


 , Abhishek Chauhan 2,*
, Abhishek Chauhan 2,* , Anuj Ranjan 2, Rajpal Srivastav 3, Ritu Chauhan 4, Vivek Narayan Singh 5, Tanu Jindal 2
, Anuj Ranjan 2, Rajpal Srivastav 3, Ritu Chauhan 4, Vivek Narayan Singh 5, Tanu Jindal 21 Amity Institute of Environmental Sciences, Amity University, 201313 Noida, Uttar Pradesh, India
2 Amity Institute of Environmental Toxicology, Safety and Management, Amity University, 201313 Noida, Uttar Pradesh, India
3 Amity Institute of Biotechnology, Amity University, 201313 Noida, Uttar Pradesh, India
4 Department of Biotechnology, Graphic Era (Deemed to be University), 248002 Dehradun, Uttarakhand, India
5 Department of Environment Protection, Shriram Institute for Industrial Research, 110007 Delhi, New Delhi, India
Abstract
Antibiotic resistance is a contemporary public health issue that poses significant environmental and public health concerns. The presence of antimicrobial-resistant (AMR) microbes has been reported across media irrespective of geography and landscape. This study aimed to analyze the antibiotic susceptibility of Bacillus subtilis obtained from the Indian Sector of the Southern Ocean (39°19′ S, 57°30′ E to 66°38′ S, 76°22′ E).
Bacillus subtilis was revived and cultured on Mannitol Yolk Polymyxin Agar. Antibiotic susceptibility was assessed via the agar well diffusion assay against 10 therapeutically significant antibiotics. Whole-genome sequencing was performed to identify the presence of AMR genes. A total of 12 AMR genes were identified via the Comprehensive Antibiotic Resistance Database (CARD). A comparative genomics approach was employed to investigate the global distribution of AMR genes from 2014 to 2024.
Antibiotic susceptibility testing indicated complete resistance to metronidazole, while the isolates remained susceptible to ampicillin, doxycycline, tetracycline, ciprofloxacin, norfloxacin, cefixime, azithromycin, meropenem, and cotrimoxazole. Whole-genome sequencing and comparative analysis identified 12 AMR genes, including aadK, vanT (within the vanG cluster), ykkC, ykkD, vanW (within the vanI cluster), FosBx1, qacJ, qacG, tet(45), vanY (within the vanM cluster), and blt. The observed resistance mechanisms included antibiotic efflux, target modification, and enzymatic inactivation. Comparative genomic analysis of 15 closely related strains revealed variability in the distribution of AMR genes, with B. subtilis strain MB415 carrying all 12 resistance genes.
The detection of antibiotic-resistant B. subtilis in the Southern Ocean suggests potential anthropogenic influences on microbial communities, underscoring the need for continuous surveillance of AMR in remote marine environments to prevent its proliferation and mitigate its ecological consequences.
Keywords
- antimicrobial resistance
- Bacillus subtilis
- antibiotics
- comparative genomics
- resistant genes
Drug resistance or antimicrobial resistance (AMR) has become an urgent global issue that poses serious risks to both the environment and public health. The excessive and unregulated use of antibiotics has resulted in their widespread presence in ecosystems, particularly in soil and water. Recognizing its severity, the United Nations Environment Programme (UNEP) has identified AMR as one of the six most pressing environmental concerns, alongside climate change, ecological degradation, and water scarcity. According to experts, this is one of the greatest problems humanity will encounter in the decades that follow [1]. The World Health Organization (WHO) has also emphasized the growing threat of AMR in pathogenic microbes and has been actively raising awareness of this issue. In a report published in May 2024, the WHO classified 15 families of antibiotic-resistant bacteria into three priority levels, namely, critical, high, and medium, to prioritize research and development efforts for new antibiotics. Among the most concerning pathogens identified are Acinetobacter baumannii (resistant to carbapenems), Enterobacterales (resistant to third-generation cephalosporins and carbapenems), Salmonella typhi (fluoroquinolone resistant), Pseudomonas aeruginosa (carbapenem resistant), nontyphoidal Salmonella (fluoroquinolone resistant), and Staphylococcus aureus (methicillin resistant), all of which present significant public health risks [2]. In this context, various studies have documented the emergence of AMR in numerous pathogenic microorganisms worldwide, resulting in resistance to several broad-spectrum antibiotics [3, 4, 5].
The Southern Ocean, bordering southern China by the Antarctic continent [6], surrounds one of the remaining untouched regions on Earth. This continent is home to a vast array of marine life and significantly contributes to global climate regulation through the albedo effect [7]. Maintaining pristine conditions is essential for preserving critical oceanic currents that support marine ecosystems and influence global climate patterns. However, human-associated microorganisms have been introduced into this isolated habitat as a result of increasing human activities, such as the construction of research stations and tourism. Antimicrobial resistance (AMR) is thought to be low in these remote areas despite the small number of people living there. However, because of their possible danger to biodiversity, the presence of AMR bacteria in aquatic environments, especially in marine ecosystems, has drawn increasing attention in the last 10 years. Marine waters and sediments close to these areas are impacted by aquaculture, industrial processes, and municipal garbage discharge, which frequently include multidrug-resistant microorganisms [8]. The prevalence of AMR genes is significantly greater in environments impacted by human activity and wildlife than in remote locations. In contaminated ecosystems, human activity directly contributes to the persistence and spread of antibiotic-resistant microorganisms [9]. Additionally, Article 5 of Annex III of the Protocol on Environmental Protection to the Antarctic Treaty permits the discharge of liquid waste and sewage into ocean waters [10, 11]. This practice poses a substantial risk of introducing mobile genetic elements and antibiotic-resistant bacteria (ARB) linked to human activity into Antarctic waters, potentially facilitating their spread to local wildlife [12].
Understanding the extent to which human activities contribute to the dissemination of antibiotic resistance genes (ARGs) and AMR is crucial, particularly in remote environments [13]. These isolated regions offer valuable model ecosystems for investigating the fundamental processes of AMR evolution, including its de novo emergence, acquisition, and transmission. Furthermore, microorganisms that thrive in such extreme conditions have naturally developed resistance to various environmental stressors, including radiation, high salinity, nutrient scarcity, and low temperatures [14]. Research has demonstrated that human interventions can alter natural microbial communities, leading to the selection of antibiotic-resistant strains against multiple antibiotics [15, 16, 17, 18]. For example, an analysis of Antarctic soil samples revealed the presence of 79 ARG subtypes associated with 12 distinct antibiotic classes, with polypeptide and multidrug resistance genes being the most prevalent. The composition, abundance, and potential for horizontal gene transfer of ARGs in these microorganisms differed significantly from those reported in activated sludge, poultry waste, and swine feces. Notably, ARG subtypes such as bacA, ceoB, dfrE, mdtB, amrB, and acrB were detected at higher frequencies than other ARG variants in Antarctic soils. This study further revealed that approximately 60% of ARGs are resistant through efflux mechanisms, whereas only approximately 16% are likely to be located on plasmids [19].
In a study by Calisto et al. [20] in 2024, they examined three Pseudomonas spp. isolates, C1-4-7, D2-4-6, and M1-4-11, from Antarctic soil, and all the isolates exhibited resistance to ampicillin, amoxicillin-clavulanic acid, cefuroxime, cefotaxime, ertapenem, trimethoprim, chloramphenicol, and bacitracin. Additionally, two-thirds of the isolates were resistant to ceftriaxone, sulfafurazole, and cotrimoxazole. In another recent study focusing on the most human-impacted areas of the Antarctic Peninsula, an analysis of 137 fresh and cloacal fecal samples from various bird and marine mammal species revealed antibiotic resistance in 80% of the isolates. Most of these resistant bacteria belong to the gastrointestinal microbiota, including Enterobacteriaceae and Enterococcus species. The study confirmed that antibiotic-resistant bacteria were prevalent among both penguins and pinnipeds in the region [12].
Bacillus spp. are utilized in several biotechnological applications, including probiotic dietary supplements for people and inoculants for animal feed, owing to their capacity to enhance the immune system [21, 22], and they generate antibacterial chemicals that suppress harmful germs [23, 24]. There is increasing public health concern over the potential of microbial cultures utilized as dietary supplements or in food production to serve as sources for the transmission of antibiotic resistance genes. Concern over the spread of antibiotic resistance genes is increasing, and little is known about their resistance to different antibiotics. In a study by Adimpong et al. (2012) [21], 85 strains of Bacillus species, including Bacillus subtilis subsp. subtilis (n = 29), Bacillus licheniformis (n = 38), and Bacillus sonorensis (n = 18), were recovered from starters used to produce Sudanese bread. The MICs of eight antibiotics were calculated for these bacteria, where the strains were all resistant to streptomycin but susceptible to gentamicin, vancomycin, and tetracycline [24]. Two different methods are used by B. subtilis cells to adapt their membranes to low temperatures. One is when Bacillus subtilis is cultivated aerobically, it uses the fatty acid desaturase gene (Des) to fluidize its membrane when exposed to a decrease in temperature. Des requires molecular oxygen to function, and its expression is controlled by DesK-DesR, a two-component system. Des transcription increases with decreasing temperature and decreases when membrane fluidity is restored [25]. In addition, an increase in low-melting-point anteiso-branched FAs (mostly a-15:0 and a-17:0) is used in long-term membrane adaptation to efficiently fluidize the membrane [26, 27].
The detection of AMR and ARGs in the farthest ocean and pristine environments raises concerns about potential threats to preserved biodiversity, environmental stability, and human health. With these concerns, the present study aimed to assess the presence of AMR bacteria isolated from the Indian sector of the Southern Ocean within the Antarctic Circle. The research involved profiling the isolate against ten commonly prescribed antibiotics, characterizing them through 16S rRNA sequencing and whole-genome sequencing, and conducting a comparative genome-wide analysis with closely related strains to examine the ARG distribution and resistance mechanisms reported between 2014 and 2024.
The Bacillus strain utilized in this study was originally isolated from water samples collected during the 10th Indian Scientific Expedition to the Southern Ocean (39°19′ S, 57°30′ E to 66°38′ S, 76°22′ E). For the present study, the strain was stored at –80 °C and cultured in buffered peptone water, followed by the addition of mannitol yolk polycyxin agar, a nutrient-rich medium conducive to Bacillus sp. growth at 37 °C. A compound microscope (Model no. Olympus BX41, Olympus Corporation, Tokyo, Japan) was used to view the form, size, edge, color, elevation, and surface texture of a colony, and biochemical tests, such as Gram staining, catalase tests, urease tests, coagulase tests, and glucose fermentation, were performed.
The antibiotics tetracycline, ampicillin, norfloxacin, doxycycline, ciprofloxacin, metronidazole, azithromycin, cefixime, cotrimoxazole and meropenem were used in the study procured from Sigma Aldrich (Bangalore, India). The agar well diffusion method was used [28] at a concentration of 5 µg/mL.
For bacterial characterization, DNA extraction was carried out via the Xploregen Soil Kit (manufactured by Xploregen Discoveries Pvt Ltd., Bangaluru, India). An ~1.5 kbp segment of the 16S rRNA gene was amplified via universal primers adopted from a previous study [29]: 16S forward (5′–GGATGAGCCCGCGGCCTA–3′) and 16S reverse (5′–CGGTGTGTACAAGGCCCGG–3′), and polymerase chain reaction (PCR) was conducted. Each PCR (50 µL total volume) contained 1 µL of bacterial DNA template, 30 µL of water, 10 µL of Taq DNA polymerase (3 U/mL), 4 µL of dNTPs (2.5 mM), 10 µL of 10X assay buffer, and 2 µL of each 16S forward and reverse primer (10 pM). The resulting PCR amplicons were purified to remove contaminants and subsequently analyzed via agarose gel electrophoresis. DNA sequencing was performed via the BDT V3.1 Cycle Sequencing Kit (Thermo Fisher Scientific, Mumbai, Maharashtra, India) and an ABI 3130xl Genetic Analyzer for both forward and reverse sequencing reactions. The genomic sequence of the strain was determined via the National Centre for Biotechnology Information (NCBI) database, and BLAST-n (BLAST+ 2.15.0) was employed to identify closely related strains [29, 30, 31].
For whole-genome sequencing, a Kapa Hyper Plus Kit (Roche Dia, Cape Town, South Africa) was used, and the extracted DNA samples were fragmented via the KAPPA fragmentation system (Roche Dia, Cape Town, South Africa). The fragmented DNA underwent end repair and A-tailing via the Hyper plus ERAT enzyme mixture (Roche Dia, Cape Town, South Africa). Adapter-ligated samples were subjected to library amplification via Illumina primers, followed by purification with AMPure beads (Beckman Coulter, Brea, CA, USA). The libraries were quantified via the Qubit dsDNA High Sensitivity Assay Kit (Fisher Scientific (Waltham, MA, USA). Sequencing was performed on the Illumina HiSeq 4000 platform (Illumina, San Diego, CA, USA), and genome assembly was conducted via Unicycler software v. 0.4.8 [32]. The annotation of the assembled genome was performed via the online tool Rapid Prokaryotic Genome Annotation (PROKKA) v. 1.14.6 [33]. The whole-genome sequence of Bacillus subtilis was submitted to the GenBank database, NCBI with SOI-28 as the strain name and accession number JBDUTP000000000. The phylogenetic tree was constructed by retrieving whole-genome sequences of Bacillus subtilis via the online database Pathosystems Resource Integration Center (PATRIC), which was last accessed on August 6, 2024. Additionally, Reference Alignment-Based Phylogeny Builder (ReaLPHY) v1.12 [34], which utilizes Bowtie2 (v2.2) for sequence alignment, was employed to construct the phylogenetic tree [35].
BLASTn analysis was performed on the isolated B. subtilis SOI-28 to evaluate its genetic similarity with other strains. A total of 10,763 high-quality genome sequences of B. subtilis isolates submitted between 1887 and 2024 are available in the Pathosystems Resource Integration Center (PATRIC) database. Among these, 233 sequences were from isolates submitted between 2014 and 2024. The good-quality assembled FASTA sequences for these 233 B. subtilis strains were retrieved, and 62 genomes were further selected on the basis of specific criteria, including sequencing platforms (Illumina MiSeq, HiSeq, and NovaSeq) and assembly methods (Unicycler, SPAdes, and Shovill). The traces of AMR genes in the retrieved genomes, including 15 closely related and high-quality genomes identified from the phylogenetic tree, were examined via the Comprehensive Antibiotic Resistance Database (CARD) 3.3.0. Only strict and perfect hits were considered to ensure result accuracy [36]. Additionally, a comprehensive genome-wide comparison was conducted via the Circular Gene Viewer (CGView) online tool (last accessed on August 22, 2024) to analyze genomic similarities and differences. Particular attention has been given to the distribution and spread of resistance genes among B. subtilis strains [37, 38].
Antibiotics are introduced into the environment through pharmaceutical industrial waste [39] and via human and animal excreta from individuals undergoing antibiotic treatments [40]. Traces of antimicrobial residues in water pose significant environmental and public health risks, as these compounds can accumulate in aquatic organisms. Additionally, the widespread use of antimicrobials in the fisheries, poultry, and dairy industries contributes to antibiotic contamination, creating potential pathways into various food chains and, ultimately, the human body [41]. Aquatic environments serve as reservoirs for antibiotic-resistant microorganisms, facilitating their proliferation and dissemination. Furthermore, resistance genes can integrate into naturally occurring bacterial ecosystems, increasing the risk of horizontal gene transfer among human, animal, and environmental microbiomes [42].
Morphologically, the strain exhibits characteristics typical of B.
subtilis, forming cylindrical rod cells, with individual cells measuring
approximately 0.7 to 3.0 µm long. Colonies appear opaque in a
creamy-white color on nutrient agar and develop a characteristic purple color on
Mannitol Yolk Polymyxin Agar due to mannitol fermentation. Biochemically, the
strain is gram-positive, displays positive catalase activity, and is coagulase
positive. These features confirmed the identity of the isolate as a cold-adapted
strain of Bacillus subtilis capable of thriving in the unique conditions
of the Southern Ocean (Table 1). Antibiotic susceptibility testing for
Bacillus subtilis SOI-28 was performed via the agar well diffusion
method against ten commonly prescribed antibiotics. The results indicated that
the isolate displayed resistance to certain antibiotics. The average diameter of
the inhibition zones for B. subtilis SOI-28 was measured at a
concentration of 5 µg/mL for each antibiotic. These findings suggest
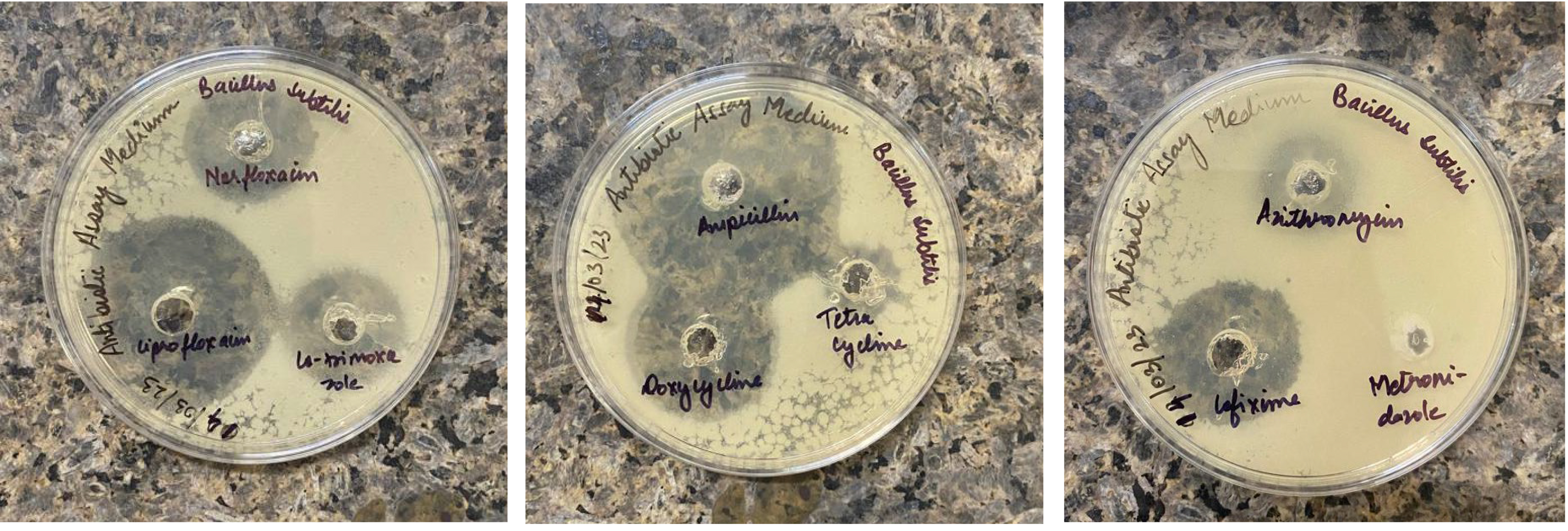
that B. subtilis SOI-28 exhibited 100% antibiotic resistance to
metronidazole (Fig. 1). The susceptibility patterns of B. subtilis
SOI-28 were as follows: ampicillin (zone—44.4 mm)
| S. No. | Morphological characteristics | Observed characteristics | Biochemical characteristics | Observations |
| 1. | Colony shape | Rods | Indole test | Negative |
| 2. | Margin | Irregular | Catalase test | Positive |
| 3. | Size of colony | 0.7–3.0 µm long | Urease utilization | Negative |
| 4. | Elevation | Flat | Citrate utilization | Positive |
| 5. | Surface | Rough & opaque | Coagulase test | Positive |
| 6. | Color | Purple-pink color on MYPA | Glucose fermentation | Positive |
| 7. | Motility | Nonmotile | Mannitol fermentation | Positive |
| 8. | Spore formation | Spore forming | Fructose fermentation | Positive |
| 9. | Gram Staining | gram-positive | - | - |
MYPA, Mannitol Yolk Polymyxin Agar.
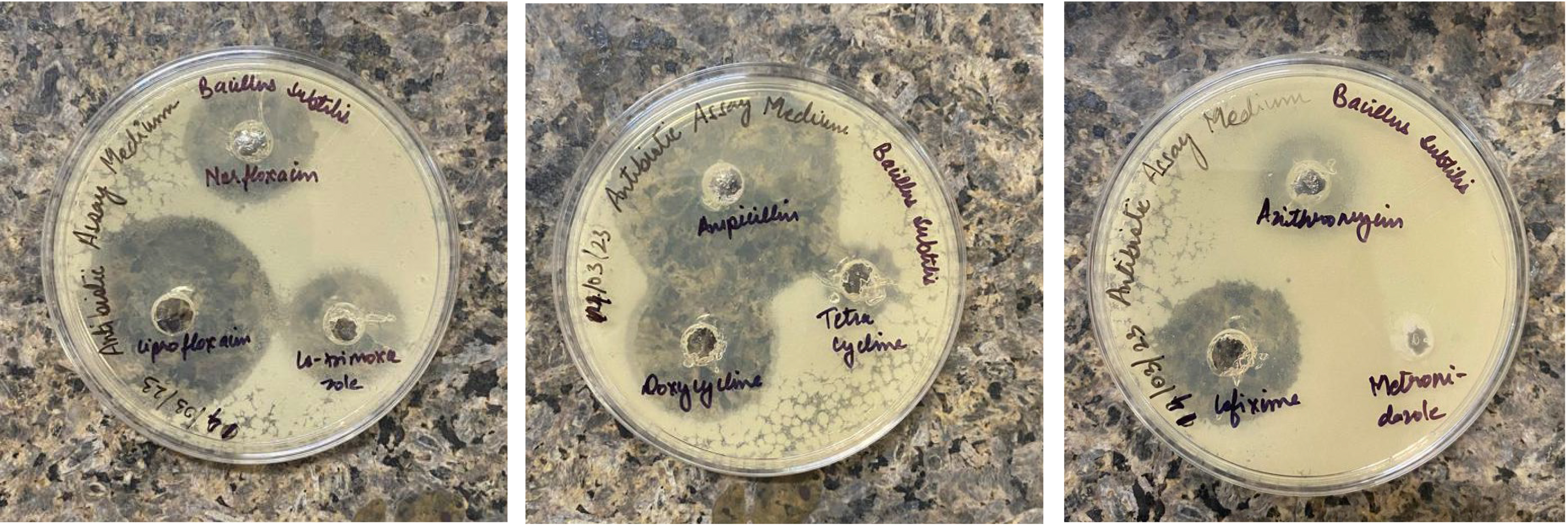 Fig. 1.
Fig. 1.
Zone of inhibition (in mm) for Bacillus subtilis SOI-28. This image depicts the zone of inhibition for B. subtilis SOI-28 (NCBI Accession Number: JBDUTP000000000) against ten therapeutically used antibiotics obtained by agar well diffusion. The inhibition zones are cefixime, 20.6 mm; metronidazole, 0 mm; cotrimoxazole, 16.4 mm; norfloxacin, 20.2 mm; doxycycline, 36.2 mm; ciprofloxacin, 34.6 mm; ampicillin, 44.4 mm; tetracycline, 10.2 mm; azithromycin, 10.4 mm; and meropenem, 28.6 mm.
Whole-genome sequencing (WGS) data for Bacillus subtilis SOI-28, along with genomes retrieved from the PATRIC database, were utilized for phylogenetic analysis. The whole-genome-based phylogenetic assessment revealed that B. subtilis SOI-28 exhibited close genetic similarity to the reference strain SHOB00000000 Bacillus subtilis A1, which was isolated from the maize rhizosphere in South Africa, and JBCNOU000000000 Bacillus subtilis SMAGNO135, which was obtained from sugarcane leaves, stems, roots, and rhizosphere soil in China (Fig. 2).
 Fig. 2.
Fig. 2.
Whole-genome sequence-based phylogenetic tree for Bacillus subtilis SOI-28. The figure shows the phylogenetic tree generated using the RealPHY tool based on whole-genome alignment of 62 Bacillus subtilis genomes, including the isolate SOI-28 and was further visualized and annotated using the Interactive Tree of Life (iTOL) tool. Bacillus subtilis SOI-28 is highlighted within the tree to indicate its phylogenetic position.
The Bacillaceae family is widely distributed across diverse natural
environments, with habitats ranging from soil and sediment to extreme and
unconventional locations, including clean rooms at the Kennedy Space Center,
vaccine production facilities, and even human blood [43, 44, 45].
Bacillus species are rod-shaped, aerobic bacteria capable of staining as
either gram-positive or gram-negative bacteria and are widely recognized for
their ability to produce resilient spores, which opposes cold, heat and common
disinfectants, enabling survival in diverse ecosystems [46]. The
Bacillus genus includes more than 200 species, most of which are
nonpathogenic and have been widely employed in biotechnological and industrial
processes [47, 48]. Only a small number of species in this genus are known to be
pathogenic to humans and animals [49]. Numerous studies have reported the
presence of Bacillus species in deep-sea environments [50, 51, 52, 53, 54]. Furthermore, members of the Bacillaceae family have been found in
marine and freshwater environments, human and animal systems, activated sludge
and diverse food sources, including fermented products. They have also been
detected in inhospitable settings such as hot solid and liquid systems (e.g., hot
springs and compost), salt lakes, and salterns [55, 56, 57, 58]. While
Bacillus species are capable of producing heat-resistant spores, they
are typically not associated with human infections. Notably, Bacillus
subtilis and Bacillus velezensis have garnered considerable attention
in the food industry because of their established safety and competitive ability
against other microorganisms in natural environments, which may influence
microbiota selection [59]. The whole-genome sequence of Bacillus
subtilis SOI-28 revealed that the strain had 43.44% Guanine-Cytosine (GC) content and a genome
size of ~3,990,483 bp. genome coverage—422.38
Table 2 in the PATRIC metadata provides detailed information on 15 Bacillus subtilis strains, including their strain names, isolation sources, sequencing platforms, assembly methods, genome sizes, and GC contents. The strains were isolated from diverse sources, including the maize rhizosphere (n = 1), cell culture (n = 1), commercial dietary supplements (n = 2), insect gut (n = 1), fish gut (n = 1), plant leaves (n = 2), cleanroom floors (n = 3), soil (n = 2), ocular surface of a healthy individual (n = 1), and sourdough (n = 1). In accordance with the PATRIC database, these genomes were sequenced primarily via the Illumina NovaSeq platform, with some also processed via Illumina MiSeq and HiSeq. The genome sizes of these 15 strains ranged from 3.9 Mb to 4.6 Mb, with the GC content varying between 42.56% and 43.85%.
| S. No. | Strain | GenBank Accession No. | Genome Size (bp) | Contigs | GC Content (in %) |
| 1. | Bacillus subtilis strain A1 | SHOB00000000 | 4,060,396 | 22 | 43.75% |
| 2. | Bacillus subtilis 2896 | JANLFH000000000 | 4,291,432 | 54 | 43.05% |
| 3. | Bacillus subtilis strain DS3_4 | JAHFZW000000000 | 4,000,557 | 65 | 43.79% |
| 4. | Bacillus subtilis strain DS6_4 | JAHFZV000000000 | 4,134,799 | 142 | 43.56% |
| 5. | Bacillus subtilis CB00893 | JAXKII000000000 | 4,215,674 | 42 | 43.35% |
| 6. | Bacillus subtilis strain 179-I 4D1 HS | JAHHXX000000000 | 3,996,990 | 27 | 43.72% |
| 7. | Bacillus subtilis strain NSS | WVRO00000000 | 4,021,428 | 150 | 43.70% |
| 8. | Bacillus subtilis strain MB415 | MQSR00000000 | 4,266,089 | 244 | 43.09% |
| 9. | Bacillus subtilis strain 179-C6.1 HS | JAHHZA000000000 | 4,211,076 | 14 | 43.35% |
| 10. | Bacillus subtilis strain 179-J 10A2 HS | JAHHXN000000000 | 3,995,942 | 12 | 43.76% |
| 11. | Bacillus subtilis TP111 | JAXIVG000000000 | 4,174,638 | 26 | 43.47% |
| 12. | Bacillus subtilis EYE_54 | JAHXNR000000000 | 4,172,904 | 16 | 43.41% |
| 13. | Bacillus subtilis OSU1015-21 | JARKHW000000000 | 4,639,106 | 346 | 42.56% |
| 14. | Bacillus subtilis strain MH1 | PYBK00000000 | 4,091,387 | 60 | 43.49% |
| 15. | Bacillus subtilis SMAGNO135 | JBCNOU000000000 | 3,910,489 | 163 | 43.85% |
*. PathoSystems Resource Integration Centere (PATRIC) Bioinformatics Resource Center (Chicago, IL, USA). GC stands for Guanine-Cytosine content.
The ecosystem of Antarctica is fragile and relatively simple due to its extreme climate and nutrient scarcity, supporting only a limited number of resilient species [60, 61]. Despite these harsh conditions, the region is home to remarkable megafauna, including whales, seals, penguins, and albatrosses, which migrate over vast distances [62]. However, environmental changes and human activities pose significant threats to this unique biodiversity, with humans acting as vectors for diseases and antibiotic-resistant bacteria [63, 64, 65, 66]. The widespread presence of antibiotic residues in the ocean, largely due to human activities, may accelerate the emergence of antibiotic-resistant Bacillus species, posing serious risks to human health [67]. Bacillus subtilis is one of the most extensively studied gram-positive bacteria, primarily serving as a model for cell differentiation and industrial applications. However, its potential virulence remains relatively unexplored [54]. Human activities contribute to the introduction of pharmaceutical residues and ARGs into marine ecosystems through wastewater discharge from healthcare facilities, agriculture, and aquaculture [68, 69, 70]. Many aquatic bacteria possess mobile genetic elements that facilitate the horizontal transfer of ARGs, potentially spreading resistance traits among environmental, commensal, and pathogenic bacteria, including Staphylococci, Bacillus spp., and Escherichia spp. [71]. Research on deep-sea environments has revealed the significant presence of spore-forming Bacillus species. One study reported that nearly 90% of bacterial isolates from deep-sea sediments were associated with Bacillus [72]. Similarly, findings from deep-sea floor samples collected at Site C0020, which is off the Shimokita Peninsula in Japan, revealed that spore-forming gram-positive bacteria from the Bacillales order were dominant in the microbial community [73]. In addition to sediment and water, Bacillus species have also been detected in marine organisms, including sponges, ascidians, and crabs, further underscoring their adaptability and ecological significance in oceanic environments [74].
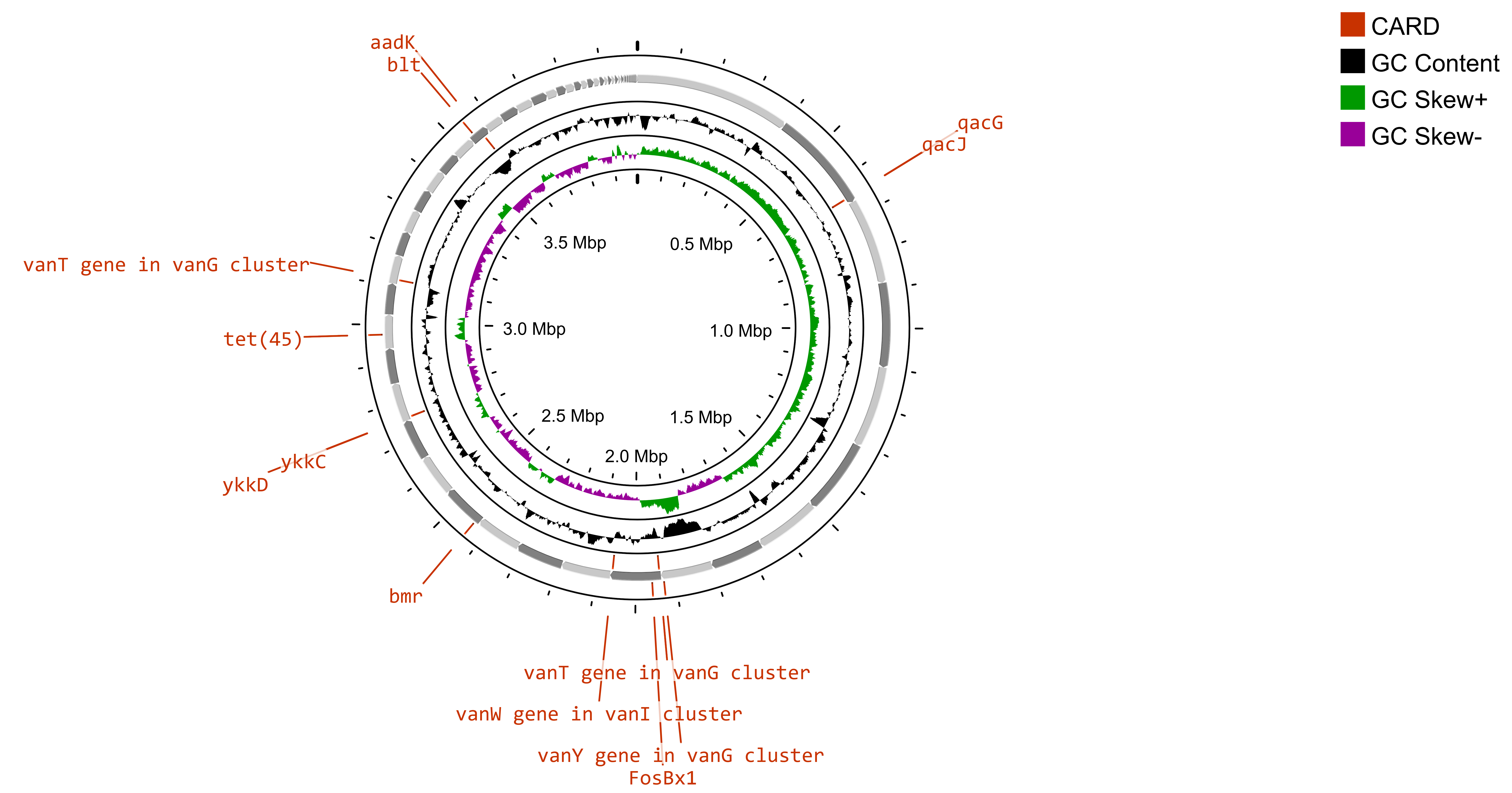
A study has revealed a limited presence of antibiotic resistance in the North Sea, particularly in areas where offshore oil and gas platforms are located [75]. Additionally, previous research has revealed that ballast water from ships can serve as a vehicle for the spread of AMR bacteria and ARGs [76, 77]. The traces of antibiotic resistance genes (ARGs) in Bacillus subtilis SOI-28 were identified via the Comprehensive Antibiotic Resistance Database (CARD), which considers only perfect and strict hits. Bacillus subtilis SOI-28, which was isolated from the Indian Sector of the Southern Ocean, exhibited complete phenotypic resistance to metronidazole, a nitroimidazole drug caused by the presence of nim gene(s). The analysis identified a total of 12 ARGs—blt, qacG, qacJ, and vanY in the vanM cluster; vanT in the vanG cluster; FosBx1 and vanW in the vanI cluster; and ykkD, ykkC, tet(45), and addK spanning multiple drug classes and resistance mechanisms (Supplementary Table 1) in which nim gene(s) were not identified. AMR genes have antibiotic resistance mechanisms, such as antibiotic efflux, antibiotic target alteration and antibiotic inactivation. Gene mapping of B. subtilis SOI-28 revealed three key gene clusters: {qacG, qacJ}, {ykkC, ykkD}, and {vanY (vanM cluster), VanW (VanI cluster)}. Furthermore, the vanT gene from the vanG cluster was found in two distinct locations within the genome, separated by 6,542 base pairs (Fig. 3). In the present study, the identification of AMR genes was conducted via the protein homolog model, which detects protein sequences by comparing their similarity to curated reference sequences via predefined BLASTP bitscore cutoffs. This model applies to any gene that confers resistance solely by its presence in an organism, such as a beta-lactamase gene located on a plasmid. The inclusion of a reference sequence and a bitscore cutoff ensures accurate detection through BLASTP. However, the phenotypic AMR results of Bacillus subtilis SOI-28 were not 100% in accordance with the genotypic results of Bacillus subtilis SOI-28 (Table 3), as Bacillus subtilis SOI-28 showed signs of phenotypic resistance to metronidazole, although no known mutations or resistance genes were found. On the other hand, the tet(45) gene is linked to tetracycline resistance, and the blt gene is linked to fluroquinolone resistance (ciprofloxacin), even though Bacillus subtilis SOI-28 shows phenotypic sensitivity to tetracycline and ciprofloxacin antibiotics. These results show 70% concordance and 30% discrepancy between the phenotypic and genotypic results.
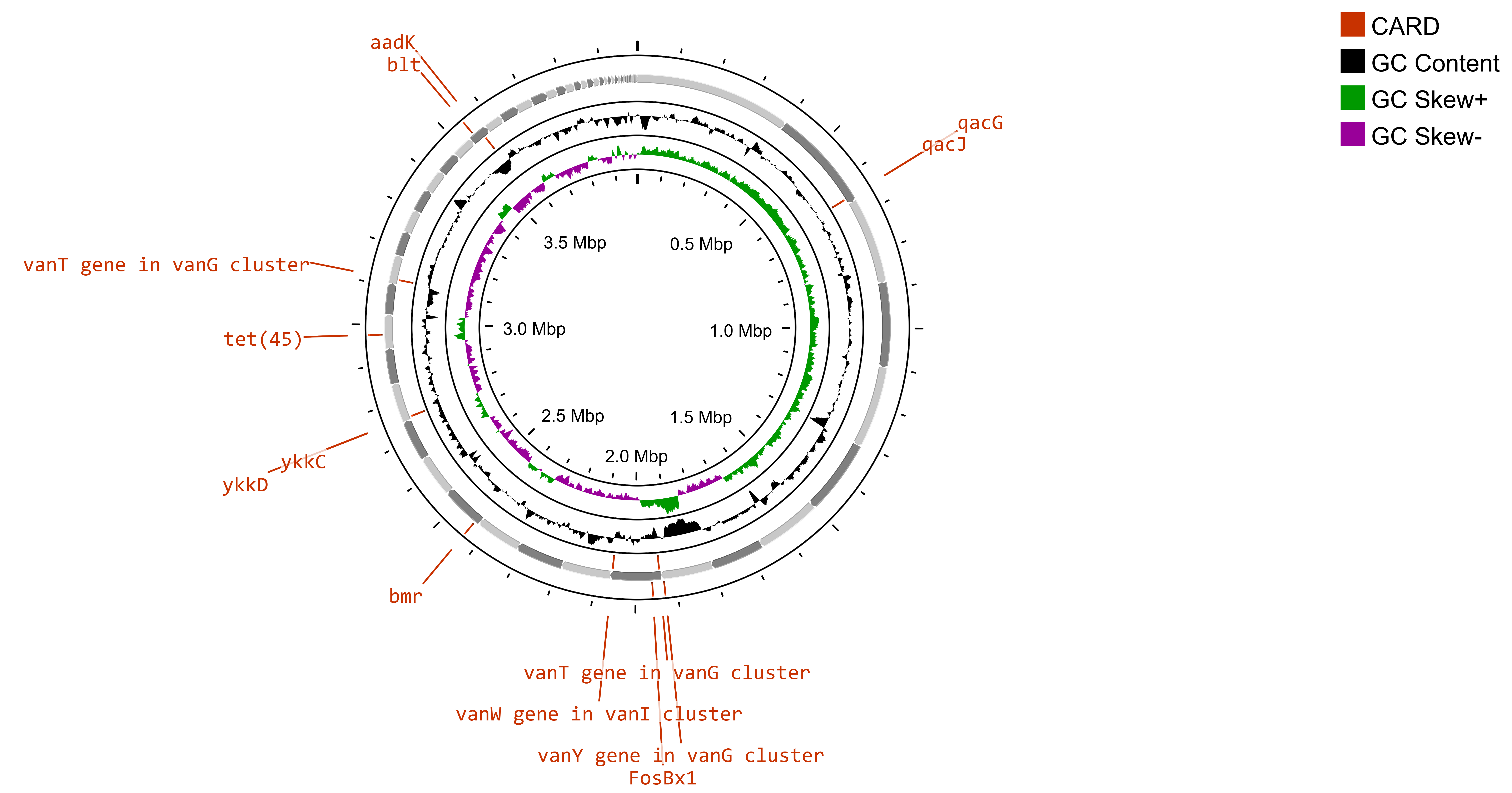 Fig. 3.
Fig. 3.
Circular gene map of AMR genes in Bacillus subtilis SOI-28. The figure shows the presence of 12 ARGs- blt, qacG, qacJ, and vanY in the vanM cluster; vanT in the vanG cluster; FosBx1 and vanW in the vanI cluster; and ykkD, ykkC, tet(45), and addK in Bacillus subtilis SOI-28 (NCBI accession number: JBDUTP000000000) constructed using Circular Genome (CG) viewer.
| S. No. | Drugs class | Antibiotics | Phenotypic Resistance (Bacillus subtilis SOI-28) | Genotypic Resistance (Bacillus subtilis SOI-28) |
| 1. | Cephalosporin | Cefixime | No | No |
| 2. | Quinolone | Norfloxacin | No | No |
| 3. | Penicillin | Ampicillin | No | No |
| 4. | Tetracycline | Doxycycline | No | No |
| 5. | Protein inhibitor | Tetracycline | No | Yes |
| 6. | Macrolides | Azithromycin | No | No |
| 7. | Fluoroquinolone | Ciprofloxacin | No | Yes |
| 8. | Sulphonamides | Cotrimoxazole | No | No |
| 9. | Nitroimidazole | Metronidazole | Yes | No |
| 10. | Carbapenem | Meropenem | No | No |
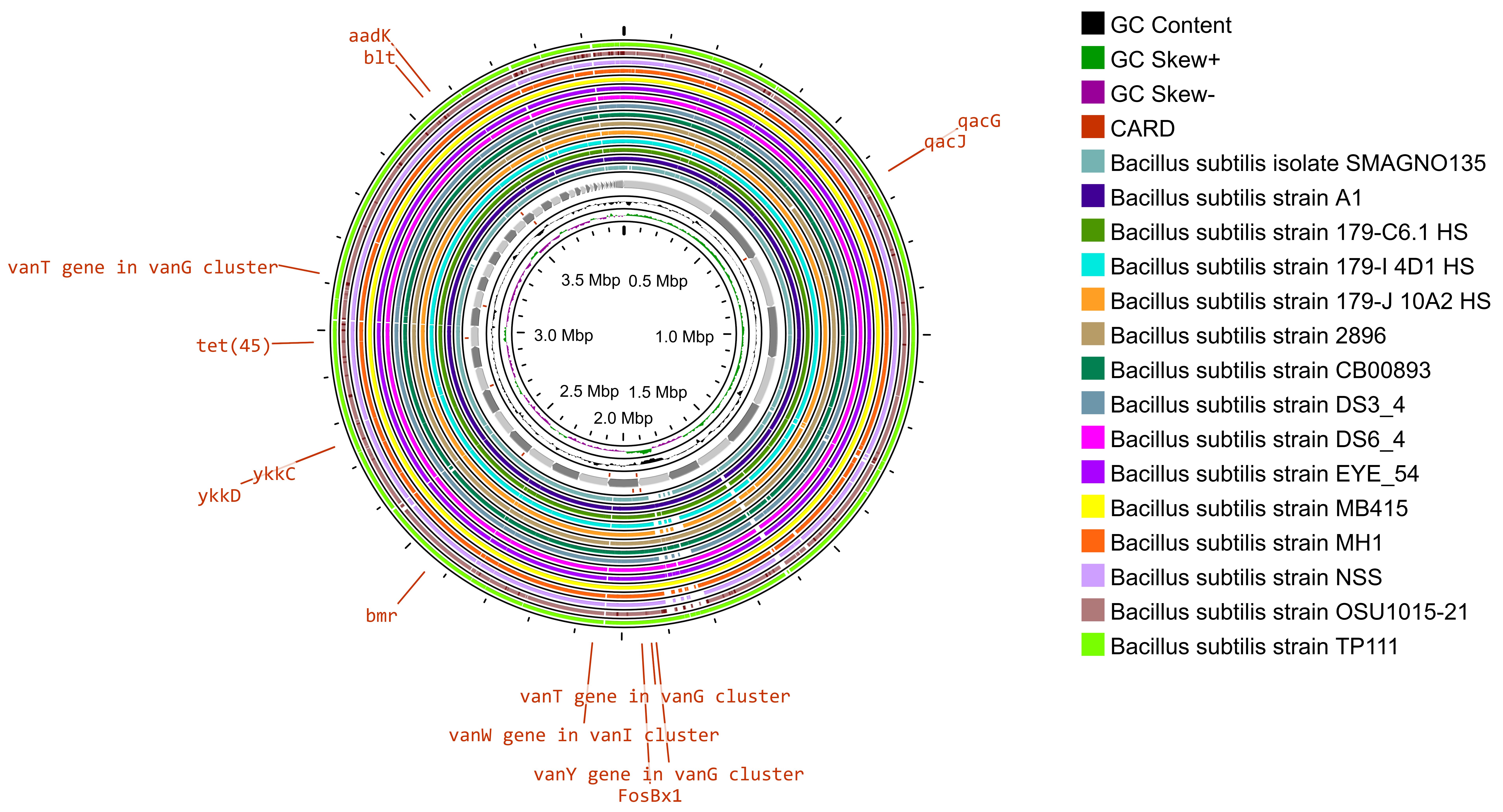
The whole-genome sequence of Bacillus subtilis SOI-28 (reference genome) along with 15 good-quality whole-genome sequences of Bacillus subtilis strains retrieved from the PATRIC database were used for comparative analysis (Fig. 4).
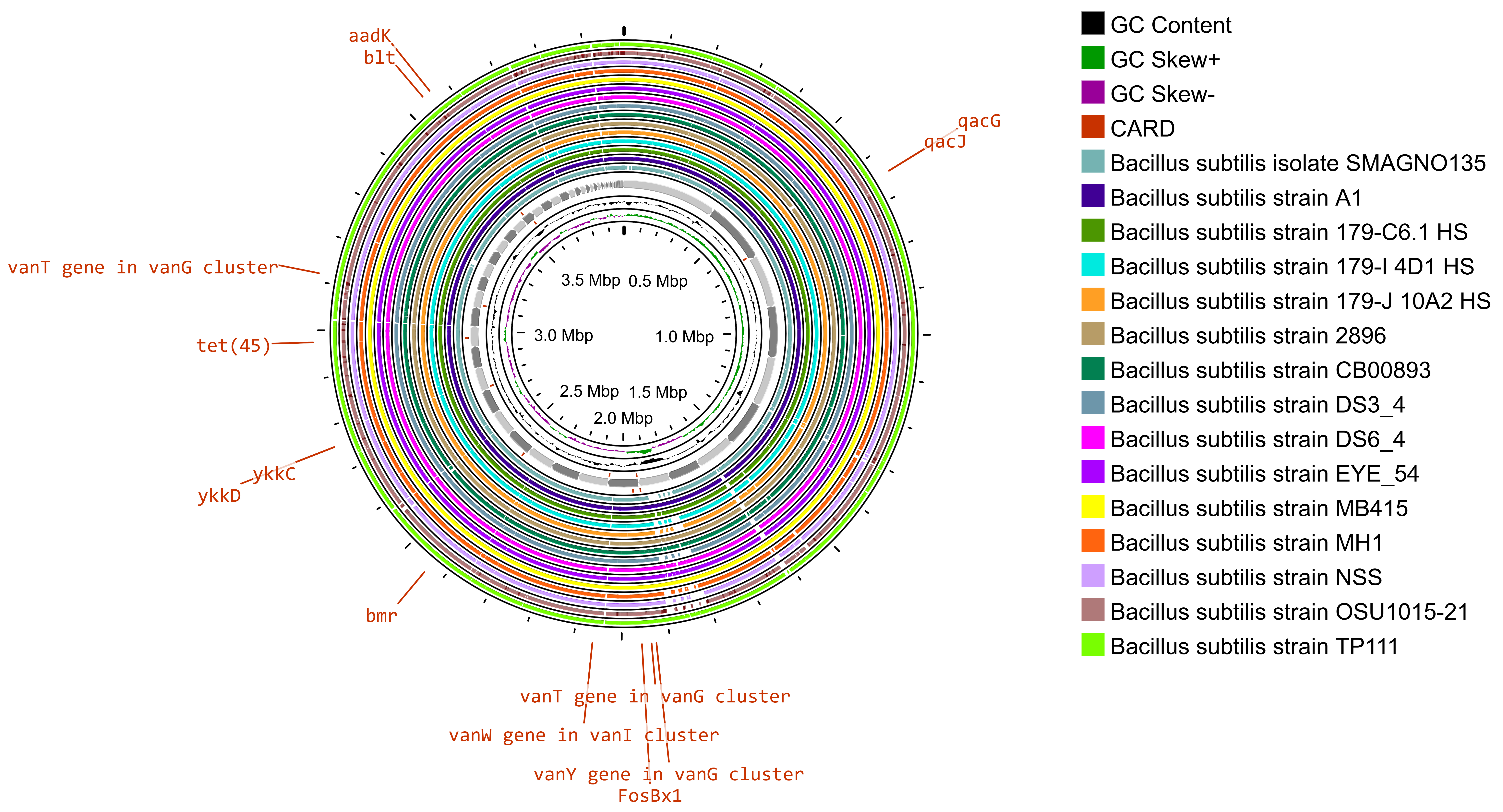 Fig. 4.
Fig. 4.
Gene map providing a comparative analysis of Bacillus subtilis SOI-28 against other Bacillus subtilis genomes. The innermost ring represents the reference genome sequence of B. subtilis SOI-28. The next 15 concentric rings illustrate regions with sequence similarity, as determined through BLAST comparisons between Bacillus subtilis SOI-28 (query genome) and 15 selected B. subtilis genomes. The outermost ring highlights the antibiotic resistance (AMR) genes identified in the reference genome on the basis of the comprehensive antibiotic resistance database (CARD) results.
Heatmapping was performed to analyze the distribution of AMR genes in 15 selected genomes. Through CARD database analysis, Bacillus subtilis SOI-28 was found to have twelve antibiotic resistance genes, which were selected for AMR profiling across the other 15 genomes. The highest resistance genes found were aadk 100% (n = 15); the vanT gene in vanG cluster 100% (n = 15) in two regions; ykkC 100% (n = 15); ykkD 100% (n =15); the vanW gene in vanI cluster 100% (n = 15); FosBx1 100% (n = 15); qacJ 100% (n = 15) and qacG 100% (n = 15) followed by tet(45) 66.67% (n = 10); and the vanY gene in vanM cluster 53.34% (n = 7) and blt 40% (n = 6). The findings indicated that the genomes included at least nine (n = 3) and a maximum of twelve (n = 1) AMR genes (Fig. 5).
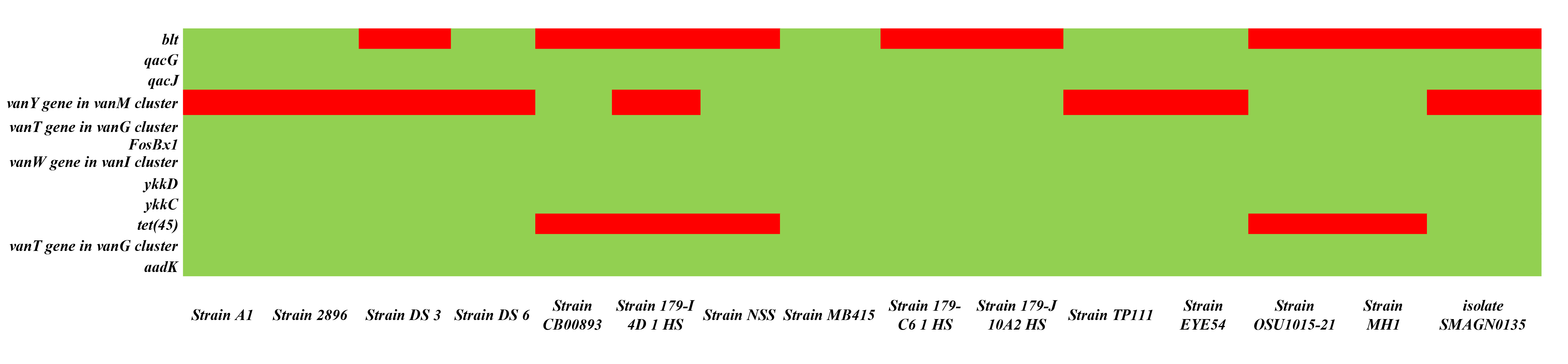 Fig. 5.
Fig. 5.
Heatmap depicting the presence of 12 AMR genes, including blt, qacG, qacJ, and vanY in the vanM cluster; vanT in the vanG cluster; FosBx1 and vanW in the vanI cluster; ykkD, ykkC, and tet(45); and addK across different Bacillus subtilis strains. The green color depicts the existence of the gene, and the red color depicts the absence of the gene (The X-axis represents the different Bacillus subtilis strains, and the Y-axis represents AMR genes present in Bacillus subtilis SOI-28).
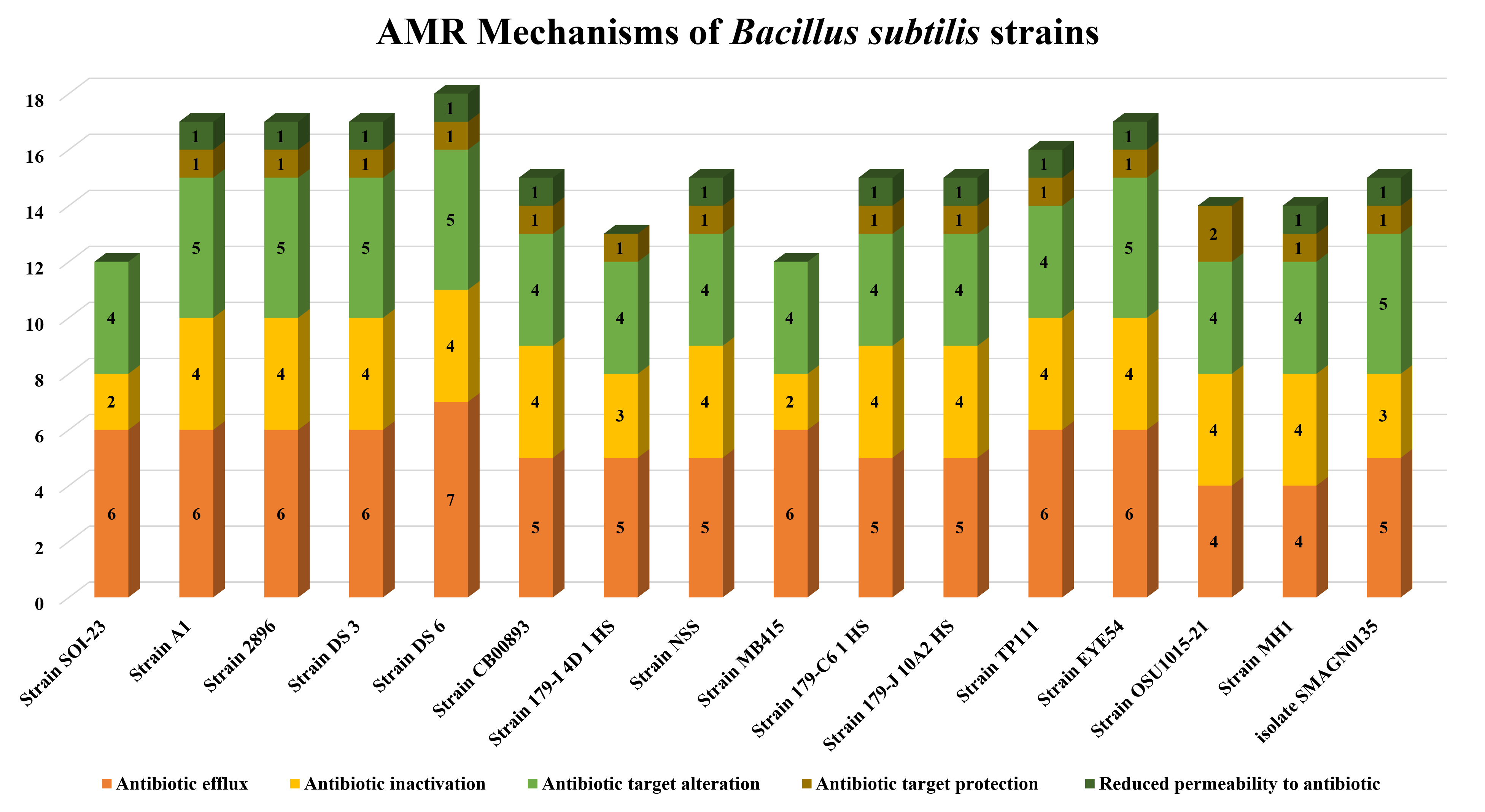
According to the CARD database results, the most common drug resistance mechanisms are antibiotic efflux (100%, n = 15), antibiotic inactivation (100%, n = 15), antibiotic target alteration (100%, n = 15), antibiotic target protection (93.34%, n = 14), and reduced permeability (80%, n = 12) (Fig. 6). The primary gene families associated with the antibiotic efflux resistance mechanism are the MFS and the SMR antibiotic efflux pump. Similarly, in the case of antibiotic alterations, vanY, the glycopeptide resistance gene cluster, vanT, and vanG were the top gene families observed across all the genomes (n = 15). The AMR genes identified in Bacillus subtilis SOI-28 included blt, qacG and qacJ; the vanY gene in the vanM cluster; the vanT gene in the vanG cluster; FosBx1; the vanW gene in the vanI cluster; ykkD and ykkC; and tet(45) and aadK, which were screened for their prevalence among 15 PATRIC-retrieved genomes. The B. subtilis strain MB415 was found to have all twelve AMR genes belonging to different antibiotic classes, representing 6.67% of the studied genomes.
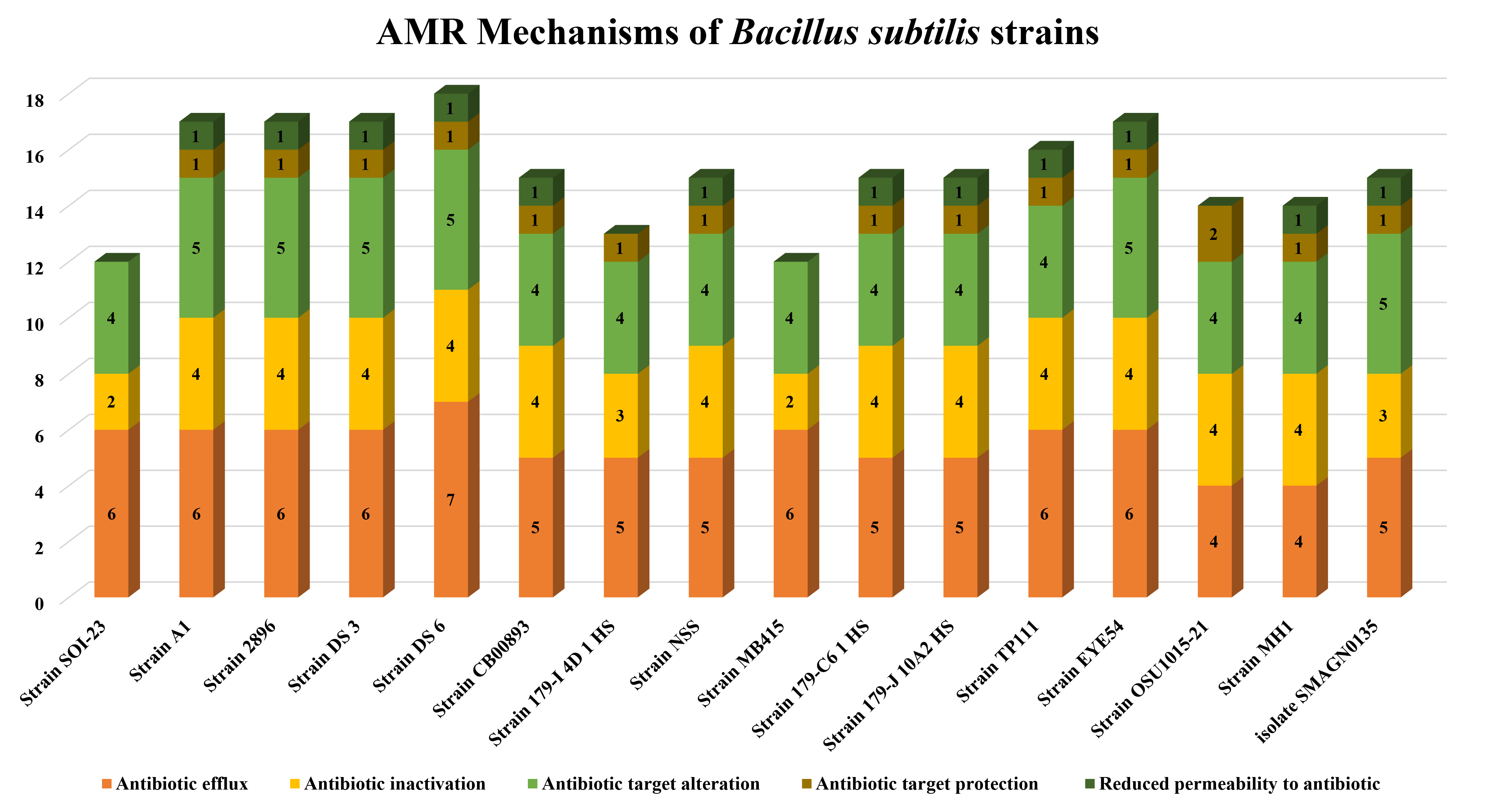 Fig. 6.
Fig. 6.
AMR genes variation. The figure shows variations in the presence of AMR genes with antibiotic resistance mechanisms- antibiotic efflux; target alteration, replacement and protection; and antibiotic inactivation present in Bacillus subtilis SOI-28 (reference sequence) across 15 different Bacillus subtilis genomes retrieved from PATRIC (The X-axis represents the different Bacillus subtilis strains, and the Y-axis represents the number of AMR genes).
Saikat et al. [78] in 2024 conducted a comprehensive phenotypic characterization and whole-genome analysis of Bacillus subtilis BDSA1 obtained from polluted river water in Dhaka, Bangladesh, and the findings revealed that B. subtilis BDSA1 exhibited resistance to streptomycin, cefotaxime, and ceftriaxone while remaining susceptible to imipenem, meropenem, trimethoprim, gentamycin, and ampicillin. Similarly, in 2021, Cardoso et al. [79] conducted a study to isolate and characterize bacterial species from deep water samples collected from three artesian wells, and the susceptibility profile was assessed against antinicrobials. This study identified four bacterial isolates: Bacillus pumilus, Bacillus subtilis, and two strains of Bacillus megaterium. All the isolates demonstrated sensitivity to all the tested antimicrobials, including amikacin, vancomycin, meropenem, oxacillin, imipenem, cefoxitin, and sulbactam with ampicillin. These findings suggest that microorganisms from natural environments tend to be inherently sensitive to antibiotics, reinforcing the ecological role of these bacteria in antimicrobial interactions [79].
Similarly, in an investigation by Haque et al. [80] in 2024, the phenotypic and genotypic resistance profiles of Bacillus spp. were evaluated, and the phenotypic analysis revealed that all Bacillus isolates (n = 191) were 100% sensitive to gentamicin. However, reduced sensitivity was observed for levofloxacin, ciprofloxacin, clindamycin, amoxicillin-clavulanic acid, azithromycin, tetracycline, nitrofurantoin, cotrimoxazole, and erythromycin. The isolates exhibited complete resistance to penicillin, amoxicillin-clavulanic acid, cefixime, ceftriaxone, vancomycin, and cotrimoxazole. A distinct species-specific pattern was evident in both the phenotypic and genotypic resistance profiles. The genotypic analysis revealed the presence of multiple resistance genes, with bla1 detected in 71.5% of the isolates, tetA in 33%, erm1 in 27%, blaTEM in 13.1%, blaCTX-M-1/blaCTX-M-2/sul1 in 10.1%, blaSHV in 9.6%, and qnrS in 4.1%. Notably, the bla1 gene was present in all the isolates that were resistant to penicillin. Among the 191 isolates analyzed, 89.6% were classified as multidrug resistant (MDR), with the MDR prevalence reaching 100% in isolates from diarrheal cases, 90.3% in food-derived isolates, and 88.7% in animal feed-derived isolates [80]. AMR in psychrophilic bacteria has received relatively little research attention to date, and B. subtilis isolated from the Southern Ocean has not yet been studied.
The presence of AMR Bacillus subtilis in the Indian sector of the Southern Ocean, Antarctica, highlights the potentially alarming intrusion of anthropogenic contaminants into one of the most pristine environments on Earth. The detection of AMR bacteria and associated resistance genes (ARGs) in this remote ecosystem suggests multiple pathways of pollution, including human activities at research stations, wastewater discharge, and the long-range transport of resistant microbes through ocean currents and migratory species. The persistence and dispersal of AMR in such isolated regions pose significant ecological risks, potentially disrupting native microbial communities and altering biogeochemical cycles. To mitigate this emerging threat, strict biosecurity measures must be enforced, including enhanced decontamination protocols for research personnel and equipment, improved waste management strategies, and rigorous monitoring of microbial resistance threats in Antarctic ecosystems. Investigating the pathways through which AMR spreads in aquatic environments and the potential for horizontal gene transfer among different bacterial species is essential for understanding its broader ecological impact. Regular monitoring of antibiotic-resistant organisms in drinking and potable water supplies is critical to safeguarding public health and minimizing AMR-related infections. Additionally, international cooperation is crucial for establishing comprehensive AMR surveillance programs and promoting responsible antibiotic use worldwide. These proactive interventions are essential for safeguarding the ecological integrity of Antarctica and preventing the unchecked spread of AMR in the Southern Ocean.
The widespread use of antibiotics in the healthcare, poultry, fisheries, and dairy industries has contributed to their environmental dissemination, not only in local water bodies but also in distant and pristine marine ecosystems. The emerging threat of antibiotic resistance in these remote regions poses a significant ecological threat. Previous studies reported antimicrobial resistance in various Bacillus species, such as B. cereus, B. pumilus, and B. subtilis. In our study, the isolate B. subtilis, strain SOI-28, was approximately 0.7–3.0 µm in length and had an opaque, creamy-white appearance on nutrient agar and a pink color colony on MYPA. Biochemically, the isolate was gram-positive, catalase-positive, and coagulase-positive. Antibiotic susceptibility testing revealed complete phenotypic resistance to metronidazole, a nitroimidazole-class drug, and susceptibility to cefixime, tetracycline, ampicillin, azithromycin, cotrimoxazole, meropenem, doxycycline, ciprofloxacin and norfloxacin. However, genotypic analysis revealed the absence of nim gene(s), which are responsible for metronidazole resistance, and the presence of the ykkD, ykkC and tet(45) genes, which are responsible for tetracycline resistance. Genome-wide analysis of Bacillus subtilis SOI-28 revealed twelve resistance genes, namely, the aadK and vanT genes in the vanG cluster; the ykkC, ykkD, and vanW genes in the vanI cluster; and the FosBx1, qacJ, qacG, tet(45), and vanY genes in the vanM cluster and the blt. Additionally, this study revealed the presence of twelve resistance genes in other closely related B. subtilis strains, with genomes containing a minimum of nine AMR genes and a maximum of twelve AMR genes. Among the resistance mechanisms identified, antibiotic efflux and target modification were the most prevalent, while antibiotic inactivation was also observed. Our results highlight the need for AMR monitoring in distant maritime areas and the possible human effects on microbial resistance in these settings. The identification of AMR genes in our Bacillus subtilis isolate SOI-28 suggests potential pathways for resistance gene transfer, which can affect marine biodiversity and pose unexpected risks to public health through ecological and food chain interactions. The current study also highlights the need for continued research on AMR mechanisms, its introduction into untouched environments, and its evolutionary behavior, which is crucial for controlling the spread of resistance and minimizing its effects on both environmental and human health.
This research paper presents a comprehensive overview of the experiments conducted and the findings obtained only from the current study. The whole genome sequence of the resistant Bacillus subtilis strain SOI-28 has been deposited in the National Centre for Biotechnology Information (NCBI) GenBank database under the accession number JBDUTP000000000. Additional details are provided in Table 1 of the Supplementary Material. Further details can be obtained upon reasonable request from the corresponding author.
Conceptualization: SS, AC, AR and VNS; methodology, data collection and original data analysis and data presentation: SS, AC, AR, RS, RC and TJ; writing, reviewing and editing: SS, AC, AR, VNS and TJ. All authors have read and agreed to the final version of the manuscript. All authors contributed to editorial changes in the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
According to Indian regulations and policies, this study does not involve human or animal subjects; therefore, ethical committee approval is not necessary.
The authors are grateful to the Director, NCPOR, Goa, India for supporting our participation in the 10th Indian Scientific Expedition to the Southern Ocean. The authors express their sincere gratitude for this support.
This research was conducted with the financial assistance of the Ministry of Earth Sciences (MOES), Govt. of India (MOES/REACHOUT/CNA/2022).
All the authors declear no confilict of interests. Dr. Anuj Ranjan, given his role as the Editorial Board member in the journal FBE, had no involvement in the peer-review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Baohong Zhang.
The writers used ChatGpt to proofread their work for grammatical and spelling errors. The writers take full responsibility for the publication’s content after using this tool, reviewing and editing it as necessary. The tool was used in the introduction and discussion sections.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBE38809.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.