
Frontiers in Bioscience-Landmark (FBL) is published by IMR Press from Volume 26 Issue 5 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.

1 Department of Rheumatology and Clinical Immunnology, University of Lübeck, Lubeck, Germany
2 Department of Statistic, Federal University of Pernambuco, Recife, Brazil
3 CellTrend GmbH, Luckenwalde, Brandenburg, Germany
Abstract
G protein-coupled receptors (GPCRs) form a most diverse family of integral membrane proteins that mediate homeostatic and pathological processes, most notably by orchestrating cell distribution throughout the body, their infiltration, and time of presence in inflamed tissues. Here we discuss loss-of-orientation-effects in GPCR-mediated cell trafficking and migration and their impact on the phenotype of autoimmune diseases. In this context, we provide a systemic and integrative view of the contribution of abnormal GPCR expression as well as the levels of natural ligands and functional autoantibodies to the phenotype of autoimmune diseases. Currently, several studies propose that functional autoantibodies (including those targeting GPCRs) constitute an exclusively pathogenic or pathognomonic phenomenon. Here we reinforce the need of revising this point of view, and suggest that functional autoantibodies primary play a role in normal human physiology, while dysregulation of their functions causes autoimmune disease. Because patients with autoimmune diseases still suffer from severe morbidity and mortality rates, we consider expanding our knowledge on (patho)physiological roles of GPCR as a prerequisite for the development of novel specific therapeutic modalities.
Keywords
- G protein-coupled receptors
- Autoimmune Diseases
- Autoantibodies
- Review
G protein-coupled receptors (GPCRs, also known as 7-transmembrane receptors) comprise the largest superfamily of integral membrane proteins with approximately 1000 different members. These receptors are involved in signal transduction responding to a variety of stimuli such as neurotransmitters, chemokines, nucleotides, Ca2+, and hormones (1–3). GPCRs are ubiquitously expressed by non-immune (e.g. endothelial cells and fibroblasts), innate (e.g. neutrophils, monocytes, macrophages, dendritic cells) and adaptive immune cells (lymphocytes), as well as more selectively in tissues (e.g. endothelin receptor expression in the lungs). They mediate a variety of physiological functions ranging from regulation of immune responses to signal transduction in the nervous system in response to a variety of ligands such as peptides, nucleotides, lipids, ions, photons and others (4).
Dysregulation of GPCR function plays a role in various pathological conditions such as cancer, infectious diseases and autoimmune diseases. Particularly in inflammatory and autoimmune diseases, a pivotal role of GPCRs in determining the spectrum of clinical manifestations is suggested by various examples. For instance, C-X-C chemokine receptors type 3 (CXCR3) (5), CXCR4 (6), C-C chemokine receptor type 2 (CCR2) (7) and CCR5 (5) mediate the recruitment of leukocytes to the central nervous system (CNS) in multiple sclerosis; gender-specific exacerbation of systemic lupus erythematosus (SLE) in women is associated with the activity of sex hormone receptors (estrogen receptor and gonadotropin-releasing hormone receptor) (8, 9); vascular GPCRs, such as angiotensin II type 1 receptor (AT1R) and the endothelin-1 type A receptor (ETAR) determine the extent of vasculopathy in systemic sclerosis (SSc) (10–12); and muscarinic acetylcholine receptors (mAChR) (13) influence the abnormal processing of the amyloid precursor protein (APP) during the formation of amyloid deposits in Alzheimer's disease (AD).
In addition to abnormalities of GPCRs and their ligands, autoantibodies targeting GPCRs that alter their function also play a crucial role in several diseases. The first observations of these pathogenic mechanisms were made in the last century when functional autoantibodies started to be characterized in patients with Chagas' disease (14, 15), allergic rhinitis, and asthma (16). Since then, several studies have demonstrated that functional GPCR-autoantibodies trigger a variety of immunopathological mechanisms such as those demonstrated in the context of cardiovascular diseases (17) and allograft rejection (18). In addition, immunobiological concepts in the pathogenesis of rheumatic diseases encompassing the synergy of such GPCR-autoantibodies with natural small molecule receptor ligands (10,19,20), have been also characterized.
Here we provide a systemic interpretation of the synergy between GPCR expression, the levels of GPCR natural ligands and GPCR autoantibodies in homeostatic regulation and its disruption in autoimmune diseases. We discuss the pivotal role of this triad in orchestrating cellular trafficking and migration throughout the host, thereby contributing to the development of different disease phenotypes.
The permanent control of leukocyte circulation throughout the body involves the activation of several GPCRs (Figure 1). Therefore, it is reasonable to assume that dysregulation of subgroups of GPCRs contribute to disease phenotypes by causing abnormal cellular infiltration into inflamed tissues and modulation of the residence time of immune cells at these sites. In response to exogenous and endogenous factors, GPCRs (21–27) emit signals to the cytoskeleton and to adhesion molecules (e.g. integrins), thereby promoting cell trafficking and cell migration. They do so under homeostatic as well as pathological conditions when signals from pathogen-associated molecular pattern (PAMPs) and damage-associated molecular patterns (DAMPs) induce the inflammatory response and trigger signal cascades such as those elicited by the interaction between chemokine/chemokine receptors that coordinate the recruitment of leukocytes.
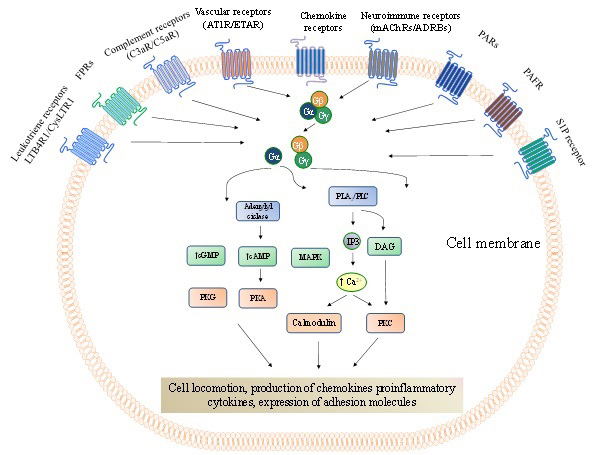
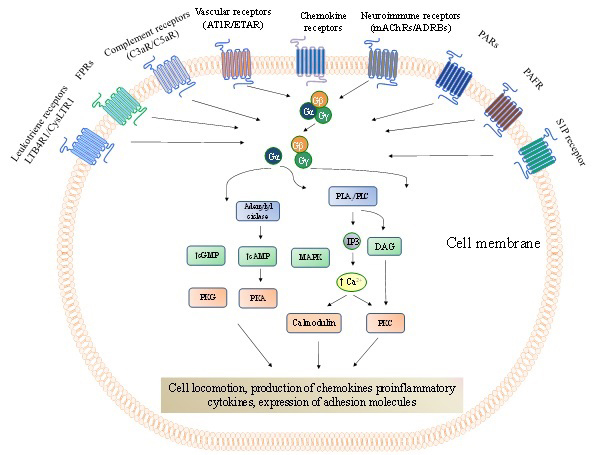 Figure 1
Figure 1G protein-coupled receptors (GPCRs) that mediate cell trafficking and migration throughout the body. Chemokine receptors are the most abundant GPCRs that directly modulate cell locomotion with approximately fifty representors. However, other GPCR representors are not of less importance than the chemokine receptors. The GPCRs signalling pathways have been remarkably revised and depicted elsewhere (124,125). In brief, following the ligand binding to a GPCR, it undergoes conformational changes leading to the exchange of guanosine diphosphate (GDP) for guanosine triphosphate (GTP) on the α-subunit of heterotrimeric G proteins (composed byα-, β-, andγ-subunits). This process starts the activation of different downstream signalling pathways involving, among others, second messengers (adenylyl and guanylyl cyclases, phosphatidylinositols), protein kinases (PKG, PKA and PKC), phospholipases (PLA2 and PLC), Rho GTPases (RhoA) and increased intracellular Ca2+concentration. These mechanisms are necessary to regulate the dynamics of the actin cytoskeleton and cell-matrix adhesion complexes to promote cell locomotion during cell trafficking and migration. cAMP, cyclic adenosine monophosphate;cGMP, cyclic guanosine monophosphate; DAG, diacylglycerol;FPRs, formyl peptide receptor; IP3, Inositol trisphosphate; PAFR,platelet-activating factor receptor;PARs,protease-activated receptors;RhoGEF, Rho family guanine nucleotide exchange factors; S1PR,sphingosine 1-phosphate receptor.
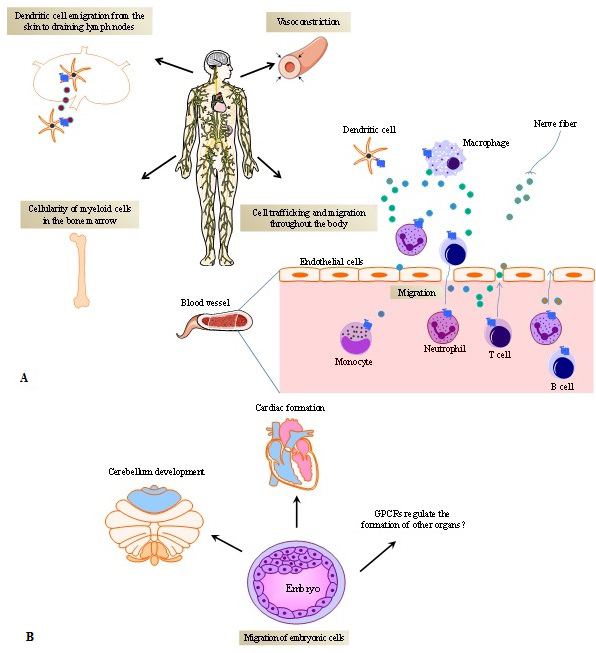
From the perspective of the micro-environment encompassing cell-to-cell contact-induced activation in secondary lymphoid organs, GPCR-ligand interactions contribute significantly to the fate of the immune response. GPCRs coordinate cell attraction induced by chemotactic gradients and the time of interaction between T cells and antigen presenting cells (APCs) (28). Namely, the interplay between T cells and APCs requires a stable T cell receptor (TCR) recognition of antigen/human leukocyte antigen (HLA) complexes. When chemokine receptors expressed by T cells are sequestered at the immunological synapse, this will result in increased T cell-APC engagement due to reduced T cell attraction by other chemokine sources. For instance, CCR5 and CXCR4 translocate to the T-cell/APC immunological synapses, thus favouring T cell proliferation and differentiation while decreasing T cells chemotactic attraction from other locations in the body (29). From a systemic viewpoint, GPCRs regulate the localization and distribution of immune cells throughout the body, as they induce hematopoietic stem cell mobilization (30), maintain homeostatic neutrophil counts in blood (31), promote monocyte immigration from bone marrow into inflamed skin, promote dendritic cell (DCs) emigration from the skin to draining lymph nodes (32) and T cell homing into lymph nodes (33), contributing to cellularity of immune cells in several organs and organogenesis (34, 35) (Figures 2A and B).
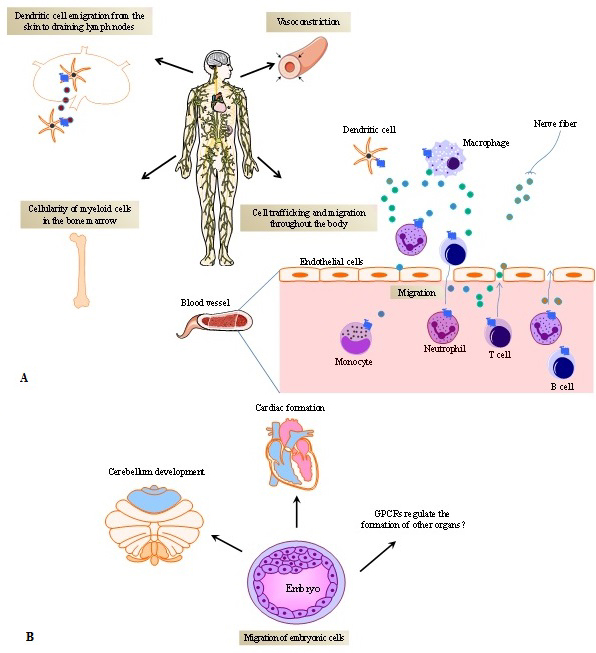 Figure 2
Figure 2G protein-coupled receptors (GPCRs) and their ligands promote the dynamics of cell trafficking and migration. A) GPCRs mediate the cell distribution throughout the body. Chemoattractants or other cell modulators released by different cells including immune and non-immune cells (e.g endothelial cells and tissue-resident cells) in different organs, bind to GPCRs, orientating the cell to reach their destiny. In physiological conditions, cells appropriately infiltrate into tissues and keep their regular time course of cell-cell interactions. Dysregulation of GPCR expression and abnormal production of GPCR natural ligands (e.g. chemokines, leukotrienes, neuro-immune modulators, etc.) break this natural dynamic, influencing the spectrum of disease phenotypes. The different boxes within the Figure represent the different effects of GPCRs. The little circles represent GPCR ligands. B) GPCRs expressed on embryonic cells and the production of GPCRs ligands promotes organ formation.
In this context described above, the biochemical modulation of adhesion molecules by GPCRs comprises an essential step of the biological dynamic. Selectins and integrins enable leukocyte rolling along the surface of endothelial cells and homing (36) towards specific tissue compartments. A particular characteristic of integrins involves their ability to rapidly and reversibly regulate cellular homing through different affinity states without significant alteration of cell surface expression. Chigaev and co-authors (37) have monitored the integrin affinity in real-time by demonstrating that inside-out signalling through Gαi-coupled GPCRs in a cAMP-dependent manner regulates the affinity of very late antigen-4 (VLA-4, also called α4β1-integrin; expressed on leukocytes and hematopoietic stem cells) to the vascular cell adhesion molecule-1 (VCAM-1) expressed on endothelial cells. During this process, GPCRs such as CXCR4 and the urotensin 2 receptor UTS2R inhibit the autophagy-mediated reduction of integrin expression on the cell surface by repressing the formation of pre-autophagosomal vesicles from the plasma membrane (38), allowing integrin-dependent cell adhesion to the extracellular matrix and subsequent diapedesis.
Fine-tuning of cell trafficking is essential for appropriate cell distribution during development processes such as embryogenesis and organogenesis. At these development stages, GPCRs play an important role (Figure 2B). A fundamental insight into this mechanism has been gained by elucidating the role of CXCR4 and its natural ligand (CXCL12, also called stromal cell-derived factor 1, SDF-1) (39) in regulating the development of immune and non-immune organs. In mice, homozygous disruption of Cxcr4 gene is frequently lethal. Alternatively, the animals present impaired B cell lymphopoiesis and myelopoiesis. Moreover, deletion of Cxcr4 results in the disruption of the formation of the cerebellar neuronal layer and the cardiac septum during fetal development (35). Cxcl12 knockout mice exhibit an analogous phenotype (34).
However, cell distribution within the body is not only regulated by chemotactic GPCR, but also by vascular GPCR which modulate cell trafficking at various physiological levels (Figure 1). Thus endothelin receptors (ETAR and ETBR) and angiotensin II receptors (AT1R and AT2R), which are under the control of the potent vasoconstrictors endothelin-1 (ET-1) and angiotensin II (ang II), respectively, modulate actin polymerization, thereby regulating cytoskeletal remodelling in vascular smooth muscle cells and consequently controlling blood flow throughout the body tissues (40, 41). Endothelin and angiotensin receptors are also expressed by immune cells and play several roles in the regulation of different immunological mechanisms, including cell migration. Stimulation of endothelin and angiotensin receptors by their ligands induce migration of myeloid cells (e.g. neutrophils and monocytes) by a mechanism involving the activation of various mitogenic kinases including protein kinase G (G-kinase), proto-oncogene tyrosine-protein kinase Src, p38-mitogen-activated protein kinase (MAPK), and extracellular-regulated protein kinase (ERK 1/2) (42, 43). In addition, several reports show that activation of endothelin and angiotensin receptors regulates immune cell infiltration in various tissues by modulating the chemokine/cytokine milieu therein. Signalling through endothelin and angiotensin receptors activates the transcription factors nuclear factor kappa B (NF-kB) and activator protein 1 (AP-1), thereby increasing the production of related chemokines (e.g. monocyte chemoattractant protein-1 or MCP-1 and cytokine-induced neutrophil chemoattractant-1 or CINC-1) and pro-inflammatory cytokines (IL-6, TNF-α), which promotes leukocyte accumulation in organs such as lung, kidney and brain (44–48).
The pathogenesis of autoimmune diseases involves multiple intrinsic (e.g. genetic) and extrinsic (e.g. environmental) factors. Natural occurring loss-of function mutations in GPCRs are known to cause primary immunodeficiencies (49). However, so far, no case of autoimmune diseases has been associated with inherited loss- or gain-of-function mutations in GPCRs. To find such mutations would represent a unique opportunity to study the role of GPCRs in the pathophysiology of autoimmunity in vivo. On the other hand, GPCR dysregulation due to extrinsic factors is implied in the pathogenesis of various autoimmune diseases, which poses a rapidly evolving field of research. In this context, the loss of fine-tuned regulation of GPCR-mediated cell trafficking and migration contributes to autoimmune diseases by altering normal cellular infiltration of tissues and by modulating the time course of mutual interaction of immune cells at tissue sites of inflammation.
The interleukin (IL)-8/IL-8 receptor (IL-8R) axis represents a prototypical ligand/GPCR interaction involved in the development of inflammatory and autoimmune diseases. Two IL-8Rs exist: CXCR1 (the high affinity IL-8 receptor or IL-8RA) and CXCR2 (the low affinity IL-8 receptor or IL-8RB), which have 77% identity in their amino acid sequence. Macrophages and other non-immune cells, e.g. endothelial, epithelial and airway smooth muscle cells can produce IL-8, a chemokine that predominantly regulates the traffic and migration of neutrophils (50). For this reason, high IL-8 levels in inflammatory and autoimmune diseases are associated with massive mobilization of neutrophils toward inflammation sites. At these sites, neutrophils cause deleterious effects by releasing lysosomal hydrolases, reactive oxygen species (ROS) and neutrophil extracellular traps (NETs), following the uptake of immune complexes and under the influence of different inflammatory mediators (51, 52).
The involvement of IL-8/IL-8R in the development of immune-mediated diseases have been extensively reported. IL-8-dependent recruitment of neutrophils to synovial fluids of patients with seropositive rheumatoid arthritis (RA) renders this immune cell type one of the most abundant in inflamed joints at sites, where cartilage destruction and bone erosion occurs (53). In the murine model of monoarticular antigen-induced arthritis (AIA), CXCR1 and CXCR2 blockers reduce disease severity by inhibiting neutrophil recruitment to the joints, while reducing the capacity of the cells to adhere on synovial microvessels (54). The functional homologues of human IL-8 in mouse (CXCL1 and CXCL2) also have potent neutrophil chemotactic activity and their levels are significantly increased in skin lesions of experimental epidermolysis bullosa acquisita (EBA) (55). Elevated levels of IL-8 in patients with psoriasis induce epidermal accumulation of neutrophils in psoriatic lesions (56), while in subjects with SSc, enhanced IL-8 concentration in serum correlates with internal organ damage (especially lungs) (57). In SLE patients, IL-8 concentration in exhaled breath condensate and broncho-alveolar lavage fluid denotes a useful biomarker for SLE activity and pulmonary fibrosis (58). Each of these reports demonstrates an association between increased levels of IL-8 and enhanced neutrophil trafficking and migration towards inflammation sites, but the authors of these works did not assess the expression of CXCR1 and CXCR2 on neutrophils. Hu and colleagues (59) demonstrated in patients suffering from active anti-neutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis (AAV), that, despite increased serum levels of IL-8, neutrophils expressed low levels of CXCR1/2, which impaired their migration capacity. Therefore, this observation was associated with increased neutrophil adhesion to the glomerular endothelium, which accumulated in vessel walls during persistent inflammation in AAV.
CXCR3 is a chemokine receptor mainly expressed on activated T helper (Th) 1 cells, which are important source of interferon-γ (IFN-γ). The CXCR3 ligands (CXCL9, CXCL10, and CXCL11) are IFN-γ-inducible chemokines produced by immune and tissue-resident cells such as fibroblasts, keratinocytes, epithelial and endothelial cells. Consequently, these cells promote local attraction of Th1 cells and amplify their response (60). In patients with autoimmune diseases, Th1 cell activation is increased at inflamed sites and this enhances the local activity of phagocytes through the activation properties of IFN-γ. This cytokine is pleiotropic, and among others, it modulates the transcriptome of phagocytic cells, increasing their capacity to undergo potent oxidative burst (61). These reagents chemically modify amino acids, generating neo-epitopes which plays a role in patients with autoimmune diseases such as SLE and RA (62). Dysregulation of CXCR3 and CXCR3 ligands is commonly found in patients with SLE and RA. High expression of CXCR3 on CD4+ T cells present in blood and urine of SLE patients constitutes a biomarker for acute nephritis flares (63), while Th1 cells from RA patients express increased levels of CXCR3 in synovial tissue (64). Moreover, in patients with cutaneous lupus erythematosus, the number of infiltrating CXCR3+ T lymphocytes in skin lesions is associated with enhanced local production of CXCR3 ligands (65). Similarly, dysregulation of CXCR3 and/or CXCR3 ligands are found in other inflammatory/autoimmune disorders such as in progressive multiple sclerosis lesions in which the number of CXCR3+ Th1 cells is increased when compared to unaffected white matter from patients and healthy subjects (5).
The pleiotropic chemokine receptor CXCR4 represents an important partner of CXCR3, with which it forms heteromer complexes (66). CXCR4 blockade reduces the activation of T and B cells in nephritic mice leading to decreased autoantibody production and inflammation, prolonging lifespan (67). In SLE patients having activity index (SLEDAI) scores >10 and suffering form class IV lupus nephritis (LN) or active neuropsychiatric SLE, the expression of CXCR4 on B cells is remarkably increased as compared with patients with less severe clinical features. Moreover, in both mice and humans, the severity of the renal pathology is directly correlated to the up-regulation of the CXCR4 ligand (CXCL12) in renal tubules and glomeruli (67).
In addition to the above mentioned, several other GPCRs show a critical involvement in the development of autoimmune diseases. The CXCL13/CXCR5 axis has been reported to influence the development of SLE by regulating the trafficking and infiltration of B-and double-negative T cells into inflamed organs like kidneys (68) where the cells participate in the production of immunoglobulin and pro-inflammatory cytokines, respectively. Similarly, other GPCRs subgroups also affect the pathogenesis of autoimmune diseases. Increased levels of anaphylatoxins are known to influence the development of rheumatic diseases. Typical examples for this mechanism are the interactions of C3a and C5a with C3aR and C5aR receptors in RA (69) and SLE (70).
Another important involvement of GPCRs in the pathogenesis of autoimmune diseases occurs through the communication between the nervous and immune systems (71). Studies of neuro-immune receptors such as mAChR demonstrate that administration of atropine (a mAChR antagonist) to mice attenuates inflammatory responses in tissues by inhibiting leukocyte migration (24). However, several reports show that a positive or negative regulation of migration induced by mAChR receptors depends upon the cell population that are involved and the receptor subtypes (M1, M2, M3, M4 or M5) that are thereon activated (72–74). This is also true for β-adrenergic receptors (ADRBs), which participate in the development of autoimmune diseases (75) and display sexual dimorphism since their activation promotes chemotaxis of neutrophils in females, but not in males (76). Gender bias in autoimmune disease is a finding which needs to be further explored in order to elucidate neuro-immunological mechanisms that possibly contribute to the prevalence of autoimmune diseases in women more than men (77).
In the last decade, a variety of other GPCRs, were recognized to also play various roles in the modulation of inflammation, including the regulation of cell migration, hence, influencing the phenotype of autoimmune diseases. This applies to GPCRs such as PAFR (78, 79), PARs (80, 81), sphingosine 1-phosphate (S1P) receptor (82, 83), leukotriene receptors (84, 85), or N-formyl-methionyl-leucyl-phenylalanine (FMLP) receptors (Figure 1), as well as several other chemokine receptors (86). It stands to reason that these receptors are also dysregulated in rheumatic diseases but this notion remains poorly investigated. It will be important to establish the particular contribution of each GPCR subgroup during abnormal cellular infiltration into inflamed tissues and their influence in the time course of residence of the immune cells in these tissues. Reaching this goal would open new avenues to a better understanding of disease phenotypes and of overlapping syndromes presented by patients with different autoimmune diseases (87).
Several major contributions to our understanding of the pathophysiology of autoimmune diseases arise from understanding the interplay of a large number of GPCRs and their natural ligands (88, 89). Comparatively, little is known about the contribution of autoantibodies directed towards GPCR, since so far only a small number of GPCR-autoantibodies have been investigated in these diseases (90). All the autoantibodies directed against GPCRs that we discuss here belong to the IgG class and have been characterized as functional autoantibodies. I.e. they are able to bind to cell receptors and trigger the activation of intracellular signaling pathways (agonist autoantibodies) or inhibit signal transduction pathways (antagonist autoantibodies) that a natural ligand normally uses to control a particular cellular function (91–93).
Vascular injury is an early event in SSc (94) and several lines of evidence demonstrated the involvement of agonistic autoantibodies toward well-known vascular receptors such as AT1R and ETAR in the pathogenesis of SSc. In addition to their expression on non-immune cells (e.g. endothelial cells and fibroblasts), AT1R and ETAR are also expressed by innate and adaptive immune cells (11). In response to their natural ligands ET-1 and ang II, AT1R and ETAR trigger potent vasoconstrictor effects on endothelial cells (vasoconstriction and cytokine production). In addition, AT1R and ETAR regulate fibroblast responses (e.g. production of collagen) and orchestrate several mechanisms of immune responses including the regulation of cell traffic and migration (95–97). Riemekasten et al. (10) reported for the first time the presence of increased serum levels of autoantibodies targeting AT1R and ETAR in patients with SSc. These autoantibodies trigger cellular effects that resemble and synergize the effects induced by the natural ligands ET-1 and ang II (10,11,19,98–102). For instance, patients with SSc can suffer from pulmonary arterial hypertension (PAH) due to pathological synergistic effects of ET-1/ang II and anti-ETAR/AT1R on endothelial cells that result in abnormal vasoconstriction (103).
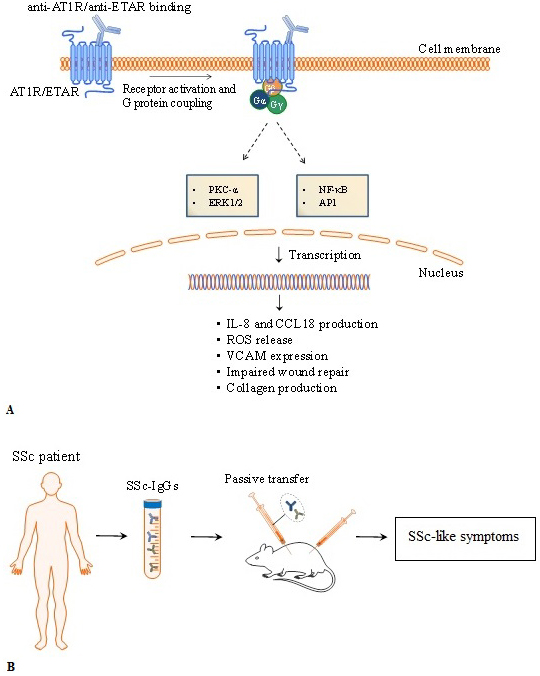
Autoantibodies targeting AT1R and ETAR activate signalling pathways involving pleiotropic proteins such as ERK and transcription factors NF-κB and activator protein 1 (AP-1) (10, 18), as well as regulate the levels of intracellular Ca2+ concentration ((Ca2+)i) (103). In vitro and in vivo data (e.g. passive transfer of IgG containing high levels of anti-ETAR and anti-AT1R from SSc patients to mice) demonstrate that autoantibodies directed against ETAR and AT1R induce the production of collagen by skin fibroblasts, ROS release by neutrophils, and reduced endothelial wound repair (12) (Figure 3). Furthermore, the skin of patients with SSc shows a pro-adhesive phenotype that promotes myeloid cell adhesion via adhesion molecules like the vascular cell adhesion molecule-1 (VCAM-1) (104). Autoantibodies targeting AT1R and ETAR from patients with SSc induce endothelial cells to express VCAM-1 and to produce IL-8, leading to increased neutrophil migration into inflammation sites (12). Anti-AT1R and anti-ETAR autoantibodies also promote the release of CC chemokine ligand 18 (CCL18) by peripheral blood mononuclear cells. CCL18 is a chemoattractant for T, B and natural killer (NK) cells and one of the most highly expressed chemokines in human chronic inflammatory disorders (11,105–107). Serum level of CCL18 is a predictive marker for lung disease progression and mortality in SSc (108). Thus, by promoting the recruitment of immune cells to inflammation sites, anti-AT1R and anti-ETAR autoantibodies play a pivotal role in the immune exacerbation presented by patients with SSc.
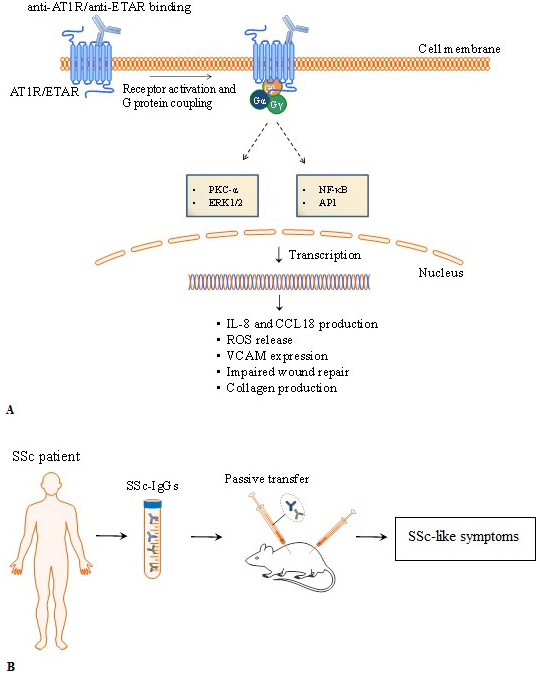 Figure 3
Figure 3Overview of pathophysiological mechanisms triggered by anti- angiotensin II type 1 receptor (anti-AT1R) and anti-endothelin-1 type A receptor (ETAR). Experimental evidences in vitro and in vivo of autoantibodies directed against AT1R and ETAR. A) Anti-ETAR and anti-AT1R autoantibodies from SSc patients trigger biological processes by activating protein kinase C (PKC) and mitogen-activated protein kinases (extracellular signal-regulated kinases or ERKs). Anti-AT1R and anti-ETAR induced-activation involve pleiotropic transcription factors such as nuclear factor-κB (NF-κB), activator protein 1 (AP-1). Passive transfer of autoantibodies from SSc patients induce SSc-like symptoms in mice. IL-8, interleukin-8; CCL18, chemokine (C-C motif) ligand 18, VCAM-1, vascular cell adhesion molecule-1; TGF-β, transforming growth factor β.
Several evidences have demonstrated neural and immune system interactions through complex and physiological relevant mechanisms including those orchastrating cell migration (109–111). In this context, autoantibodies directed against GPCRs have been identified. Functional antagonistic autoantibodies that target M3 have been associated with the development of gastrointestinal dysmotility (112, 113). Gastrointestinal dysfunction is frequently developed by patients with SSc (114) and it is possible that antagonistic autoantibodies toward M3 inhibit neurotransmission leading to a blockade of the gastrointestinal tract smooth muscle contraction (115). However, since mAChRs also affects the migration of immune cells (24), future studies characterizing the effects of anti-M3 autoantibodies or autoantibodies directed against the other four mAChRs subtypes (M1, M2, M4 and M5) on the regulation of immune cells trafficking and migration will open new eveneus to better understand the physiology of the neuroimmune interface.
G protein-coupled receptors (GPCRs) and their implications in the regulation of homeostasis and contribution to pathogenesis of autoimmune diseases are evolving areas. In particular, the appearance of GPCRs such as chemokine receptors and others during the evolution of vertebrates (116) provided essential steps to gain an increased level of biological complexity that allows the fine-tune regulation of cell trafficking upon environmental or intrinsic (e.g. metabolic) changes. Namely, several GPCRs play a pivotal role in the regulation of cell distribution throughout the body and the loss-of-orientation in GPCR-mediated cell trafficking and migration is an essential mechanism that determines the phenotype of autoimmune diseases. Therefore, we provided here a systemic view of receptors, ligands and functional autoantibodies in this context. However, while the role of soluble factors traditionally call “GPCRs natural ligands” such as chemokines are extensively investigated, the role of anti-GPCRs antibodies in the regulation of human homeostatic processes remain to be investigated.
It is noteworthy that functional autoantibodies targeting GPCR (117) as well as other receptors such as PDGFR (118, 119) and others (120–123) are present in sera from healthy subjects, although in lower concentrations when compared to patients with autoimmune-mediated disorders. Therefore it is important to determine whether functional autoantibodies targeting GPCRs are important for normal physiological and homeostatic processes. Studies determining quantity and quality of autoantibodies targeting GPCRs in healthy subjects according to sex, age and environmental factors would yield valuable information for future investigations. To reach this goal will change the way we understand the biological relevance of anti-GPCR autoantibodies and will breakdown frontiers in our understanding about biological sciences, which will allow to reinterpret the mechanisms of human autoimmunity.
The authors acknowledge DFG (grant, RI 1056-11-1/2), Actelion Pharmaceutical GmbH Germany, GSK, CellTrend for financial support. We acknowledge all patients and their families for their participation in our studies. We thank Dr. Antje Mueller from the Department of Rheumatology, University of Lübeck for critical reading of the manuscript. We acknowledge the support and funding from the intramural grant of the University of Lübeck, German Systemic Sclerosis Network (DNSS), DFG grant RI 1056-11-1/2, Actelion Pharmaceutical GmbH Germany, GSK, CellTrend, Scleroderma Foundation, and Mirjam Lichy Foundation.