




 , Luis Vitetta 1,2,*
, Luis Vitetta 1,2,*1 Research Department, Medlab Clinical, 2015 Sydney, Australia
2 Faculty of Medicine and Health, The University of Sydney, 2006 Sydney, Australia
Abstract
Inflammatory bowel diseases (IBD) are characterized by chronic inflammation and damage of colonocytes with etiology of genetic, epigenetic and environmental factors. MicroRNA-223 (miR-223) has been found to be increased in both IBD patients and animal colitis models. However, contentious opinions relevant to the roles of miR-223 in IBD have been reported. Notwithstading that most studies have described that miR-223 has anti-inflammatory effects, several reports have progressed a pro-inflammatory view. In this review, we summarise both the anti-inflammatory and pro-inflammatory effects of miR-223 on key molecules in inflammatory responses in both animal models and in patients diagnosed with IBD and objectively discuss the possible basis for the discrepancies.
Keywords
- miR-223
- IBD
- inflammation
- inflammasome
- gut microbiota
Inflammatory bowel diseases (IBD) including ulcerative colitis (UC) and Crohn’s
disease (CD) are common and complex conditions that are difficult to cure. UC
often starts from the rectum and extends to part or whole colon in a continuous
manner with mucosal damage and clinical manifestations are blood diarrhea and
abdominal pain [1]. CD is characterized by transmural damage, which is an
immune-related abnormality and could occur in any part of the gastrointestinal
tract [2, 3]. Multiple factors including genetic, epigenetic, environmental and
intestinal microbiota dysregulation have been attributed to the causes of IBD
[4]. The main characteristic of IBD is the chronic inflammation that ensues with
increased levels of pro-inflammatory cytokines interleukin-6 (IL-6), IL-12,
IL-17, IL-23, IL-1
As proinflammatory responese play crucial roles in the pathogenesis of IBD, anti-inflammatory therapies have been developed [5]. The standard regimens include 5-aminosalicylates, stroids and biologics. The commonly used biologics are anti-TNF-alpha infliximab, anti-integrin vedolizumab and anti-IL6/Janus-Kinase (JAK) signaling Torfacitinib [1]. These have been effective in some patients but response rate is still low, i.e., treatment outcomes are limited and efficacy is often suboptimal. IBD not only decreases quality of life but also increases the risk of colon cancer [6]. Therefore, the requisite is to further investigate the mechanisms of the disease as a prelude to searching for novel therapeutic regimens for curative treatments. Further understanding of each altered factor in IBD could facilitate the development of effective therapeutic approaches for IBD. Newly developing therapeutic approaches have been broad such as signalling pathway inhibitors, gut microbiota modulation and targeting microRNAs (miRs) [7, 8]. For examples, anti-IL-23 antibodies risankizumab, mirikizumab, ustekinumab, anti-Janus-Kinase1 (JAK1) signaling small molecule filgotinib have been in phase2/3 clinical trials [1, 9, 10, 11]. The interactions of these approaches could be intertricate through modulation of inflammatory responses.
The pathogenesis of IBD is still not well elucidated; as such the complexity presents a collective of interactions between genetic defects, immune dysregulation and environmental factors such as perturbations of the gut microbiota [12, 13]. It has been recognised that a dysregulated gut microbiota (gut dysbiosis) could be a crucial causal factor, which together with other factors initiates and or promotes inflammatory disease development. In IBD, the abundance of bacteria from the phyla Firmucutes is reduced while members from the phyla Proteobacteria and Bacteroidetes have an increased abundance [14, 15, 16]. Sokol et al. [14] reported that the Firmucutes to Bacteroidetes ratio was decreased in IBD patients. There was also decreased abundance of the beneficial bacterial genus Bifidobacteria [14] Machiels et al. [15] also identified that butyrate-producing bacteria Fecalibacterium prausnitzii and Roseburia hominis, both from the Firmucutes phyla, were decreased. Frank et al. [16] showed that Enterobacteriaceae family from the phylum Proteobacteria was outgrown. These changes could cause increased intestinal permeability and inflammation [17]. The mechanisms are associated with altered commensal bacterial metabolites such as bile acids, short-chain fatty acids and tryptophan metabolites [18]. Indeed, the decrease in abundance of butyrate-producing bacteria F. prausnitizii and R. hominis [19, 20, 21, 22] could cause decreased levels of butyrate, which is important in anti-inflammatory mechanism and intestinal permeability [23].
With the exception of the involvement of multiple pro-inflammatory signalling pathways, miR alterations have been extensively studied in IBD. Endogeneous miRs have a base length of 19–24 nucleotides that are noncoding single stranded RNAs, which regulate gene expression by binding to the 3’-untranslational regions (3’-UTRs) of mRNAs and degrade the mRNAs [24].
Many miRNAs have been found to be altered in IBD and are associated with the
pathogenesis of IBD [25, 26]. To this, a review has reported on the expanding
research evidence that supports that miRs promote post-transcriptional regulation
of intestinal proinflammatory responses, an important step in IBD cellular
turnover [27]. Wang and colleagues (2018) [28] review has highlighted studies that
have shown that entities such as miR-142-3p, miR-320, miR-192, and miR-122 can
target the Nucleotide-binding oligomerization domain-containing protein 2 (NOD2)
gene [27], an IBD-relevant autophagy and immune system function gene. This
genetic dependent effect modulates autophagy in IBD. Other entities such as
miR-142-3p, miR-93, miR-106B, miR-30C, miR-130a, miR-346, and miR-20a were
reported to regulate inflammation by targeting the autophagy gene (i.e.,
ATG16L1) via several different molecular pathways. For example, miR-196
can downregulate the IRGM gene [28], that codes for a protein that plays
an important role in immunity [29]. In addition, miR-196 suppresses autophagy by
inhibiting the accumulation of LC3II [28]. The review also reports that
endoplasmic reticulum stress response molecular pathways with miR-665, miR-375,
and miR-150 modulate autophagy by regulating the unfolded protein response with
an important role in IBD intestinal fibrosis. Relevance to the release of anti–
or pro–inflammatory cellular factors in autophagy-related pathways, miR-146b,
miR-221-5p, miR-132, microRNA-223 (miR-223), miR-155, and miR-21 have been
reported to regulate NF-
Given that IBD is an immune-mediated digestive/intestinal system inflammatory disease that includes Crohn’s disease (CD) and ulcerative colitis (UC), among the various miRs, miR-223 has a central role. MiR-223 is a crucial regulator of innate immunity, involving myeloid differentiation and neutrophil and macrophage functionality [30], while being highly expressed in neutrophils that effectively limits the activation and function of neutrophils in inflammatory diseases [30]. Uncontrolled immune cellular activation (i.e., myeloid activation) can lead to detrimental consequences in inflammatory disease. Hence, miR-223 serves as a negative feedback mechanism controlling excessive innate immune responses in the maintenance of myeloid cell homeostasis [30]. In addition to inflammatory diseases Yuan et al. [30] have also reported on the involvement of miR-223 in acute respiratory distress syndrome and IBD. An important highlight of current research efforts is the reported therapeutic benefit, that miR-223 has to dampen inflammatory targets as well as the potential treatment to control excessive innate immune responses during mucosal inflammations [30, 31, 32]. Therefore in this review, we summarize the current progress of the role of miR-223 in the pathogenesis of IBD in association with the gut microbiota and discuss the contentious findings.
The increased miR-223 levels in colonic tissues and blood samples have been well
demonstrated in both IBD patients and animal models. Several clinical studies
showed that many miRs were altered in the samples from patients with UC and CD.
Among them miR-223 levels were highly increased [24, 33, 34, 35]. Wang et al.
[35] investigated the blood levels of miR-223 in patients with UC and CD
and found that miR-223 were increased in both UC and CD, which were correlated
with inflammatory indicator C-reactive protein levels. Wu et al.
[24] found that miR-223 levels in CD samples obtained from colonoscopic pinch
biopsies were 8.6 folds of that in samples from healthy controls. The clinical
findings of increased miR-223 in IBD have been confirmed in various animal
colitis models. In the IL-10
Studies with animal colitis models have demonstrated that increased levels of
miR-223 reported a protective effect on colitis. Zhang et al.
[37] demonstrated that administration of miR-223 agomir alleviated DSS-induced
colitis while antagomir aggravated the colitis. Neudecker et al.
[38] reported that deficits of miR-223 aggravated DSS-induced colitis in mice
that expressed an miR-223 gene mutation (miR-223
Many studies have reported that the roles of increased miR-223 levels in the pathogenesis of IBD are mediated by the direct effects of miR-223 on key molecules in multiple signalling pathways. MiR-223 either promotes or inhibits inflammation, leading to controversial opinions about the roles of MiR-223 in IBD. In a recent review miR-223 was reported to be an effective regulator of immune cell differentiation as well as a regulator of inflammatory processes of molecular regulatory networks and treatments for inflammatory diseases this being especially and highly relevant for IBD [32].
NLRP3 (Nucleotide-binding oligomerization domain, Leucine rich Repeat and Pyrin
domain-containing protein 3) inflammasome is a defense mechanism to microbial
infections [40]. It is activated by microbial-associated molecular patterns
(MAMPs) and damage-associated molecular patterns (DAMPs) to elicite innate immune
responses. MAMPs and DAMPs bind to and activate pattern recognition receptors
(PRRs) such as toll-like receptors (TLRs) and nucleotide-binding oligomerization
domain-containing-2 (NOD2), resulting in activation of the NF-
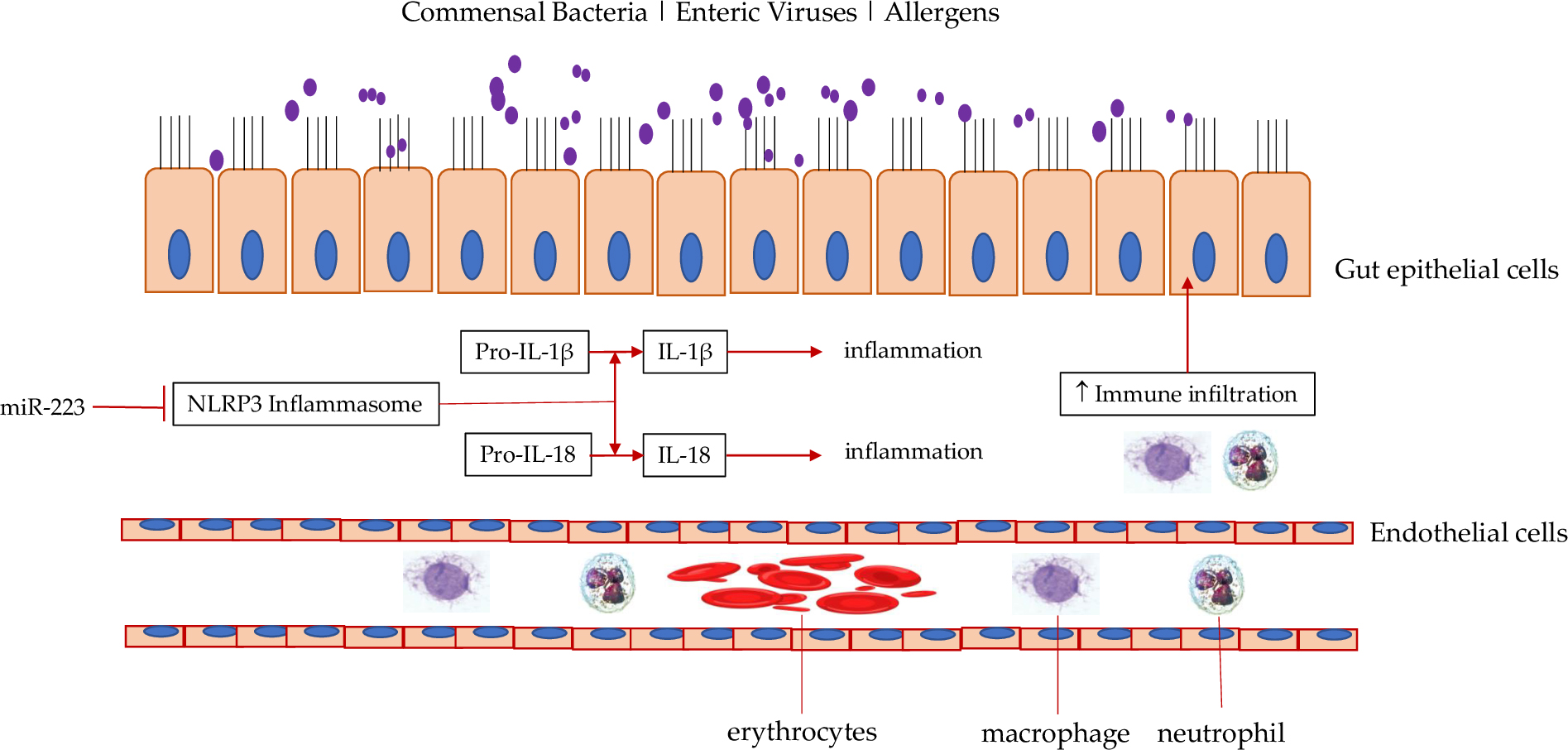 Fig. 1.
Fig. 1.Effect of miR-223 on inflammasome in IBD. MiR-223 can block
NLRP3 inflammasome, which is pro-inflammatory by converting pro-IL-1
An uncontrolled and excessive activation of NLRP3 has been reported to be a
major pathogenetic factor of many inflammatory diseases including IBD and
knockout of NLRP3 improved these diseases [40, 42]. The anti-inflammatory effect
of miR-223 could be explained by its inhibitory effect on NLRP3. MiR-223 reduces
NLRP3 expression by binding to its mRNA 3’-UTR, leading to degradation of NLRP3
mRNAs [41]. Correspondingly, increased miR-223 reduces NLRP3 inflammasome and
pro-inflammatory cytokines IL-1
Inhibition of inflammasome in IBD provides a therapeutic effect. Isoflavone
formononetin can inhibit the NLRP3 inflammasome to reduce intestinal inflammation
in a DSS-induced colitis mouse model [24]. Rev-ErbA proteins are members of the
nuclear receptor family of intracellular transcription factors. As such these
proteins regulate the colon clock, and have been found to suppress NLRP3 via
inhibition of NF-
NF-
The effects of miR-223 on NF-
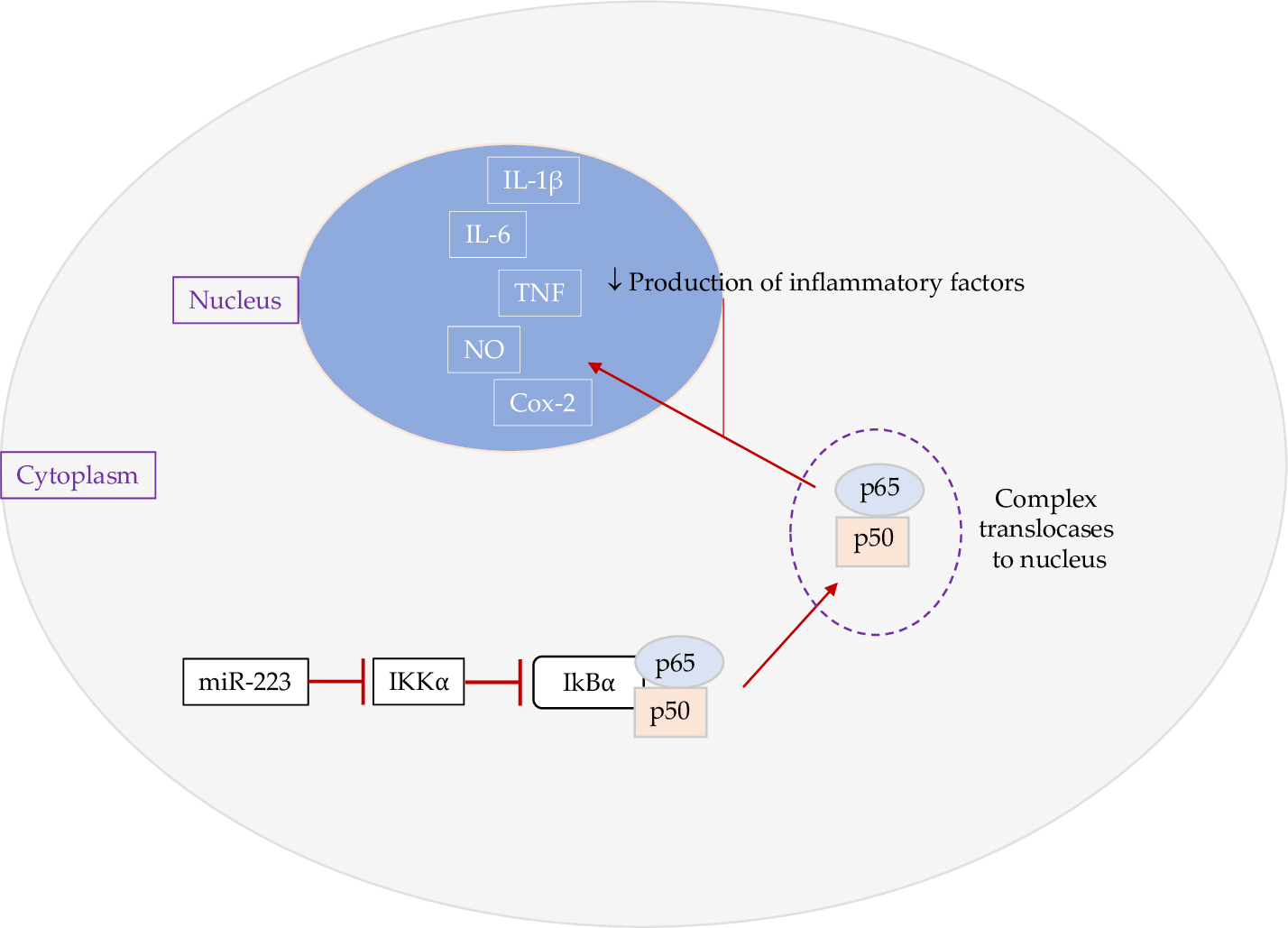 Fig. 2.
Fig. 2.Effect of miR-223 on NF-
IL-6/STAT3 (signal transducer and activator of transcription 3) pathway has been demonstrated to play key roles in the pathogenesis of IBD [54]. It increases macrophage and T cell survivial, proliferation and differentiation and thus increases the production of pro-inflammatory cytokines. Therefore, the IL-6/STAT3 pathway has been targeted for the treatment of IBD. So far, Torfacitinib, an inhibitor of IL-6 downstream signalling molecule JAK, has been approved by the FDA for the treatment of IBD [55, 56].
MiR-223 has been shown to inhibit the IL-6/STAT3 pathway in a mouse colitis
model induced by DSS [37]. MiR-223 agomir decreased MPO, TNF-
The PI3K/Akt pathway has been extensively studied and reported to participate in the regulation of multiple downstream cellular processes [58]. The downstream activation signaling of corresponding effector molecules serve important fuctional activities in the cell cycle, in cellular growth and proliferation. Increased activation of the PI3K/Akt pathway is proinflammatory and oncogenic. It down-regulates anti-inflammatory cytokines such as IL-10 and upregulates pro-inflammatory cytokines such as IL-12, IL-6 and IL-17. Moreover we note that the effect on IL-10 has been shown to exert immunosuppressive functions to reduce tissue damage caused by excess and uncontrolled inflammatory effector responses, especially in microbial host defences [59]; an important effect during the resolution phase of infection/inflammation that is directed at maintaining homeostasis to intestinal bacteria. Several studies showed that the activation of the pathway in IBD and the target therapy against the pathway has been tested in animal models [60, 61, 62].
In TNBS-induced colitis, proinflammatory cytokines IL-6, TNF-
MiR-223 has been shown to be increased in DSS-caused inflammation and AOM/DSS-induced colorectal cancer [63]. The miR-223 levels in colon cancer cells were inversely correlated with the activity of PI3K/Akt. The target mRNA was identified as IGFR (insulin-like growth factor 1 receptor), an upsteam signalling molecule of the PI3K/Akt pathway. As the activation of the PI3K/Akt pathway plays a key role in promoting colitis-associated colorectal cancer [64, 65], inhibition of the pathway by increased miR-223 could have protective effect. Importantly, this study compared the miR-223 levels in myeloids and epithelial cells, which provides initial clue for the cell-context effect of miR-223.
FOXO proteins belong to the Forkhead family of transcription factors, which
regulate many physiological processes. MiR-223 has been shown to inhibit FOXO in
colonocytes, leading to increased activity of NF-
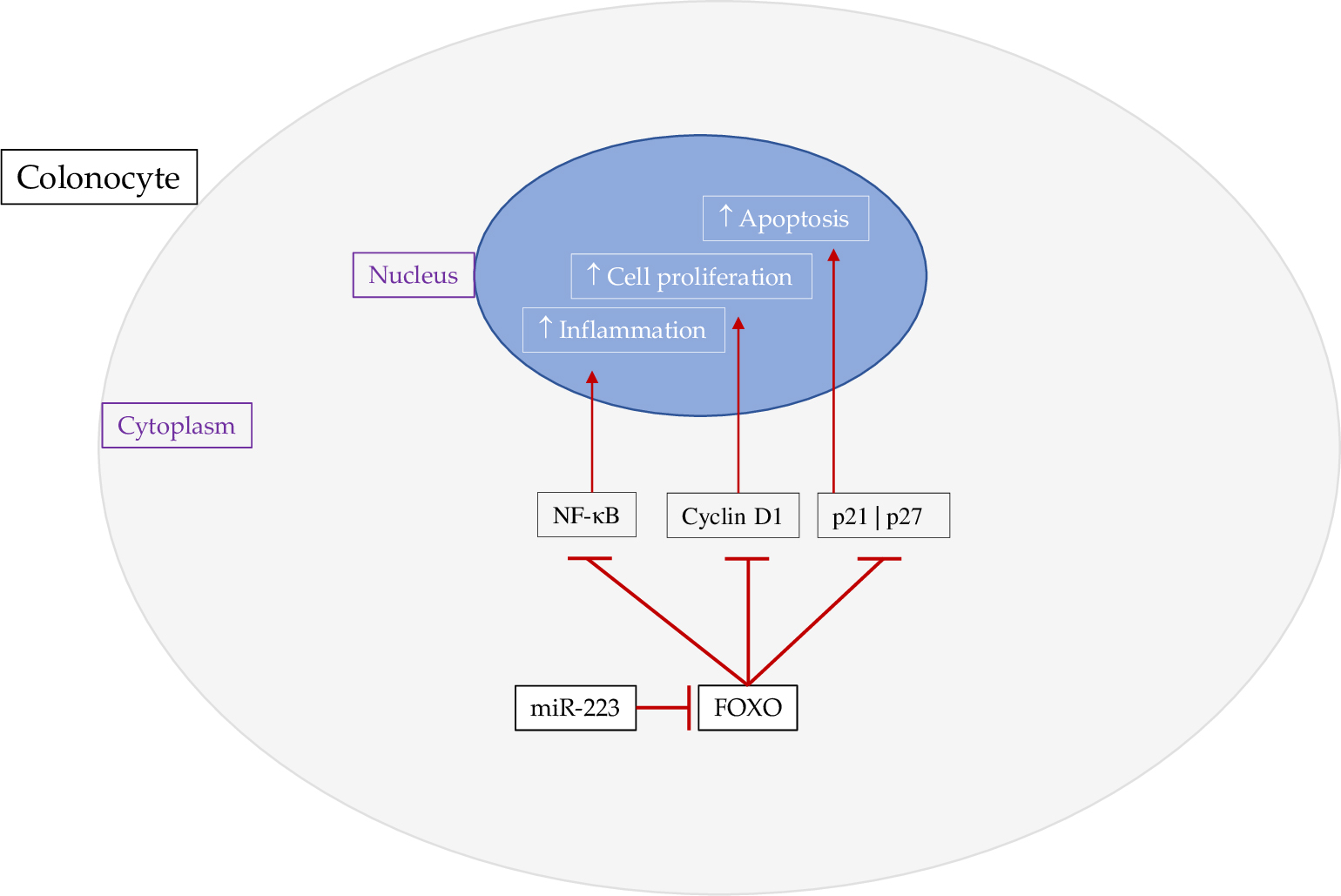 Fig. 3.
Fig. 3.Effect of miR-223 on FOXO. MiR-223 targets FOXO in colonocytes,
resulting in increased NF-
Knockout of FOXO4 aggravated TNBS-induced colitis with increased
CCL5, TNF-
Elevated miR-223 can decrease intestinal barrier integrity through inhibition of claudin8 (CLDN8), a tight junction backbone protein, in TNBS-induced colitis and thus increase translocation of LPS from the lumen to the intestinal mucosa, leading to LPS-induced systemic inflammation [39]. MiR-223 regulates CLDN by targeting its mRNA 3’-UTR to decrease CLDN8 protein expression [39]. Wang et al. [39] revealed that inhibition of miR-223 increased CLDN8 and thus facilitated tight junction formation and increased intestinal barrier integrity. In Caco-2 cells, siRNA silencing of CLDN8 decreased trans-epithelial electrical resistance while over-expression of the protein increased tight junction integrity and function [72]. A recent study confirmed that mast cell-derived miR-223 exosomes reduced the expression of tight junction proteins CLDN8, ZO-1 and occludin in colonic epithelial cells [73]. The miR-223 containing exosomes increased the permeability in monolayers of Caco2 cell cultures. In an animal model of TNBS-induced colitis, antagomir of miR-223 reduced gut permeability as well as gut inflammation, indicating a proinflammatory effect of miR-223 in this colitis model [35].
The intestinal microbiota is disturbed in IBD and modulation of the gut microbiota can facilitate IBD recovery. The commensal bacterium Roseburia intestinalis flagellin has been shown to ameliorate colitis and miR-223 could be a mediator [74]. The bacterium increased miR-223 levels and inhibited NLRP3 inflammasome. Another butyrate-producing bacterium F. prausnitizii has also been shown to have protective effects in DSS-induced colitis with improved weight loss, diarrhea, bloody stools and colon shortening as well as decreased mucosal proinflammatory cytokines [75]. However, it is not known if the mechanism in mucosal immunity and IBD pathogenesis, involves an increased expression of miR-223, and as such this remains largely unexplored. A recent study though has investigated fecal micro-RNAs in Crohn’s disease IBD [76]. Specifically this small cohort with patients diagnosed with Crohn’s disease (CD) has identified hundreds of different human miRNAs from faecal samples. Of these a total of 150 fecal miRNAs from controls and patients were significantly detected [76]. In a multivariate analyses the study showed that those patients diagnosed with high CD activity had a sharp and distinct miRNA profile. Consequently, miR-223 and miR-1246 were specific from other isolated fecal miRNAs. Moreover, in those patients with active ulcerative colitic (UC) the study reported that significantly higher levels of miR-223 and miR-1246 over that in controls. Furthermore, in those patients diagnosed with Clostridium difficile infections higher levels of fecal miR-1246 over miR-223 were reported.
What has recently been reported is that myeloid derived miR-223 regulates intestinal inflammation via an inhibitory action on the inflammasome NLRP3 [38]. The endogenous noncoding miRNAs exert regulatory inflammatory functions in the intestines [38]. Neudecker et al. [38] reported that miR-223 limits intestinal inflammation by constraining the NLRP3 inflammasome.
It has been shown that multi-strain probiotics through the administration of Lactobacillus plantarum, L. paracasei, L. rhamnosus, L. acidophilus, L. reuteri and Bifidobacterium animalis, B. lactis, B. bifidum can increase reinforce intestinal barrier integrity and functionality [77, 78, 79, 80]. This has been accompanied by decreased miR-223 levels by these probiotic species. Probiotic bacteria can also improve infllamtion which can lead to decreased miR-223 which reduces miR-223 upregulation as a protective response to inflammation. It is also possible that probiotics may also reduce inflammation via other mechanisms such as the anti-inflammatory effects of butyrate [23]. This effect linked with cross-feeding interactions by probiotic acetate producers from the Bifidobacteria genus with intestinal commensal butyrate producers such as F. prausnitzii [81]. Interestingly a recent review has reported how aberrant signaling in the PI3K/Akt pathway has been associated with a wide variety of human diseases [82]. In contrast though the authors confirmed and highlighted that a large body of experimental in vitro and in vivo evidence suggests that the beneficial contribution of probiotics to modulate the PI3K/Akt signaling pathway in gastrointestinal and metabolic diseases, skin diseases, allergy, salmonella infections, and the aging process [82]. Hence, a causal relationship emerges between the benefical effects of probiotics and their metabolites on the components of the PI3K/Akt signaling pathway and human disease.
The effects of miR-223 in DSS colitis models and the TNBS-induced colitis model
have been contentious, although miRNA-223 has been well demonstrated to be
significantly increased in both types of animal models of colitis. This may
indicate that miR-223 may exert effects that are dependent on the severity of
colitis. TNBS-induced colitis is a more robust form of inflammation than
DSS-induced colitis [83]. TNBS causes colitis through T-cell autoimmune responses
and DSS by direct toxicity to colonic epithelial cells [84]. TNBS haptenize
colonic autologous or microbiota proteins to elicit immune responses, leading to
infiltration of CD4
In immune cells, the anti-inflammatory effects of miR-223 is supported by
targeting several key signalling proteins in proinflammatory signalling pathways.
The anti-inflammatory effect of miR-223 on proinflammatory cells namely,
neutrophils and macrophages have been well demonstrated [30]. MiR-223 decreases
both neutrophil and macrophage activation through targeting NLRP3 [87]. Knockout
of miR-223 presents with increased neutrophil infiltration to the lung with
highly activated status indicated by increased CXCL2, CCL3 and IL-6 [88]. In LPS-
stimulated macrophages, miR-223 is decreased, leading to activation of the STAT3
signalling pathway, causing increased IL-6 and IL-1
The detrimental effect of miR-223 on TNBS-induced colitis has been based on
CLDN8 and FOXO alterations. MiR-223 can directly target CLDN8 mRNAs to reduce
CLDN8 protein transcription levels, leading to increased intestinal permeability
[39]. This causes the translocation of exdotoxin and bacteria, aggravating
inflammation. MiR-223 has also been shown to target FOXO, leading to increased
production of proinflammatory cytokines. We posit that miR-223 may be unable to
reduce activation of CD4
In reprise, miR-223 has opposite effects on the inflammatory profiles in the DSS- and TNBS- induced colitis models. In DSS-induced colitis, over expression of miR-223 exerts an anti-inflammatory effect. This could be possibly explained by its inhibitory effects on innate immune cells such as macrophages and neutrophils through inhibition of the NLRP3 inflammasome activity and proinflammatory signalling pathways. In TNBS-induced colitis, miR-223 is pro-inflammatory by targeting FOXO and CLDN8 in colonocytes. Further studies are warranted to elucidate the mechanisms of the effects of miR-223 in IBD, particularly in individual cells to explain its roles in different types of IBD. The accumulation of such knowledge could facilitate the clinical use of miR-223 modolators in personalized treatment of IBD.
NF-
JC—searching literature, creating conceptions together, preparing first draft. LV—supervsing, building co-conceptualization, critical revision.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest. JZC is serving as one of the Guest editors of this journal. We declare that JZC had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to BHZ.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.