






 , Ruchika Bhargav 1, Aliaa Mousa 1, Sabahat Bokhari 1,*
, Ruchika Bhargav 1, Aliaa Mousa 1, Sabahat Bokhari 1,*
1 Department of Cardiology, Rutgers Robert Wood Johnson University Hospital, New Brunswick, NJ 08901, USA
Abstract
Hypertrophic cardiomyopathy (HCM) is a multifaceted genetic disorder characterized by left ventricular hypertrophy (LVH) in the absence of alternative causes, with an estimated prevalence ranging from 1 in 200 to 1 in 500 individuals. Since HCM was first characterized in 1869, a plethora of pathogenic mutations have been identified, while ongoing research continues to elucidate the various pathophysiological mechanisms present in individuals with HCM. Comprehensive physical examination findings and multimodality imaging techniques have become crucial for accurately diagnosing and risk stratifying HCM patients. Meanwhile, recent advancements in research have contributed to a more refined definition and heightened recognition of HCM, prompting further investigations into targeted therapeutic strategies. This evolution in understanding provides alternative treatment options for patients, moving beyond traditional approaches such as myectomy or septal ablation. This review aims to systematically explore the genetic and pathophysiological underpinnings of HCM, as well as the application of multimodality imaging in identifying patients at risk for sudden cardiac death (SCD). The discussion also examines contemporary management strategies for HCM, specifically highlighting novel therapies targeting the molecular mechanisms involved in this disease.
Keywords
- hypertrophic cardiomyopathy
- sudden cardiac death
- alcohol septal ablation
- surgical myectomy
- mavacamten
- aficamten
The understanding of hypertrophic cardiomyopathy (HCM) as a distinct clinical entity has developed over a century, beginning with early morphological descriptions and culminating in recognizing its genetic basis. HCM is a genetic disease of the myocardium, which is characterized primarily by left ventricular hypertrophy (LVH) that is not due to alternative systemic, cardiac, or metabolic etiologies.
The earliest descriptions of HCM date back to Henri Liouville in 1869, who characterized the disease as “cardiac subaortic stenosis” after he discovered massive concentric LVH to 3.5 cm and left ventricular outflow tract obstruction in an autopsy study of a 75-year-old female [1]. Throughout the late 19th and early 20th centuries, case reports and pathological studies documented unexplained myocardial hypertrophy, but these observations remained isolated and lacked understanding of the underlying disease mechanisms [2, 3]. In 1958, Teare and colleagues provided a comprehensive description of familial cases characterized by massive septal hypertrophy, myocyte disarray, and sudden cardiac death, marking the first recognition of HCM as a hereditary disease [4]. Teare’s studies emphasized that the condition was characterized by familial clustering and involved disorganized myocardial architecture with myocyte disarray [4].
HCM is now recognized as a relatively common inherited cardiac disorder with a worldwide distribution. Recent epidemiological studies, supported by advanced echocardiography and cardiac magnetic resonance imaging (CMR), suggest a population prevalence of approximately 1 in 200 to 1 in 500 individuals [5]. From a morphological perspective, HCM is characterized by the restriction of hypertrophy to the myocardium. While it often exhibits asymmetric septal hypertrophy, it can also present with concentric or localized hypertrophy, including apical involvement. The disease may progress from compensated hypertrophy to restrictive cardiomyopathy and end-stage heart failure due to myocardial remodeling and fibrosis. The clinical diagnosis considers genetic and phenotypic features, emphasizing the importance of integrating imaging, electrocardiographic, genetic, and clinical data for an accurate diagnosis [6, 7, 8].
From a genetic standpoint, HCM was initially solely considered to be an
autosomal dominant disorder and determined to be caused by pathogenic variants in
sarcomere protein genes, including MYH7 (
However, there is increasing data regarding autosomal recessive inheritance, especially in populations where consanguinity is more prevalent. Pathogenic variants are responsible for approximately 30%–40% cases of HCM. Less prevalent causal genes such as MYL2, MYL3, CSRP3, and TRIM63 have been implicated in more homozygous cases [10]. Table 1 (Ref. [10]) highlights the common genetic mutations and their prevalence.
| Gene | Protein encoded | Frequency within genotype—positive individuals |
| MYBPC3 | Myosin-binding protein C | 40%–50% |
| MYH7 | Beta-myosin heavy chain | 35%–40% |
| TNNT2 | Troponin T | 7%–15% |
| TNNI3 | Troponin I | 5% |
| TPM1 | Tropomyosin | 3% |
| MYL2 | Regulatory myosin light chain | 1%–2% |
| MYL3 | Essential myosin light chain | 1% |
| ACTC1 | Actin | 1% |
| TNNC1 | Troponin C | |
| ACTN2 | Alpha-actinin-2 | |
| ALPK3 | Alpha-protein kinase 3 | ~2% |
| FHOD3 | Formin homology 2 domain containing 3 | 1%–2% |
| CSRP3 | Muscle LIM protein | |
| TRIM63 | Tripartite motif containing 63 | Unknown |
| FLNC | Filamin C | |
| FHL1 | Four-and-a-half LIM domain protein 1 | |
| PLN | Phospholamban | |
| JPH2 | Junctophilin 2 | Unknown |
Additionally, approximately 40% of patients have nonfamilial forms of HCM, which is a clinically distinct subtype and includes probands who have no identifiable genetic cause or family history of HCM. Male gender, older age, lack of asymmetric hypertrophy, and hypertension are more frequently associated with nonfamilial HCM. The nonfamilial HCM subgroup generally has a more benign clinical course with a lower rate of adverse cardiac events as compared to patients with sarcomere-positive HCM [11].
Phenotypic expression can vary significantly: a substantial number of mutation carriers may remain asymptomatic due to incomplete penetrance. Furthermore, even individuals with pathogenic mutations can show considerable variability in disease severity, age of onset, and clinical outcomes, often within the same family. The genetics of HCM involve a combination of strong-effect mutations, polygenic modifiers, and non-genetic factors, highlighting the necessity for comprehensive and nuanced genetic evaluation [5].
This paper aims to provide a comprehensive review of the pathophysiology, screening criteria, diagnosis, and advancements in the treatment of HCM.
Left ventricular outflow tract obstruction (LVOTO) represents a hallmark
pathophysiologic mechanism in HCM, affecting approximately 70% of patients with
gradients
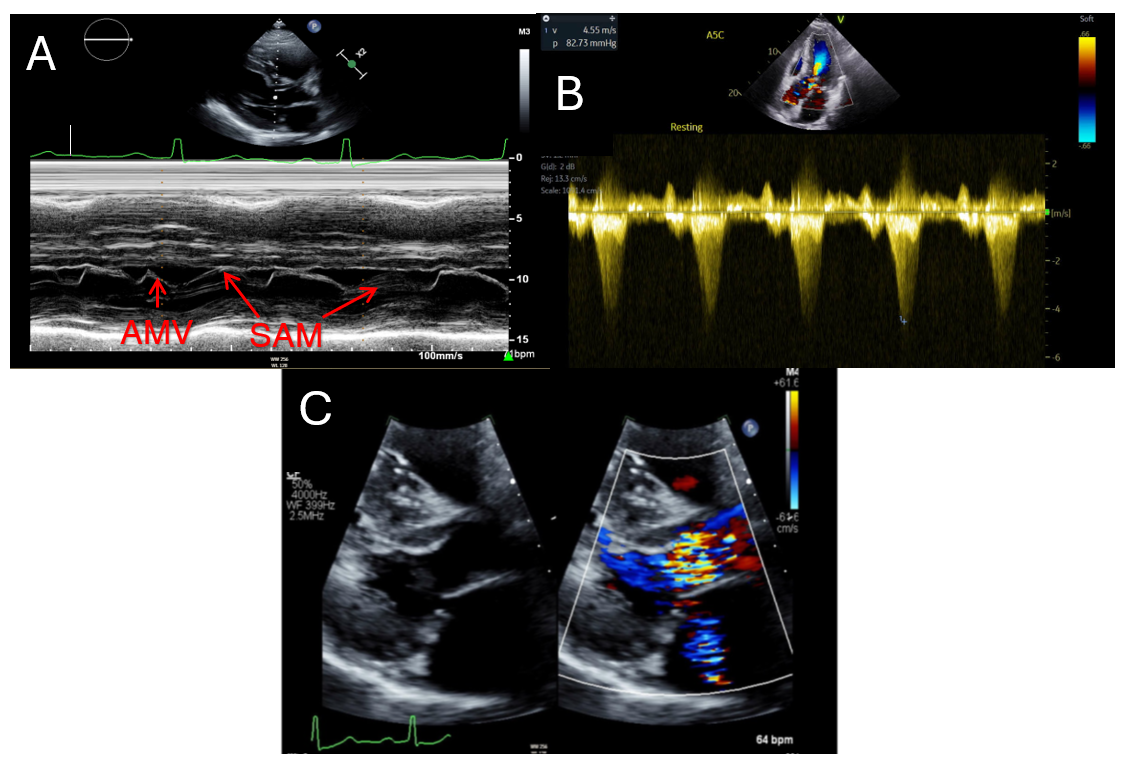 Fig. 1.
Fig. 1.
Echocardiographic findings in hypertrophic cardiomyopathy. (A) showcases a patient with systolic anterior motion of the anterior mitral valve leaflet (AMV) demonstrated by M-Mode of the parasternal long axis. (B) highlights a severely obstructed outflow tract gradient (82.7 mmHg) in an HCM patient. (C) displays mild mitral regurgitation via color seen on parasternal long axis. HCM, hypertrophic cardiomyopathy; SAM, systolic anterior motion; AMV, anterior mitral valve leaflet.
Apical hypertrophic cardiomyopathy visualized by cardiac magnetic resonance imaging. Video associated with this article can be found, in the online version, at https://doi.org/10.31083/RCM42824.
Mitral regurgitation (MR) in patients with HCM primarily occurs due to SAM of the MV, which is commonly attributed to the Venturi effect [13]. However, it can also result from intrinsic abnormalities of the mitral valve apparatus, such as excessive elongation of the anterior or posterior leaflets, and issues with the papillary muscles, including anomalous insertion or anterior displacement [2, 14]. Understanding these pathophysiological changes is crucial, as they can precede hypertrophic changes in the LV and may represent an early phenotypic manifestation of sarcomere mutations [15, 16]. For example, Velicki et al. [17] investigated patients with mutations in the MYBPC3 or MYH7 genes and found that those with MYH7 mutations exhibited more significant MV abnormalities and greater regurgitation compared to patients with MYBPC3 mutations. This genetic heterogeneity may account for the variability in valvular dysfunction observed across the HCM spectrum.
The etiology of MR also carries management implications, as study has demonstrated a significant reduction in MR after isolated septal myectomy without requiring additional MV surgery [18]. In contrast, those with intrinsic disease of the MV apparatus may require an additional MV repair procedure during septal myectomy [2, 18, 19]. Differentiation of the etiology of MR is therefore crucial for pre-procedural planning in patients undergoing septal reduction surgery. While SAM-mediated MR often presents with a posteriorly directed jet, its absence does not definitively rule in primary mitral valve disease due to its low negative predictive value [20, 21]. Conversely, central or anteriorly directed jets should prompt further investigation via transesophageal echocardiogram or cardiac magnetic resonance imaging to evaluate for structural abnormalities [2, 20].
As the disease progresses, many cellular and morphological changes drive diastolic dysfunction in HCM. This occurs due to impaired LV relaxation, increased myocardial stiffness, and left atrial myopathy [8, 20, 22]. Using induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) from healthy controls and HCM patients with diastolic dysfunction, Wu et al. [22] showed that diastolic calcium overload, slow calcium recycling, and increased myofilament calcium sensitivity collectively impaired diastolic relaxation times; thereby providing a molecular explanation for the utility of calcium channel blockers (CCBs) in the management of HCM. Myocardial stiffness in HCM, at the cellular level, results from hypertrophied and disorganized cardiomyocytes separated by interstitial fibrosis, leading to a shrinking LV cavity [6]. In severe cases, this may result in a restrictive physiology [2]. Lastly, left atrial myopathy contributes to the development of diastolic dysfunction by impairing LV filling [20]. A culmination of these three pathophysiologic mechanisms results in elevated LV diastolic pressures that increase markedly on exertion, resulting in symptoms of exertional dyspnea and exercise intolerance, especially in patients with concomitant atrial fibrillation (AF) [6].
The pathophysiology of myocardial ischemia in HCM is complex and involves multiple factors. One mechanism is coronary microvascular dysfunction (CMD), as evidenced by autopsy studies showing hypertrophy of intimal and/or medial layers of the coronary arterioles with resultant reduction in luminal cross-sectional area [23]. The consequent impairment of vasodilatory capacity leads to decreased myocardial blood flow (MBF) during periods of heightened physiological demand, such as exercise, severe hypertrophy, and significant LVOTO. Consequently, this creates a pronounced myocardial supply-demand mismatch and myocardial injury [23, 24, 25]. Therefore, if this mismatch persists, replacement fibrosis occurs and serves as an arrhythmogenic substrate associated with sudden cardiac death [24, 26].
With an estimated prevalence of 20%, AF is the most common sustained arrhythmia in patients with HCM [8]. The elevated LV filling pressures from progressive hypertrophy leading to left atrial dilation have been attributed to the development of AF. However, increasing evidence of a primary left atrial (LA) myopathy secondary to excessive fibrosis may also serve as a predisposing factor for AF [8, 27]. In contrast to atrial arrhythmias, the mechanism behind ventricular arrhythmias in HCM patients is multifaceted and involves both functional and structural abnormalities. Functionally, alterations in intracellular calcium and sodium homeostasis lead to prolonged action potentials in the diseased myocardium, increasing the susceptibility to early and delayed afterdepolarizations that can trigger ventricular arrhythmias [28, 29, 30]. Structurally, myocardial fibrosis, myocyte disarray, and CMD act as substrates for re-entry ventricular tachycardia, with precipitating factors including exercise and LVOTO [31]. Refer to section 6 for additional discussion on managing and preventing arrhythmic complications in HCM.
The mitochondrion has been of particular focus at the cellular level for HCM. Recent studies have demonstrated severe mitochondrial damage with additional downregulation of genes responsible for synthesizing creatinine kinase and adenosine triphosphate, suggesting a global energetic decompensation in HCM hearts [32, 33]. This energy deficit was further exacerbated, independent of hypertrophy or degree of fibrosis, during exercise in patients with HCM [34]. These findings underscore the importance of understanding the pathology of HCM at a cellular level to develop effective therapies.
HCM is predominantly diagnosed in an outpatient clinical setting [4, 5, 6]. Initial workup involves thoroughly assessing a patient’s personal history, obtaining a three-generational family history, and a focused physical exam. In obstructive or labile-obstructive disease variants, patients typically manifest with dyspnea, angina, presyncope, or syncope, with the latter being particularly prevalent among young athletes [6, 13, 35]. Individuals exhibiting a milder phenotype demonstrate attenuated symptomatology, with a subset remaining entirely asymptomatic [6, 13]. For these asymptomatic individuals, a meticulous physical exam and a detailed family history become instrumental in determining the necessity for additional diagnostic imaging. Cardinal examination findings include a leftward-displaced precordial impulse, brisk peripheral artery pulsations, and a pronounced fourth heart sound (S4) [6, 13]. Depending on the obstruction’s severity, a harsh mid-systolic murmur may be best heard between the left lower sternal border and the cardiac apex. A blowing high-pitched holosystolic apical murmur suggestive of mitral regurgitation may also be present [6, 13]. Lastly, augmentation of mid-systolic murmur intensity during preload-reducing maneuvers—specifically the Valsalva maneuver or orthostatic positioning—provides further diagnostic differentiation from aortic stenosis, reinforcing the diagnosis of HCM [35, 36].
The electrocardiogram (ECG) maintains its position as an indispensable screening modality for HCM, with contemporary data indicating that merely 5–10% of affected individuals present with normal electrocardiographic patterns [37, 38]. This cost-effective diagnostic instrument demonstrates utility in detecting left atrial enlargement (LAE), LVH, and Wolff-Parkinson-White (WPW) syndrome [37, 39]. These findings warrant further imaging workup if present on ECG, as they have prognostic implications. For example, an increase in left atrial diameter is associated with an increased risk of sudden cardiac death (SCD). Moreover, significant LVH with WPW is closely linked to PRKAG2 syndrome, a rapidly progressive autosomal-dominant glycogen storage disorder that carries a high risk of arrhythmias and SCD [37, 39]. Furthermore, while ECGs alone are not very sensitive for screening HCM or identifying high-risk features for SCD, recent artificial intelligence (AI) breakthroughs show promise in this area [38, 40, 41, 42]. For example, a study done by Ko et al. [38] found that AI-based ECGs can effectively screen for HCM, achieving an overall sensitivity of 87%, specificity of 91%, and a negative predictive value of 99%. Another study found similar results using AI in a significantly younger population of children and adolescents [40, 41]. Despite these technological advances in ECG analysis, definitive HCM diagnosis still necessitates supplementary multimodality imaging assessment.
Transthoracic echocardiography (TTE) constitutes the cornerstone imaging
modality in the diagnostic algorithm for HCM, with maximal LV myocardial
thickness
Furthermore, TTE offers invaluable hemodynamic assessment capabilities, enabling
non-invasive differentiation between obstruction phenotypes. Sustained elevation
of LVOT gradient (
Additionally, echocardiography can visualize causes of obstruction, such as SAM of the MV or abnormal insertion of the papillary muscles. Moreover, it aids in evaluating the extent of functional MR, identifying the presence of an apical aneurysm, and quantifying diastolic dysfunction [5, 8, 20]. Thus, comprehensive echocardiographic evaluation provides essential morphologic, hemodynamic, and functional data that guide therapeutic interventions and prognostic stratification.
CMR provides a comprehensive evaluation of HCM. It is often recommended as a
Class I indication for patients with HCM who have technically limited
echocardiographic views or in whom the diagnosis is inconclusive via TTE
[2, 45, 46]. The diagnostic criteria for HCM on CMR are similar to those used in
echocardiography [5]. However, its advantage comes from its excellent spatial and
temporal resolution and blood pool/myocardium contrast, which overcomes the
limitations of TTE [46, 47]. CMR has particularly proven valuable in identifying
areas of hypertrophy not reliably detected by echocardiography, such as the
anterolateral free wall, apex, or posterior septum. This is especially important
as roughly 20% of patients have focal HCM in one or two LV segments [46, 47].
Moreover, even in patients in whom apical HCM was detected on echocardiography,
there was a significant discrepancy between the measured apical wall thickness on
TTE vs CMR (mean difference of 1.7 mm) [48]. TTE also overestimates LV wall
thickness in 59% of patients due to off-axis imaging, right ventricular (RV)
trabeculations, or papillary muscle inclusion [48]. With TTE being operator and
reader-dependent, it is no surprise that a study found a significantly lower
interobserver variability with CMR than TTE [49]. Lastly, CMR can more accurately
identify high-risk features such as the presence of LV apical aneurysm, severe
LVH (
LVH is a non-specific finding frequently observed on TTE, making it challenging
to differentiate HCM from its phenotypic mimics. However, CMR can be used to
distinguish an athlete’s heart from HCM via the identification of focal
hypertrophy (in favor of HCM) or regression in maximal LV wall thickness by
Infiltrative cardiomyopathies that phenotypically simulate HCM demonstrate distinctive CMR signatures: cardiac amyloidosis manifests as subendocardial and transmural enhancement with relative apical sparing and characteristic simultaneous myocardial and blood nulling; Fabry disease exhibits enhancement of mid-lateral wall segments with subendocardial sparing; and Danon disease presents with severe LVH accompanied by extensive enhancement with mid-septal sparing [20]. These pathognomonic enhancement patterns provide critical diagnostic information that guides therapeutic interventions, facilitates familial genetic counseling, and enables longitudinal assessment of disease progression.
While non-invasive modalities remain the cornerstone of hemodynamic characterization in HCM, invasive cardiac catheterization assumes an essential diagnostic role in clinical scenarios where echocardiographic data proves inconclusive, technically inadequate, or is contraindicated [2, 51]. Specific indications include circumstances where Doppler echocardiography cannot differentiate between an increase in velocity profile from an LVOTO versus contamination by MR, necessitating direct pressure gradient measurement with corresponding waveform analysis [2, 51]. Additional indications for invasive hemodynamic assessment encompass patients with concomitant aortic stenosis and dynamic outflow obstruction, and those with symptomatic burden disproportionate to resting non-invasive imaging findings [2, 51]. Moreover, patients with persistent chest pain warrant catheterization to accurately rule out coronary artery disease (CAD) due to the high false-positive and negative rates associated with nuclear and echocardiographic stress testing [2]. Lastly, for patients planned for surgical myectomy (SM) or alcohol septal ablation (ASA), coronary angiography is often performed to aid procedural planning [2].
Since hemodynamic assessment of HCM is crucial for guiding management, the role of cardiac computed tomography angiography (CCTA) remains limited. As a result, it holds a Class IIb recommendation for assessing patients with suspected HCM if an echocardiogram is inconclusive and CMR is contraindicated [2]. However, due to its excellent three-dimensional resolution, CCTA can reveal morphological features of HCM to establish a diagnosis, evaluate for myocardial bridging, and visualize septal perforators within the myocardium [20].
Exercise stress testing is safe for HCM patients and provides valuable
information on their functional capacity and limitations. Studies have shown that
patients with reduced peak oxygen consumption (
Endomyocardial biopsy plays a crucial role in diagnosing HCM, especially in cases where non-invasive imaging and genetic testing do not yield definitive results. Histologically, HCM is characterized by myocyte hypertrophy with enlarged, hyperchromatic nuclei, disorganized myofiber architecture known as myofiber disarray, and interstitial fibrosis marked by increased collagen deposition [57, 58, 59]. In contrast, amyloidosis, an infiltrative cardiomyopathy, can mimic HCM on imaging but has distinct histopathological features: extracellular amyloid deposits which stain positively with Congo red and exhibit apple-green birefringence under polarized light [60]. These deposits appear as rigid, non-branching fibrils of 7–10 nm diameter on electron microscopy (EM) [61], and unlike the patchy fibrosis with myocyte disarray in HCM, amyloid infiltration results in concentric myocardial thickening. Identifying these differences is critical for appropriate treatment and prognosis [62, 63].
EM offers additional diagnostic precision by revealing ultrastructural abnormalities that are not visible via light microscopy [64]. Common EM findings include variability in mitochondrial size, swelling, cristae disruption, paracrystalline inclusions, and distinctive crystalline structures within mitochondria that suggest mitochondrial dysfunction [62]. These features help differentiate primary HCM from phenocopies caused by metabolic or lysosomal storage diseases such as Fabry disease, where EM detects characteristic lamellar inclusions [62, 65]. Such ultrastructural insights are valuable because they augment the pathological understanding and tailor patient management.
Contemporary screening guidelines for relatives of HCM probands have been meticulously delineated in the 2024 American College of Cardiology/American Heart Association (ACC/AHA) consensus recommendations [2]. Cascade genetic testing is indicated for first-degree relatives when a pathogenic (P) or likely pathogenic (LP) sarcomeric variant has been identified in the proband; conversely, in genotype-negative HCM probands, familial genetic screening yields limited diagnostic utility, although phenotypic surveillance remains imperative [2]. The screening protocol involves comprehensive risk stratification for SCD and AF and objective assessment of functional capacity [2]. Initial testing involves ECG, TTE, and genetic testing. However, additional testing for HCM mimics (PRKAG2, LAMP2, GLA, etc.) should also be performed if the affected patient meets specific disease phenotypes [2, 20, 44]. American and European guidelines vary with regard to the time of genetic testing in the pediatric population. European guidelines suggest screening after the age of 10–12 years, whereas American guidelines suggest screening children and adolescents at the time a pathogenic variant of HCM is diagnosed in a family member [2, 66]. In this population, TTE and ECG should be repeated every 1–2 years [2, 20]. For pediatric patients in whom family history is negative for a pathogenic variant, screening is recommended at any time after the family member was diagnosed, but no later than puberty. TTE and ECG should be repeated in this population every 2–3 years [2, 20].
Screening for adult family members of a proband depends on the phenotype of the
individual being tested. In HCM phenotype-positive adults, a baseline evaluation
includes SCD risk assessment by TTE, stress echocardiogram, ECG, ambulatory ECG
(to rule out AF), and CMR (to evaluate for LGE and apical aneurysm). Repeat
clinical assessment, TTE, and ambulatory ECG monitor should be performed in these
patients every 1–2 years [2]. Stress TTE or CPET should be considered in
asymptomatic adults every 2–3 years to assess for occult serial decline in
functional status [2]. In symptomatic adults, it is crucial to assess for
worsening dynamic LVOTO every 1–2 years via stress TTE (if their resting
gradient is
In adults with phenotype-negative HCM, screening intervals depend on the presence of a P/LP genotypic variant in the patient and family. For patients who don’t have a P/LP variant or have a family and personal history of a P/LP variant, it is recommended that they undergo screening with ECG and TTE (CMR if TTE is insufficient) every 3–5 years [2, 20]. Conversely, if a patient has a known family history of a P/LP variant but has no variant on genetic testing, no additional surveillance is required. Table 2 (Ref. [2, 5]) summarizes the screening recommendations adapted from the 2024 ACC/AHA HCM Guideline.
| Pediatric population | Adult population | |
| Initiation of screening [2, 5] | • If genotype-positive family, or early-onset disease in family, screening starts at the time of HCM diagnosis in the proband (preferably after 12 years of age) | • At the time of HCM diagnosis in another family member |
| • Continued surveillance from adolescent screening protocols upon transition to adult care | ||
| Screening Intervals [2, 5] | • Genotype-positive/early-onset disease families: Every 1–2 years (until 18–21 years of age) | • In phenotype-negative patients: Every 3–5 years |
| • All other pediatric cohorts: Every 2–3 years (until 18–21 years of age) | • In phenotype-positive patients: Every 1–2 years with standard diagnostic testing | |
| Imaging modalities [2, 5] | • Initial approach: Baseline 12-lead ECG, ambulatory ECG, TTE, stress TTE (to assess for dynamic obstruction) | • Initial approach: Baseline 12-lead ECG, ambulatory ECG, TTE, stress TTE (to assess for dynamic obstruction) |
| • CMR when echocardiographic findings are inconclusive | • ECG, ambulatory ECG, and TTE every 1–2 years | |
| • CMR every 3–5 years for SCD risk assessment in absence of ICD | ||
| • Exercise testing for functional status assessment every 2–3 years in asymptomatic individuals | ||
| Symptomatic individuals [2, 5] | • Exercise testing with stress TTE (if LVOT gradient | |
| • CPET for functional status assessment and consideration of advanced heart failure therapies | ||
| Genetic testing [2, 5] | • Recommended for family members of a patient with a pathogenic variant | |
| • In atypical clinical presentations of HCM, genetic testing for HCM and HCM phenocopies should be performed | ||
| • Unclear usefulness in assessing the risk of SCD | ||
| • Cascade genetic testing is not recommended for relatives of a proband without a pathogenic variant | ||
Table describing the screening guidelines for family members of a relative with HCM, adapted from the 2024 ACC/AHA guidelines. HCM, hypertrophic cardiomyopathy; ACC/AHA, American College of Cardiology/American Heart Association; CMR, cardiac magnetic resonance imaging; ECG, electrocardiogram; TTE, transthoracic echocardiography; SCD, sudden cardiac death; ICD, implantable cardioverter defibrillator; LVOT, left ventricular outflow tract.
The overall risk of SCD in HCM is relatively low and is estimated to be 0.5%
per year [20]. However, the risk of SCD should be assessed based on the
individual patient. For example, in a study of SCD among athletes of various
ages, the sports with the highest incidence of SCD include basketball, football,
and soccer. Moreover, a higher incidence rate was noted in Black male NCAA
Division I college athletes compared to their high school counterparts [67, 68].
Risk stratification for SCD is a critical component in managing patients with HCM
and has evolved considerably over the past several decades. Multiple clinical
risk markers have been identified that stratify patients according to their level
of risk for potentially life-threatening ventricular arrhythmias. These
established risk factors include: family history of SCD in first-degree or close
relatives
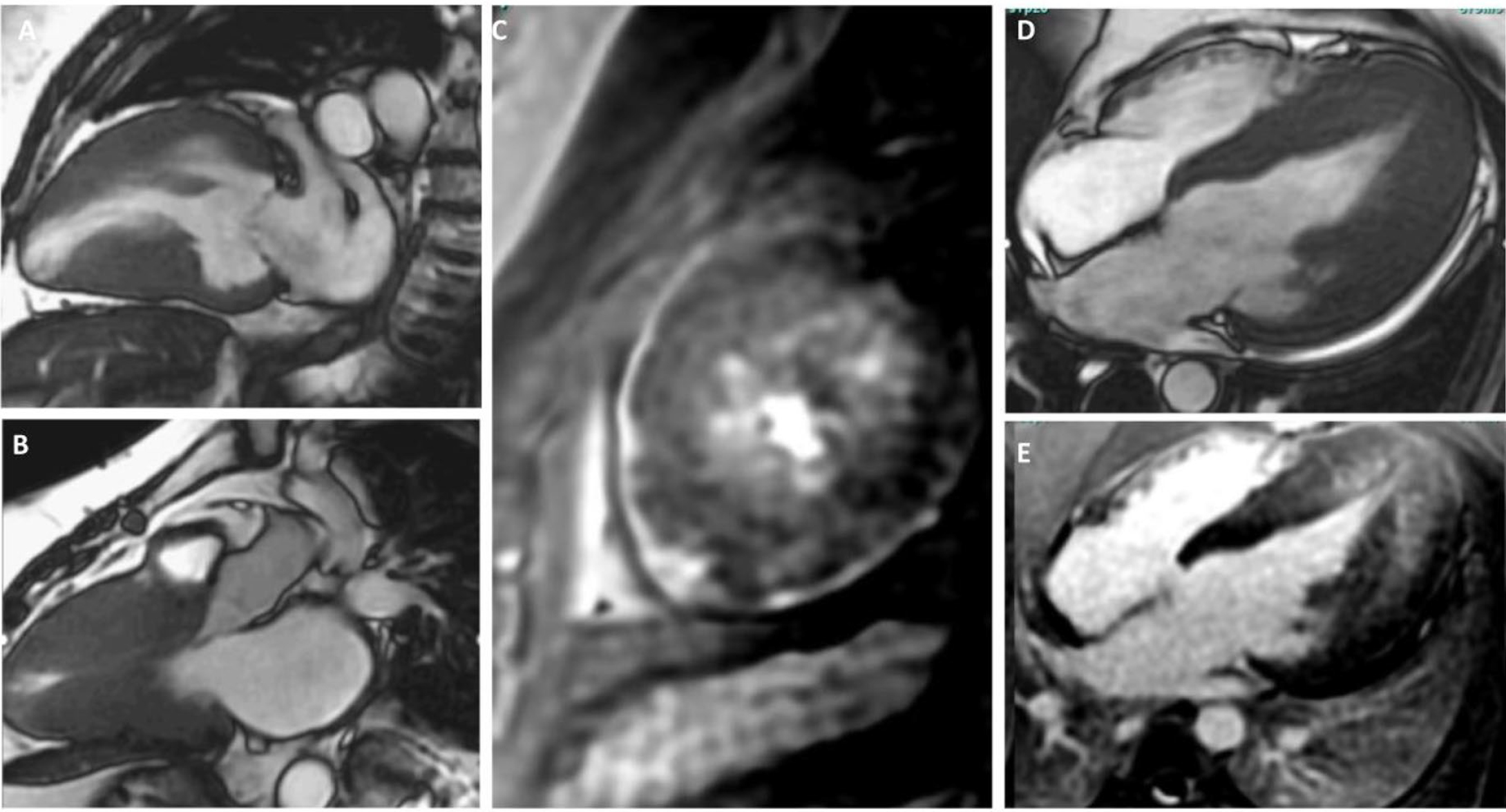 Fig. 2.
Fig. 2.
Cardiac MRI Cine SSFP images showing 2,3, and 4-chamber views highlighting additional characteristics seen in hypertrophic cardiomyopathy. (A,B,D) showcases severely hypertrophied walls. (A,B) showcase mid-ventricular hypertrophy with an aneurysmal apex (A). (E) showcases apical hypertrophy with mid-wall late gadolinium enhancement. (C) shows a short-axis view with mid-wall late gadolinium enhancement of the left ventricular apex. MRI, magnetic resonance imaging; SSFP, steady-state free precession.
Cardiac magnetic resonance imaging highlighting apical aneurysm in hypertrophic cardiomyopathy. Video associated with this article can be found, in the online version, at https://doi.org/10.31083/RCM42824.
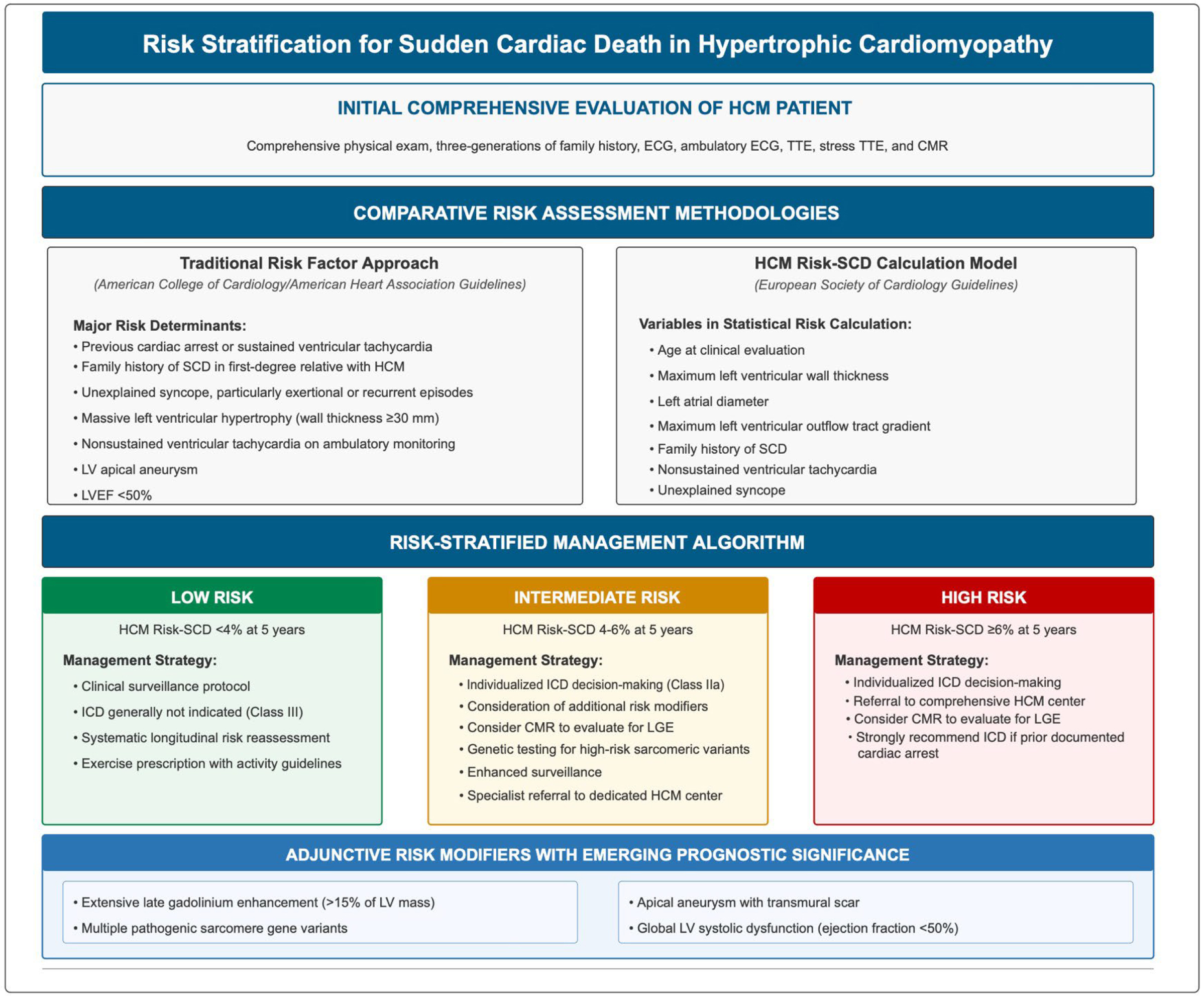 Fig. 3.
Fig. 3.
Risk stratification for sudden cardiac death in hypertrophic cardiomyopathy [74, 75]. HCM, hypertrophic cardiomyopathy; CMR, cardiac magnetic resonance imaging; ECG, electrocardiogram; TTE, transthoracic echocardiography; SCD, sudden cardiac death; ICD, implantable cardioverter defibrillator; LV, left ventricular; LVEF, left ventricular ejection fraction; LGE, late gadolinium enhancement.
Despite the current guidelines, there remains conflicting evidence regarding screening criteria for genotype-positive, phenotype-negative individuals, given the phenotypically heterogeneous nature of HCM. Family members with identical mutations have been shown to display different phenotypic expression, which indicates the potential role of exogenous factors in disease development [76]. There is evidence that these patients have impaired myocardial relaxation and impaired energy metabolism, but the clinical implication of these findings on the disease onset or severity and the risk of sudden cardiac death remains unknown, which makes screening and treatment extremely challenging [76].
ICD therapy is an effective intervention for the primary prevention of lethal ventricular arrhythmias and SCD. However, due to the complexities associated with HCM, not all patients benefit from this treatment [77].
ICD selection has evolved from advocating “all-purpose” ICD systems toward a nuanced approach predicated on individualized patient characteristics and specific clinical scenarios. Single-chamber devices and subcutaneous ICDs (S-ICDs) represent reasonable options for most HCM patients without pacing requirements [78].
S-ICDs offer particular advantages for young patients with extended life expectancy who face a heightened cumulative risk of complications related to transvenous leads. Studies from the EFFORTLESS cohort illustrate these benefits, indicating a negligible risk of bloodstream infections, a low incidence of lead fractures, and an impressive 2-year estimate of 92.7% for freedom from procedural complications compared to traditional transvenous ICDs [78]. Furthermore, findings from the PRAETORIAN trial have shown that S-ICDs are non-inferior to transvenous ICDs in terms of inappropriate shocks among general ICD candidates. As a result, S-ICDs have gained popularity among patients with HCM without additional requirements [78, 79].
The nuanced approach stems from patient-specific clinical scenarios that warrant specific device selection, such as:
1. Left ventricular outflow tract obstruction: Transvenous systems may be preferred for oHCM patients, particularly if septal reduction procedures might be required, as these interventions carry a non-negligible risk of conduction defects (10% in ASA patients, 4.4% in SM patients) necessitating pacing capabilities [80]. Moreover, changes in QRS-T morphologies after septal reduction therapies (SRT) may result in T-wave oversensing and failure to recognize the stored QRS-T template, leading to inappropriate shocks [78]. Therefore, patients with oHCM who may require SRT in the future may benefit from transvenous ICD, rather than S-ICD, therapy upfront.
2. End-stage disease: Patients who develop “burnt out” HCM, characterized by advanced heart failure with reduced ejection fraction, face an annual event rate of 10% for life-threatening arrhythmias [78]. Given that left bundle branch block (LBBB) is prevalent in such cases, cardiac resynchronization therapy with a defibrillator (CRT-D) may offer more advantages than S-ICDs. This is especially relevant in patients exhibiting severe interventricular septal fibrosis on CMR, as this condition heightens concerns about potential heart block that may necessitate pacing [78].
3. Pediatric patients: High complication rates (approximately 9.5% per year) have been documented in children and adolescents with HCM, primarily involving inappropriate shocks (41%), lead malfunction, and lead stretching [78, 81]. As a result, S-ICDs may be a better alternative in this population. However, a significant limitation is their large size in relation to the transvenous ICD and the body size of the child (due to concern for device erosion in smaller patients) [2, 78, 81].
Non-vasodilating beta-blockers (BBs) are regarded as first-line therapy for
symptomatic oHCM [2, 35, 82]. They work by increasing LV filling time and volume
(negative chronotropy) while decreasing the contractile force (negative
inotropy), attenuating dynamic LVOTO and mitigating associated symptoms
[2, 35, 82]. Although these drugs are widely used, there is a surprising lack of
studies assessing their effectiveness, with the majority being underpowered and
not randomized [82]. For example, all the studies on BBs found significant
variability in LVOT gradient reduction, limited improvement in symptoms, and a
stark proportion of non-responders, raising the need for larger randomized trials
to better understand the role of these medications, especially in non-responders
[82]. BB dosing is individualized, with medications uptitrated until a patient
achieves symptomatic relief. However, if patients continue to have suboptimal
response despite maximum dosing, non-dihydropyridine CCBs, such as verapamil or
diltiazem, can be substituted [2, 35, 82]. CCBs also work by providing negative
inotropic and chronotropic effects; however, they pose the risk of exacerbating
outflow gradients in some patients. In fact, verapamil is specifically
contraindicated for patients with severely elevated resting gradients (
Regarding treating asymptomatic carriers, it is crucial to continue close clinical surveillance to monitor for symptom onset or for the presence of additional risk factors, which might warrant further management. Pharmacotherapy should be utilized to provide symptomatic relief and improve quality of life, but the role of medical therapy in asymptomatic carriers is currently not recommended and remains a topic of ongoing investigation. Overmedicalization of asymptomatic carriers poses an additional financial and psychosocial burden on these patients.
Disopyramide serves as an additional treatment option for patients with oHCM who remain symptomatic despite first-line pharmacotherapy [2]. Because it can enhance conduction through the atrioventricular (AV) node, potentially facilitating rapid ventricular rates during AF, disopyramide should only be administered with BBs or CCBs [2].
As a class I antiarrhythmic agent, disopyramide possesses potent negative inotropic effects. Recent studies have demonstrated that it influences multiple ion channels, reduces the inward calcium current, and stabilizes the ryanodine receptor. This stabilization subsequently leads to diminished calcium release from the sarcoplasmic reticulum. Furthermore, disopyramide inhibits the late inward sodium current, which is often elevated in HCM and contributes to early and late afterdepolarizations. This mechanism may help mitigate the risk of ventricular arrhythmias in these patients [83, 84].
Disopyramide’s cost-effectiveness allows better access for patients of lower socioeconomic backgrounds, unlike the significantly more expensive mavacamten discussed in section 7.3 [76, 77, 78]. Fig. 4 (Ref. [2, 85, 86, 87, 88, 89, 90]) summarizes the overall management of patients with symptomatic HCM.
 Fig. 4.
Fig. 4.
Evidence-based management algorithm for symptomatic hypertrophic cardiomyopathy [2, 85, 86, 87, 88, 89, 90]. HCM, hypertrophic cardiomyopathy; CMR, cardiac magnetic resonance imaging; ECG, electrocardiogram; TTE, transthoracic echocardiography; LVEF, left ventricular ejection fraction; LVOT, left ventricular outflow tract; NYHA, New York Heart Association; FDA, Food & Drug Administration; LGE, late gadolinium enhancement.
Mavacamten is a first-in-class reversible cardiac myosin inhibitor that, unlike negative inotropes, exhibits a novel mechanism of action through selective modulation of the sarcomeric contractile apparatus [91].
At the molecular level, mavacamten acts as an inhibitor of the
In the landmark phase 3 EXPLORER-HCM randomized controlled trial, mavacamten
demonstrated significant efficacy in HCM patients with LVOT obstruction. This
pivotal trial enrolled 251 patients with symptomatic oHCM (NYHA class II–III,
and LVOT gradient
Since the discovery of mavacamten, several trials have been conducted to
elucidate its benefits across various patient populations. The VALOR-HCM study
enrolled 112 patients who fit the criteria for septal reduction therapy, defined
by an LVOT gradient of
The EXPLORER-CN study randomized 81 Chinese patients with oHCM to mavacamten or placebo and found a significant improvement in Valsalva LVOT gradient in the mavacamten arm [95].
It should be noted that the effects of mavacamten have only been found to be beneficial in HCM patients with LVOTO, as major landmark trials (i.e., EXPLORER-HCM, VALOR-HCM) have specifically included LVOTO patients and excluded those with mid-ventricular obstruction. As discussed in section 10, the ODYSSEY-HCM trial sought to address the role of mavacamten in non-obstructive HCM patients (including those with mid-ventricular obstruction or apical HCM). However, a recent update from the study investigators reveals no significant improvement in peak oxygen consumption or patient-reported quality of life [96, 97, 98]. Altogether, these trials showcase the significant symptomatic and hemodynamic benefits of mavacamten in patients with LVOTO and underscore its potential to provide a better quality of life for HCM patients. Fig. 5 showcases the effects of mavacamten on the LVOT gradient in the same patient pre- and post-therapy.
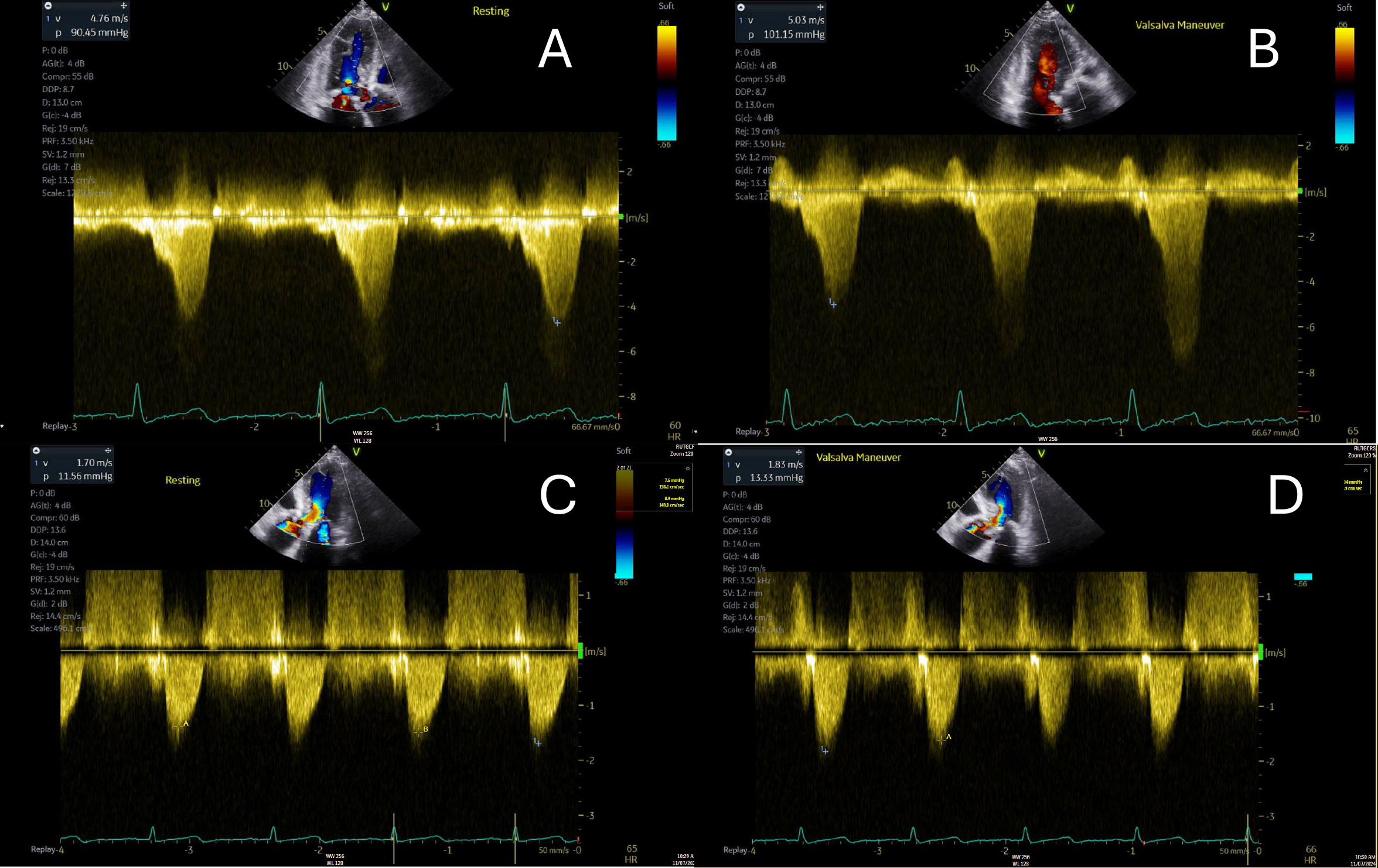 Fig. 5.
Fig. 5.
Pre- and post-mavacamten left ventricular outflow tract gradients in the same patient with hypertrophic cardiomyopathy are illustrated. (A,B) depict the pre-mavacamten gradients at rest (A) and during Valsalva maneuver (B). (C,D) present the post-mavacamten gradients at rest (C) and with Valsalva (D). Notably, there is a significant improvement post-mavacamten, with no provokable gradient observed during the Valsalva maneuver.
The principal safety concern with mavacamten is its negative inotropic effect,
manifesting as a reduction in LVEF. In the EXPLORER-HCM trial, 7 patients (5.4%)
receiving mavacamten experienced LVEF
If treatment is halted due to LVEF dropping below 50%, it is recommended to repeat TTEs every 4 weeks until LVEF exceeds 50%. Once LVEF is stable, treatment can be restarted at half the previous dose, with an uptitration after two TTEs obtained 4 weeks apart from each other show a stable LVEF [99].
Aficamten, like mavacamten, is a selective cardiac myosin inhibitor that modulates sarcomere function by reducing actin-myosin cross-bridge formation [88]. However, there are key differences between the two. Aficamten was specifically designed with a shallow dose-response curve, meaning that increases in dosage lead to only modest reductions in LVEF. This feature provides it with a broader therapeutic window compared to mavacamten [88]. Additionally, aficamten has a shorter half-life of 3.4 days, whereas mavacamten has a half-life of 7 to 9 days. This shorter half-life facilitates more rapid titration and allows for quicker reversibility after dose adjustments [85].
The SEQUOIA-HCM trial was the first major landmark trial to explore the efficacy
of aficamten among 142 of the 282 patients enrolled with symptomatic oHCM across
multiple clinically relevant endpoints [88]. At the 6-month mark, 58.5% of
patients had improvement in their baseline NYHA class by
SRTs are definitive interventions for patients with oHCM who remain symptomatic despite maximal tolerated medical therapy. These procedures should be conducted in comprehensive HCM centers (2024 ACC/AHA HCM Guideline, Class I recommendation) for optimal patient outcomes [2]. The two main types of SRT are SM and ASA.
SM is an open-heart intervention characterized by median sternotomy, cardiopulmonary bypass, aortic cross-clamping, and a transaortic approach for the excision of a segment of the IVS to alleviate the LVOTO [101, 102]. The classic Morrow procedure, introduced in the 1960s, has evolved from a straightforward excision of the basal IVS from the aortic valve annulus to a more extensive septal excision. This modern technique extends well beyond the mitral-septal contact point and involves the midventricular septum, reaching up to the level of the papillary muscles [101]. Myectomy, therefore, reliably results in the immediate and typically permanent elimination of outflow obstruction, with normalization of LV pressures and preservation of systolic function via the enlargement of the LVOT cross-sectional area and subsequent redirection of blood flow away from the anteriorly displaced MV seen in SAM. Fig. 6 and Video 3A,3B showcase the immediate results of a patient’s outflow tract gradient pre- and post-myectomy. During SM, a comprehensive evaluation via intra-, and post-operative transesophageal echocardiogram (TEE) may be done to assess the hemodynamic implications of the procedure, including the persistence of the LVOT gradient. Should such gradients remain problematic, re-establishment of cardiopulmonary bypass may be warranted to facilitate a more extensive myectomy [101].
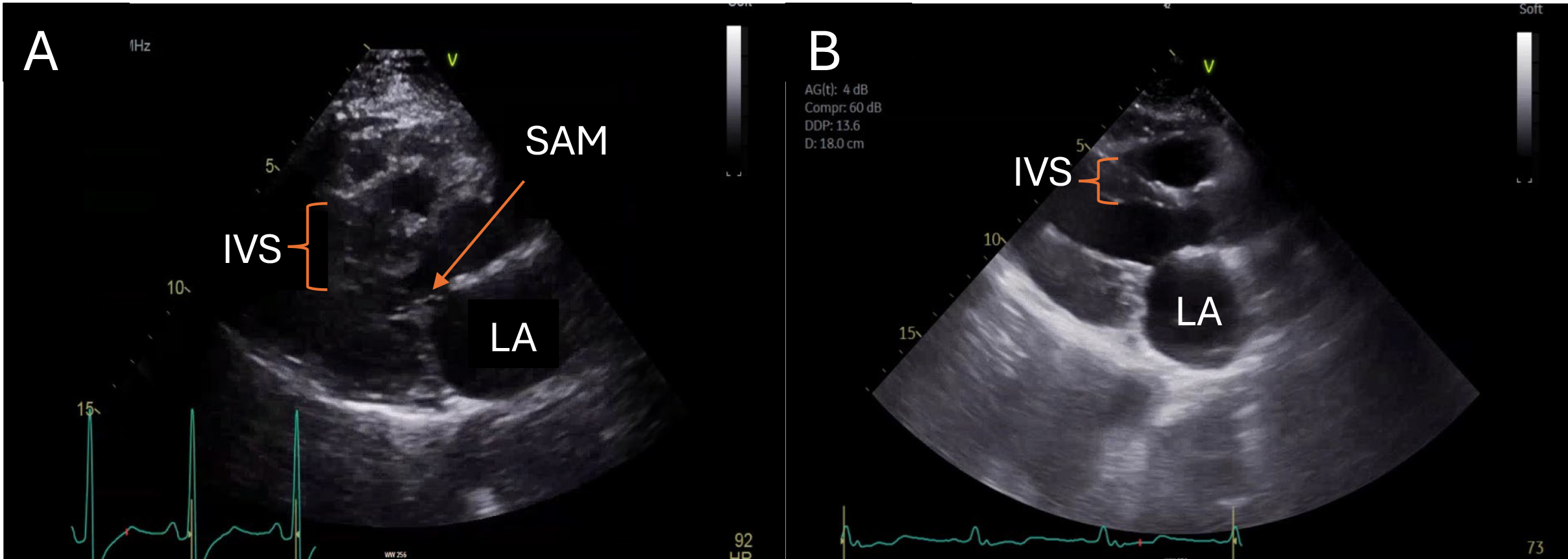 Fig. 6.
Fig. 6.
Parasternal long-axis views of a patient pre- and post-myectomy are illustrated. Systolic anterior motion of the anterior mitral valve leaflet in the setting of marked basal hypertrophy in the patient pre-myectomy (A) is no longer observed after myectomy (B). IVS, interventricular septum; SAM, systolic anterior motion; LA, left atrium.
Systolic Anterior Motion Pre- and Post-myectomy in HCM. Video 3A (still image) showcases SAM and outflow tract obstruction in a patient pre-myectomy. HCM, hypertrophic cardiomyopathy; SAM, systolic anterior motion. Videos associated with this article can be found, in the online version, at https://doi.org/10.31083/RCM42824.
Systolic Anterior Motion Pre- and Post-myectomy in HCM. Video 3B (still image) showcases post-myectomy resolution of SAM and relief of outflow tract obstruction. HCM, hypertrophic cardiomyopathy; SAM, systolic anterior motion. Videos associated with this article can be found, in the online version, at https://doi.org/10.31083/RCM42824.
Optimal candidates for SM include symptomatic (NYHA III–IV) patients with oHCM
who have additional structural diseases that require intervention. These
conditions may include anomalous insertion of the papillary muscle, severe
elongation of the anterior or posterior mitral leaflet (greater than 30 mm),
multivessel CAD, and valvular aortic stenosis [2, 101]. Additionally, patients
with AF who might benefit from intraoperative pulmonary vein isolation or a maze
procedure should also be considered for SM [2]. NYHA class II patients with
significant pulmonary hypertension secondary to LVOTO or associated MR, poor
exercise capacity, and children and young adults with severely elevated LVOT
gradients of
In general, SM, when done at comprehensive HCM centers, carries exceptional
success rates of
The importance of performing SM at comprehensive and highly skilled HCM centers is underscored when comparing the outcomes of these specialized centers with those of lower-volume facilities. For instance, data from the Mayo Clinic, which analyzed over 3000 SM procedures conducted between 1993 and 2016, revealed impressive results. Specifically, there was less than a 1% risk of post-operative death, a 2% chance of CHB that required the insertion of a pacemaker, and an iatrogenic risk of ventricular septal defect of 0.3% [107]. Additionally, 90% of severely symptomatic patients experienced an improvement of more than 2 NYHA classes, leading to a significant relief from their symptoms [107].
Similarly, Tufts Medical Center reported favorable outcomes for 482 SM procedures performed from 2003 to 2016. They recorded a post-operative mortality rate of 0.8%, with 94% of patients improving to NYHA Class I or II, owing to substantial reductions in LVOT gradients and a decrease in associated mitral regurgitation [108].
In contrast, a recent study from the National Readmission Database, which tracked patients receiving septal reduction therapy from 2010 to 2019, found that those who underwent SM at low-volume centers faced double the risk of complications, such as needing a pacemaker (11.8% compared to 6.9%). These patients also had higher rates of composite in-hospital mortality and 30-day readmissions (21.7% versus 11.8%) [109].
The significant disparities between low- and high-volume HCM centers highlight the critical need to refer patients to specialized institutions to ensure better outcomes.
ASA is a therapeutic option for adult patients with symptomatic oHCM refractory to maximal pharmacological intervention, particularly in those for whom surgical intervention is contraindicated or confers prohibitive risk due to significant comorbidities or advanced age [2]. This percutaneous interventional procedure involves the targeted delivery of alcohol via a catheter-based approach to a selectively engaged septal perforator artery, inducing a precisely controlled septal myocardial infarction [110]. Appropriate targets for ASA are determined by several factors, including the presence of basal-predominant hypertrophy, an adequate diameter of the septal artery (ranging from 1.25 mm to 2.5 mm), and the absence of concurrent surgical indications [110]. Notably, septal measurements of less than 2.5 mm are associated with a greater likelihood of benefiting from alcohol septal ablation. In contrast, cases exhibiting massive or diffuse septal hypertrophy extending into the mid-ventricle are better addressed through surgical resection, which effectively eliminates all hemodynamic gradients across the entire septal length [110].
Before initiating an ASA procedure, a transvenous pacemaker is typically positioned to mitigate the significant risk of conduction abnormalities, which ranges from 8% to 15%. The most prevalent conduction disorders observed in this context include right bundle branch block (RBBB), which occurs in approximately 50% to 70% of patients, and transient intraprocedural CHB resulting from tissue edema secondary to localized myocardial infarction [110]. Post-procedural improvements in gradients are not immediate, and the full benefits from ASA may take up to 1 year [110].
In centers with requisite expertise, ASA demonstrates a favorable procedural
safety profile with perioperative mortality rates comparable to SM at
approximately
| Advantages | Disadvantages | |
| Surgical myectomy [8, 104, 105, 110, 113, 114] | - More effective than ASA, with reduced residual outflow tract gradients | - Longer recovery time |
| - Addresses mid-ventricular and apical hypertrophy | - More invasive | |
| - Preferred for massive hypertrophy ( |
- Expertise limited to few HCM centers of excellence | |
| - Does not form myocardial scar | - Left bundle branch more likely to be damaged, posing risk of complete heart block in patients with baseline right bundle branch block | |
| - Addresses additional problems (primary mitral disease, atrial fibrillation via Maze procedure, multivessel coronary artery disease, etc.) | - Less likely to result in damage to the coronary arteries | |
| Alcohol septal ablation [8, 104, 105, 110, 113, 114] | - Shorter recovery time | - Less effective than SM |
| - Less invasive | - Higher residual outflow tract gradients compared to SM | |
| - More available than SM | - Not preferred if hypertrophy extends to mid-ventricle | |
| - Preferred in elderly patients with minimal hypertrophy | - Requires adequate septal perforator artery length in the region of interest | |
| - Comparable perioperative mortality to SM at centers of excellence | - Increased risk of ventricular arrhythmias due to scar | |
| - Higher likelihood of developing complete heart block |
ASA, alcohol septal ablation; HCM, hypertrophic cardiomyopathy; SM, surgical myectomy.
In addition to SM and ASA, there are several emerging minimally invasive techniques for HCM management. Percutaneous intramyocardial septal radiofrequency ablation (PIMSRA) was evaluated in the largest cohort study involving 200 patients with drug-refractory symptomatic oHCM, and a significant overall reduction in the LVOT gradient was noted. However, 4% of patients continued to experience exertional chest pain or dyspnea despite the procedure. Additionally, 5.5% of patients developed permanent RBBB, while 1% experienced LBBB. Notably, no patients required permanent pacemaker implantation after the procedure. These findings suggest that PIMSRA may be an effective option for treating LVOT obstruction and providing symptomatic relief [115]. Additional procedures include transapical beating-heart septal myectomy, a minimally invasive technique for septal reduction that does not require cardiopulmonary bypass or a median sternotomy. Another option is the thoracoscopic Morrow procedure, which has also been demonstrated to provide significant symptomatic relief using a minimally invasive surgical approach [116, 117]. These emerging techniques offer favorable treatment options with minimized surgical trauma, and larger studies with long-term data will be needed to compare the efficacy of the minimally invasive techniques against the traditional SM and ASA procedures.
Nonobstructive HCM is a common subset representing patients without resting LVOT gradients or dynamic outflow tract obstruction. Nonobstructive HCM physiology is generally well-tolerated, and most patients are either asymptomatic or experience minimal symptoms, such as mild exertional dyspnea, chest discomfort, or fatigue. Symptoms can stem from diastolic dysfunction leading to increased left ventricular filling pressures, microvascular dysfunction, coronary artery disease, or heart failure.
However, the management of nonobstructive HCM remains challenging since there is currently a lack of clinical trials evaluating the long-term outcomes of medical management in this subset. BBs and CCBs are used as first-line agents in treating symptomatic nonobstructive HCM since they lower heart rate and improve LV filling with decreased LV diastolic pressures. Per the 2024 ACC/AHA HCM Guideline, an oral diuretic can also be considered for symptomatic relief in volume overload conditions (class of recommendation IIa) [2]. The benefits of the treatment of asymptomatic nonobstructive HCM are not well-studied and are not recommended presently.
A minor subset (5–10%) of nonobstructive patients can progress to advanced heart failure stages (NYHA classes III/IV) with symptoms refractory to pharmacological therapy and may require consideration for heart transplant [118].
In HCM patients with systolic dysfunction, defined as a LVEF of less than 50%,
guideline-directed medical therapy (GDMT) should be initiated promptly according
to the 2022 ACC/AHA Heart Failure Guideline. HCM patients with LVEF
Previously thought to be a benign condition without a significant increase in mortality risk, recent data suggest that 1 in 3 patients with apical HCM (aHCM) may experience adverse life events such as malignant ventricular arrhythmias, SCD, and heart failure [119]. Recent data also reveals a mortality rate ranging from 0.5% to 4.8%, similar to that of typical HCM patients [119, 120]. In the absence of large randomized clinical trials, medical therapy in patients with aHCM is largely limited and primarily based on the management of classic HCM [120]. However, surgical options such as transapical myectomy and the novel transapical beating-heart septal myectomy may significantly benefit long-term survivability [120]. Lastly, while risk scores have been made to predict adverse events such as death, need for transplant, or ICD shocks, additional research is required to account for newer predictors of outcomes such as LGE [71].
The risk of stroke is significantly higher in patients with HCM, and in cases of AF, anticoagulation is highly recommended regardless of the CHADS2-VASc score. Per the 2024 ACC/AHA HCM Guideline, direct-acting oral anticoagulants are the first-line option, and Vitamin K antagonists are the second-line treatment option. Regarding management, utilizing BBs, verapamil, or diltiazem as rate control agents is acceptable. In cases of poorly tolerated AF, a rhythm control strategy can be pursued with amiodarone, which is considered an effective agent in HCM patients. Alternatively, catheter ablation can be considered in patients with severely symptomatic AF, but HCM patients have twice the risk of relapse when compared to patients without HCM, likely due to a higher degree of electrophysiologic and structural remodeling noted in these patients [121].
As per the 2024 ACC/AHA HCM Guideline, HCM patients should be counseled to engage in mild to moderate intensity recreational exercise to optimize their cardiovascular health and enhance their overall quality of life. The decision to participate in vigorous-intensity exercise, especially for athletes, relies on an individualized shared decision-making approach between the patient and an HCM expert, with extensive consideration of the potential risks and benefits. ICD implantation solely for participation in competitive sports is not recommended unless patients meet the clinical criteria described in Section 6.
Families affected by HCM bear a significant psychological burden that affects not only the diagnosed individual but also their relatives. Research has shown that asymptomatic gene-positive relatives, known as silent gene carriers, face various psychological challenges. These include shock, worry, and uncertainty about their disease status, which can lead to anxiety and depression, ranging from minimal to severe levels [122]. Additionally, relatives of silent gene carriers often seek genetic testing primarily out of concern for their loved ones rather than personal benefit [122]. Moreover, the interpretation of positive gene results among silent carriers varies widely, with some patients viewing themselves as carriers without personal risk.
In contrast, others believe they have a serious heart condition, resulting in heightened anxiety and paranoia [122]. This unhealthy thought process leads to significant behavioral changes in some patients and influences career choices, physical activity, insurance decisions, and family planning [122]. Therefore, it is essential to provide adequate counseling and clearly communicate the meaning of genetic test results and their implications to patients to alleviate any psychological burden faced by families navigating this complex disease.
The current understanding of the pathophysiology of HCM, prominently at the molecular level, continues to evolve, and the genomic database continues to expand. It is crucial to elucidate further the complex genetics in HCM, which will lead to advances in the optimization of family screening and the development of appropriate risk stratification algorithms. There is increased interest in investigating genotype-targeted therapies for HCM, such as the MyPEAK-1 study, which is a phase 1b clinical trial investigating the safety, pharmacodynamics, and tolerability of TN-201 in patients with symptomatic HCM due to mutation in MYBPC3 [10, 123]. TN-201 is a recombinant adeno-associated virus serotype 9, which contains a myosin-binding protein c transgene, and the purpose of this investigational gene therapy is to assess if delivery of a functional MYBPC3 gene to the myocardium can aid in restoring normal heart function. The use of gene editing therapies in humans presents several challenges and limitations. These include the potential for off-target effects, which may increase the risk of cancer, the immunogenicity of the delivery vector, and the need to optimize delivery to cardiomyocytes. However, the outcomes of the MyPEAK-1 trial could open the door for future genome-targeting therapies [10].
Additionally, several studies have elucidated the role of prognostic biomarkers in HCM, most notably high sensitivity troponin T and NT-proBNP, with elevated levels reflecting increased myocardial wall stress and cardiac remodeling, which has been associated with adverse outcomes in patients with HCM. Extensive prospective studies are required to explore the role of biomarkers in disease progression and classify their impact in genotype-positive, phenotype-negative family members [124, 125]. Furthermore, integration of AI in imaging is also being utilized in HCM as mentioned in Section 3.2. The ongoing studies on AI-derived imaging analysis offer a promising approach to HCM diagnosis and monitoring of disease progression [97].
On the other hand, HCM molecular pathway targeting has led to the introduction of cardiac myosin inhibitors such as mavacamten and aficamten. The ODYSSEY-HCM (A Study of Mavacamten in Nonobstructive HCM) is a phase 3 randomized, double-blind, multicenter trial that investigated the efficacy of mavacamten in nonobstructive HCM patients on the following two primary end points: change from baseline to week 48 in Kansas City Cardiomyopathy Questionnaire-23 Clinical Summary Score (KCCQ-23 CSS) and peak oxygen consumption [97]. However, early updates from the phase 3 clinical trial have not shown promising results with mavacamten administration not leading to significant improvement in quality of life or peak oxygen consumption [126]. Additionally, ACACIA-HCM (Assessment Comparing Aficamten to Placebo on Cardiac Endpoints In Adults with Nonobstructive HCM) is a phase 3 multicenter clinical trial that will evaluate the efficacy of aficamten on improvement in health-related quality of life in patients with nonobstructive HCM [127].
Furthermore, a novel cardiac mitotrope agent, Ninerafaxstat, is being investigated in the IMPROVE-HCM phase 2 proof-of-concept clinical study to assess its efficacy in treating nonobstructive HCM [128]. Ninerafaxstat targets the energy metabolism pathway and improves the energy-depleted states in HCM patients; the agent was associated with enhanced ventilatory efficiency and exercise capacity in IMPROVE-HCM. Further clinical trials are warranted in the subsets of patients with nonobstructive HCM and heart failure with preserved ejection fraction to guide appropriate management in these patients to enhance their quality of life and functional performance.
HCM is a complex clinical entity with an intricate interplay between various genetic, environmental, physiologic, and lifestyle components. There is a broad spectrum of phenotypic manifestations, and appropriate screening and risk-stratification strategies should be employed in managing these patients. Treatment involves a multi-faceted approach including a combination of lifestyle, pharmacologic, and invasive surgical techniques, and the future seems promising with several clinical studies and novel agents on the horizon.
DP and RB outlined the manuscript, conducted literature reviews, and wrote chapters in the manuscript. DP designed figures and tables in the manuscript. AM conducted literature reviews and wrote a chapter in the manuscript and contributed to a table and a figure. SB supervised, critically revised, and corrected the final manuscript. All authors contributed to the conception in the manuscript. All authors reviewed, edited, and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest. Sabahat Bokhari is serving as Guest Editor of this journal. We declare that Sabahat Bokhari had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Francesco Pelliccia.
Grammarly, an artificial intelligence tool, was utilized to help ensure the grammatical accuracy of this manuscript.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.