electrode surface (45). A variety of reports mention Nafion-coated electrodes, which enhance the selectivity for cations (DA) against anions (AA, DOPAC) (2, 3, 8, 20, 33). In another report, Broderick PA (1990) states that graphite-stearate electrode measures AA, DA and 5-HT to the potentials of 55mV, 140mV and 290mV, respectively, without detecting the interfering species of DOPAC and 5-HIAA (23). Important application of this electrode is the detection and separation of norepinephrine from dopamine (23). It is commonly accepted though, that the above modifications come at the cost of response time, providing electrodes unable to measure rapid changes in the physiological or in vivo concentrations of neurotransmitter (2, 8, 13, 20, 33). These electrodes dispose response time approaching seconds or even minutes (8). Hermans et al. (2006) pretreated carbon fiber electrodes and achieved increased sensitivity to cations (esp. DA) without considerable loss in temporal resolution (46). Another aspect we ought to consider in the present work is the correlation between the exposed surface area of the working electrode and the noise being produced or the resolution of the measurement. Briefly, if the electrode surface extends out of the area of interest, several compounds may come in contact with the electrode, thus we come up against enhanced noise (due to increased capacitance) and decreased spatial resolution, because of the detection of these residual species (10, 20). On the other hand, Budai et al. (2010) claim that excess small electrode surface produces restricted performance and low Signal/Noise (S/N) ratio (19).
Generally, scat rates from the order of uV/sec to mV/sec have been applied to voltammetric techniques (10). However, the vast available voltage range, impose effects on the capacitive noise and the amplitude of the charging current, as well as the selectivity and sensitivity (13). At slow scan rates, the capacitance at the electrode-solution surface is larger as compared to fast scan rates. Thus, for lower capacitive noise, faster scan rates are necessary (10, 41). At higher scan rates (above the order of V/sec) the charging current is magnified (2, 8, 10, 23, 41) due to its proportionality to the scan rate (2, 8, 13). Faradaic current is proportional to the square root of the scan rate (2, 8), so slower scan rates (on the order of mV/sec (2) or below the order of V/sec (10)) suppress charging current at the loss of temporal resolution (8, 10). Stamford JA (1985) mentions that scan rate affects the number of peaks measured by voltammetry, where at slower scan rates more peaks may be detected (2), as well as the intensity of the electrocatalytic effect (2).
Many chemical compounds (thiols, phenols, etc.) have oxidation potentials that fluctuate along with pH (9). This property could be useful in order to strengthen selectivity (9). Nussbaum et al. (1992) claim that under the reaction of 1-cyanobenz(f) isoindole (CBI), amines may be separated from other interfering species (9). More specifically, in the same report, it has been deduced that CBI derivatives (CBI-glutamic acid, CBI glycine, etc.) possess independent oxidation values upon pH changes, whilst phenols can be transferred at higher oxidation potentials by operating at a lower pH (9). Serifi et al. (2015), noticed a diminution at the oxidation values of a variety of species, after an increment in pH (47). For instance, norepinephrine displayed an anodic peak at 750mV (pH=4), at 320mV (pH=7.4), at 150mV (pH=9) and at -10mV (pH=11) (47). L-DOPA displayed peaks at 470mV, 250mV, 150mV and 70mV, respectively (47).
Voltammetry has been utilized throughout time, for the detection of monoamines in both in vitro as well as in vivo cases. The present section concerns unpublished results from experiments performed in the Institute of Neurochemistry, at the Philipps-Universitaet, Marburg (Lahn). In the particular experiment, carbon fiber electrodes were used of a diameter 3-30um. The electrodes were constructed in-house, following the diagrammatic form presented inFigure 3). Electrodes have been pre-treated with respect to the monoamine, or metabolite under investigation. In particular, pre-treatment included the application of a voltage in a range between 0.8.-3V. The precise electrode pre-treatment included: for DOPAC, 2.9.V for 20sec, -0.8.V for 5sec and 1.5.V for 5sec, for DA, 2.8.V for 20sec, -0.8.V for 5sec and 1.5.V for 5sec, for 5-HIAA, 3.0.V for 20sec, 2.5.V for 20sec and 2.0.V for 15sec. Pre-treatment was performed in a PBS electrolyte. In vitro measurements were performed in an electrochemical cell with an Ag/AgCl reference electrode and a tungsten wire auxiliary electrode. The differential pulse voltammetric measurements were performed using a modified Polarecord E506 (Metrohm, France), with potential sweep in a range of 0mV to +500 mV with 10 mV/sec scan rate. Resulting current was recorded on an analogue device depicting the peak of each measuring cycle (Figure 7). The presented measurements concerned the estimation of a DOPAC 20uM solution in an electrochemical cell, dissolved in PBS. Initial recording was analog and in order to be able to process the complete signal and not just the peak of oxidation, we have developed a method for signal digitization (results and method not presented). A digitized signal is presented inFigure 8). Signal digitization served as a tool for the investigation of signal properties. In particular, we have performed a Discrete-Time Fourier Transform (DTFT) with the Matlab® Computational environment (48). DTFT was performed in order to investigate signal properties beyond oxidization current (Figure 9). The DTFT of a discrete-time signal x(n) can be represented as X(ejv) (49, 50). DTFT generally is a complex function of angular frequency v and it can be written as: X(ejv)=|X(ejv)|ejX (Equation 6) (49, 50). The amplitude (|X(ejv)|) of the transformation and the phase (X(ejv)). Such approaches may contribute to the better understanding of the voltammetric signal.
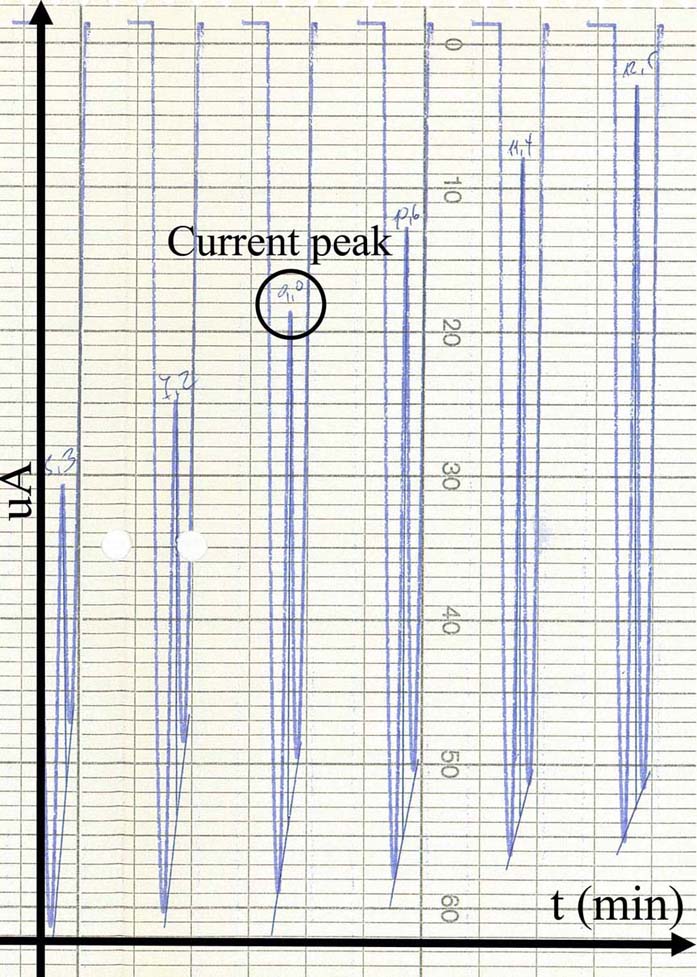 Figure 7
Figure 7An analog voltammogram recorded during an in vitro measurement in DOPAC 20uM electrolyte solution. Peaks (black circle) correspond to the optimal voltage where DOPAC (or in general any substance under investigation) is oxidized.
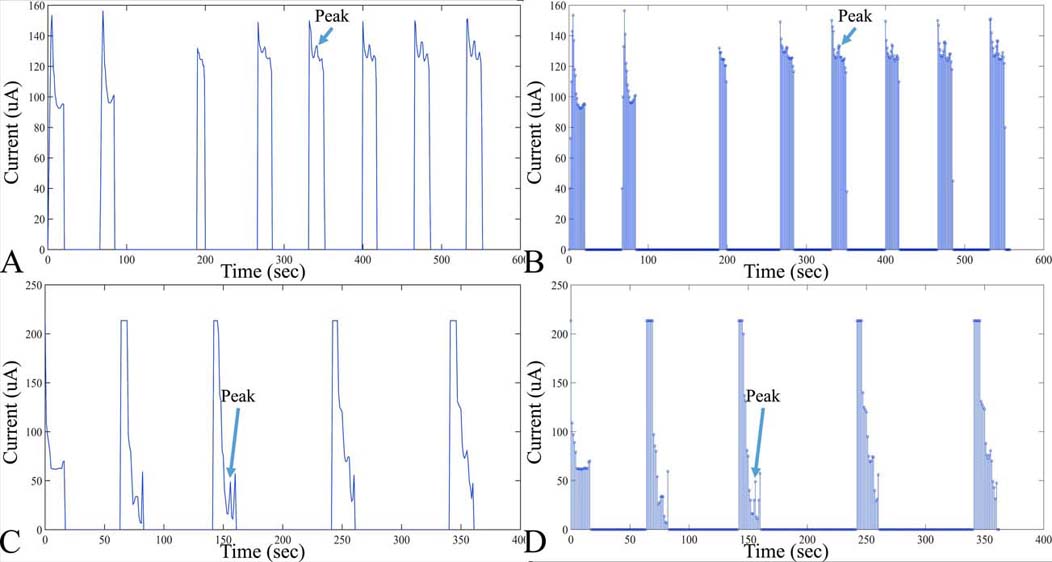 Figure 8
Figure 8Examples of digitized analog recordings of voltammetric measurements. In particular, an in vitro measurement of DOPAC is presented (A), with the respective points taken for digitization (B). Measurement conditions of voltammograms A and B. DOPAC concentration: 20 M, Working electrode diameter: 20 m, Scan rate: 10mV/sec, Scan duration: 120sec. Further on, another example of a voltammetric digitized signal is presented in C, D. In particular, measurement conditions of voltammograms include: DOPAC Concentration: 20 M, Working electrode diameter, 200um, scan rate, 10 mV/sec, scan duration, 300sec.
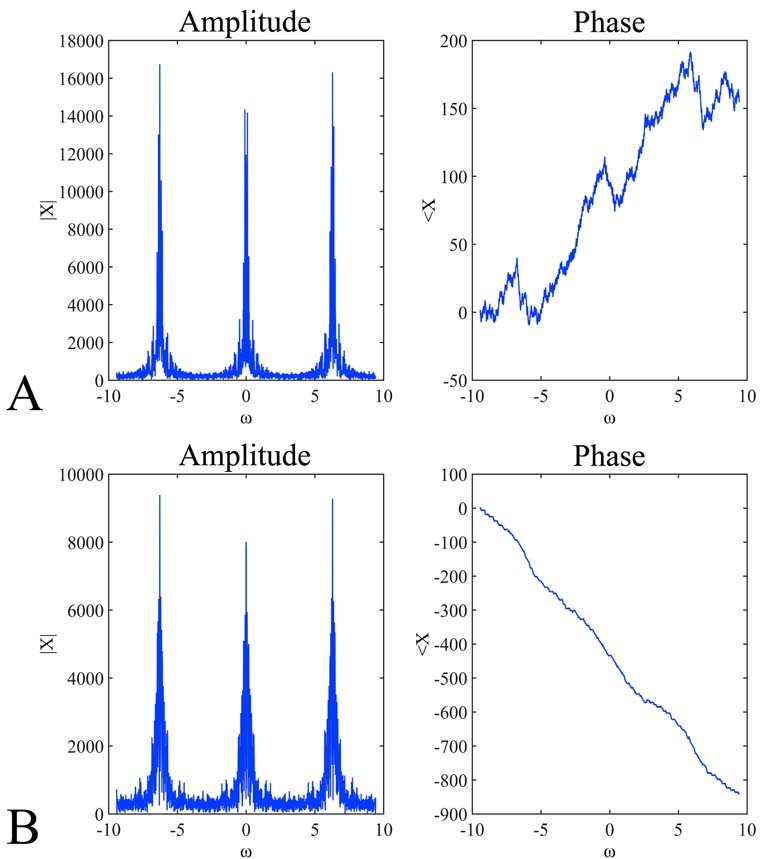 Figure 9
Figure 9Discrete-Time Fourier Transform (DTFT) of the signals presented inFigure 8). In particular, the amplitude (A, B left) and phase (A, B right) of the digitized signals presented in Figure 8A, 9B and Figure 8C, 8D respectively. The DTFT plots represent the amplitude and the phase of the function X(ejv). The angular frequency varies from -3pi to 3pi. The expected periodicity (-pi, pi) of |X(ejv)| is confirmed by the diagrams viewed in A and B left . In addition, the angle of the Fourier transform displays an approximately linear shape (A, B right).
Voltammetry, the development of the initial method of polarography, has been proven a useful tool for the investigation of monoamines. Monoamines, are essential molecules of the CNS physiology. Their detection and investigation were considered of crucial importance and at the same time their detection proved a tedious task. The main problem with monoamine detection is that their turnover is rapid and there was a need for an in vivo monoamine detection. Towards that task voltammetry proved to be an invaluable method since it facilitated the in vivo measurement of monoamines (14, 51-58). In the present work, we have attempted to review the available knowledge on voltammetric techniques as well as present some experimental results manifesting the use of in vitro monoamine detection.
Abbreviations: CNS: Central Nervous System, DPV: Differential Pulse Voltammetry, ACh: Acetylcholine, A: Adrenaline, DA: Dopamine, NA: Noradrenaline, NE: Norepinephrine, 5-HT: 5-Hydroxy Tryptamine, Tyr: Tyrosine, MAO: Monoamine Oxidase, DOPA: 5-Dihydroxy Phenylalanine, DOPAC: Dihydroxy-Phenyl Acidic Acid, Tyr: Tyrosine, GABA: Gamma Aminobutyric Acid, Glu: Glutamate, FSCV: Fast scan cyclic voltammetry, DPV: Differential Pulse Voltammetry, DNPV: Differential Normal Pulse Voltammetry, DDPV: Differential Double Pulse Voltammetry, SSV: Steady State Voltammetry, FDRV: Fast Differential Ramp Voltammetry, DPA: Differential Pulse Amperometry, SWV: Square Wave Voltammetry, AA: Ascorbic Acid, DHA: Dehydroascorbate, DOQ: Orthoquinone
Footnote:1In Hochstetler SE & Wightman RM (10) determine this distance to a few angstroms for voltammetric techniques.