1. Introduction
Atrial fibrillation (AF) is a commonly diagnosed form of sustained arrhythmia,
affecting millions of patients worldwide and increasing their risk of both heart
failure and stroke. The complex etiological basis for AF has been characterized
in great detail in recent years, highlighting roles for structural remodeling,
electrical remodeling, calcium ion handling abnormalities, and dysregulated
autonomic nervous system activity [1]. Furthermore, AF is closely linked to
hypoxia. Meanwhile, hypoxia-inducible factor (HIF)-1 functions as a
central coordinator of oxygen homeostasis within cells, and its expression in
both cardiac myofibroblasts and cardiomyocytes is thought to be relevant to AF
development [2]. This review provides a detailed overview of the functional role
of HIF-1 in AF and discusses its potential therapeutic implications,
thereby laying a foundational knowledge base to support future therapeutic
strategies against this disruptive form of arrhythmia.
2. The Pathophysiology of Atrial Fibrillation
AF is characterized by an irregular, rapid atrial rhythm that is linked to a
higher risk of heart failure, stroke, dementia, cognitive dysfunction, and
mortality [3]. These affected patients experience a reduction to overall quality
of life [3]. With the accelerating aging of the global population, AF is forecast
to impose an increasingly heaving burden on healthcare systems and societies in
the coming decades.
Electrical and structural remodeling, together with the dysfunction of the
autonomic nervous system and calcium homeostasis, are closely tied to AF
progression [4]. AF is characterized by the abnormal expression of many ion
channels, including L-type Ca2+ channels (ICa,L), late Na+ channels
(INa,L), and voltage-gated K+ channels (Kv) [5]. These changes contribute to
a reduction in the atrial effective refractory period (ERP) and action potential
duration (APD), contributing to emergent conduction disturbances that result in a
feedforward loop that fuels further AF progression through persistent electrical
remodeling [5].
Myocardial fibrosis plays a central role in atrial remodeling in affected
patients. Specifically, the formation of fibrotic tissue can physically separate
longitudinal atrial myofibers such that muscle discontinuities form, establishing
a physical barrier to local signal conduction in the atrium. Interactions between
fibroblasts and cardiomyocytes can also alter cardiomyocyte conduction
properties, in turn triggering ectopic discharges and AF [6]. Both cardiac
sympathetic and parasympathetic nervous system activity are involved in the
development of AF. AF, in turn, can alter levels of autonomic excitability,
consistent with the mutually reinforcing effects of autonomic dysfunction and AF
[7]. The loss of appropriate intracellular Ca2+ homeostasis is also linked
to AF incidence, with abnormal Ca2+ channel density, for instance, leading
to atrial APD shortening and elevated Ca2+ levels within cells. These ions
can then activate protein kinase C (PKC), engaging a downstream signaling axis
that culminates in further structural remodeling and AF onset [3].
3. The Structural and Functional Properties of HIF-1
Members of the HIF family of hypoxia-sensitive transcription factors are central
to the ability of cells to detect oxygen availability and to regulate oxygen
homeostasis [8]. HIF-1 is a member of this family that is composed of the
HIF-1 and HIF-1 subunits, which are universally expressed
across mammalian cells, with HIF-1 serving as the primary regulator of
HIF-1 activity [9].
The HIF1A gene encodes HIF-1, which is expressed at high
levels in tissues exposed to hypoxic conditions and can be rapidly activated by
exposure to severe acute hypoxia (1–2% O2) [2]. Under these conditions, it
controls anaerobic glycolytic activity or cell death [2]. Under conditions with
normal O2 levels, the hydroxylation of HIF-1 by
prolyl-4-hydroxylase (PHD) enzymes, leads to its proteasomal degradation through
a process mediated by von Hippel-Lindau protein (pVHL), an E3 ubiquitin ligase
[10]. The HIF-1 Asn-803 residue can also be hydroxylated by factor
inhibiting HIF (FIH) [11], interfering with its ability to bind to
transcriptional co-activators including CREB-binding protein (CBP)/p300 [12].
Exposure to hypoxia leads to the disruption of PHD and FIH activity, preventing
HIF-1 from undergoing the associated post-translational modifications.
It instead translocates to the nucleus and heterodimerizes with HIF-1,
after which the HIF-1-HIF-1-p300-CBP complex can bind to
hypoxia-responsive elements (HREs) in the promoters of hypoxia-responsive target
genes [13]. This process ultimately leads to the upregulation of a range of
glycolytic enzymes, vascular endothelial growth factor (VEGF), erythropoietin
(EPO), and other target genes [8, 12].
4. HIF-1 as a Regulator of Atrial Fibrillation
Acute AF is characterized by an estimated 2- to 3-fold increase in cardiomyocyte
contractile and electrical activity, resulting in greater atrial oxygen and
energy consumption [14]. Despite the significant increase in atrial blood flow
relative to the sinus rhythm, cardiomyocytes experience relatively hypoxic
conditions during AF episodes [15]. A clinical study revealed a significant
increase in myocardial HIF-1 levels in the right auricle of patients
with AF compared to those in sinus rhythm [16]. After comparing left atrial
samples from patients with paroxysmal, persistent, and permanent atrial
fibrillation to those with sinus rhythm, Xu et al. [17] found that the
expression of HIF-1 in the left atrial tissues of patients with
persistent or permanent atrial fibrillation was increased compared to those with
paroxysmal atrial fibrillation or sinus rhythm. Together, these findings suggest
that HIF-1 is readily expressed by cardiomyocytes under AF conditions,
but also progressively upregulated as the disease advances.
4.1 HIF-1 Contributes to Atrial Electrical Remodeling
Electrical remodeling is central to the pathogenesis of AF, consisting primarily
of changes in the expression and activity of a range of gap junction proteins and
ion channels that result in abnormalities to cardiomyocyte repolarization,
resting potential, excitability, and conductance [18]. The activity of
HIF-1 is crucial for coordinating this electrical remodeling process.
Sarcoplasmic reticulum Ca2+ adenosine triphosphatase (SERCA) is a major regulator of
cardiomyocyte excitation-contraction coupling. In transgenic mice expressing an
oxygen-stabilized isoform of HIF-1, the cardiomyocytes exhibit marked
reductions in SERCA 2a and ryanodine receptor 2 (RyR2) transcript levels [19].
These functional RyR2 defects can increase in the release of systolic
sarcoplasmic reticulum-derived Ca2+ together with abnormally elevated
cytosolic Ca2+ concentrations, leading to APD shortening and AF onset. While
HIF-1 primarily exerts its functions by binding to HREs and inducing
the transcription of target hypoxia-responsive genes, in some instances it can
also suppress transcription in cases of reversed HRE orientation within a gene
promoter [20].
HIF-1 can also reduce target gene expression via competitive
inhibition mediated by HRE binding [21]. Ronkainen et al. [22] noted
time-dependent reductions in cardiomyocyte SERCA 2a expression under hypoxic
conditions (1% O2), and determined that desferrioxamine (DFO)-mediated
HIF-1 activation or overexpression of a normoxia-stabilized
heterodimeric form of HIF-1 (HIF-1/VP16) was sufficient to
suppress the endogenous expression and promoter activity of SERCA 2a. This
aberrant SERCA 2a functionality, in turn, leads to higher cytosolic levels of
Ca2+ and dysregulated Ca2+ activity (Ca2+ transients), culminating
with subsequent disturbances such as delayed afterdepolarization (DAD) and AF
[3]. Notably, the maintenance of SERCA activity necessitates the expenditure of
approximately 15% of cardiac energy, suggesting that reductions in SERCA 2a
expression may represent an adaptive response aimed at reducing energy
expenditure during hypoxic conditions [22].
The Na+/Ca2+ exchanger 1 (NCX1), encoded by Slc8a1, is
another key regulator of Ca2+ homeostasis [23]. Elevated levels of NCX1
protein have been observed in patients with AF, and an overly active NCX1 can
promote action potential alternans, thereby increasing susceptibility to AF [23].
Wang et al. [24] found that FK506-binding protein 5 (FKBP5) is
significantly under-expressed in atrial samples from patients with persistent
long-term AF. They used Fkbp5 knockout (Fkbp5-/-) mice, which
exhibited increased susceptibility to AF compared to controls. This is due to the
fact that both FKBP5 and HIF-1 compete to bind with heat shock protein 90 (HSP90), and reduced
FKBP5 expression increases the stability of HIF-1. As a promoter of
Slc8a1, HIF-1 upregulates NCX1 expression. Moreover, after
treatment with an HSP90 inhibitor, the levels of HIF-1 and NCX1
proteins decreased in Fkbp5-/- mice. Most importantly, the rate of
AF induction in Fkbp5-/- mice treated with the inhibitor was
significantly lower compared to untreated Fkbp5-/- mice. These
results suggest that elevated expression of HIF-1, by enhancing its
interaction with cardiac Slc8a1, promotes the occurrence of
NCX1-mediated atrial arrhythmias.
4.2 HIF-1 Induces Atrial Structural Remodeling
Atrial fibrosis is a marker of atrial structural remodeling, characterized by
the abnormal activation, proliferation, and differentiation of fibroblasts, as
well as the excessive synthesis and irregular deposition of extracellular matrix
proteins. Atrial fibrosis can be classified into two types: reactive fibrosis and
reparative fibrosis.
Reactive fibrosis, a response to cardiac inflammation or pressure overload,
manifests as perivascular and interstitial fibrosis [25]. It is commonly
characterized by the activation of fibroblasts, which proliferate and
differentiate into secretory myofibroblasts in response to various profibrotic
stimuli. This process is typically accompanied by an upregulation of matrix
metalloproteinases (MMPs) and a downregulation of tissue inhibitors of
metalloproteinases (TIMPs). Ogi et al. [26] found that
hypoxia-associated AF features upregulated HIF-1 and VEGF, which
contributie to the enhanced expression of MMP-9. In a rabbit model of
isoprenaline-induced AF, Su et al. [27] observed high levels of
angiotensin-2, HIF-1, transforming growth factor-
(TGF-), and MMP-9 expression, while also noting a positive correlation
between HIF-1 levels and the degree of myocardial fibrosis.
Accordingly, the inhibition of HIF-1 expression resulted in
corresponding decreases in TGF- and MMP-9 expression, reducing the
degree of myocardial fibrosis and thereby supporting the ability of
HIF-1 to induce AF in part through the upregulation of MMP-9 and
TGF-. These abnormalities lead to an imbalance in the deposition and
degradation of the extracellular matrix within the vascular space and cardiac
interstitium, ultimately altering the ultrastructure of the heart.
Numerous studies have explored the mechanisms whereby HIF-1 can induce
myocardial fibrosis in AF patients. Tsai et al. [28] noted that under
hypoxic conditions, HIF-1 promotes AF by inducing phosphorylation of
c-Jun N-terminal kinase (JNK) and activator of transcription factor 2 (ATF2),
along with the concomitant upregulation of proteins associated with fibrosis.
Chen et al. [29] demonstrated that HIF-1 can enhance miR-210
expression, inhibiting regulatory T cell (Treg) function via the targeting of
FoxP3, contributing to AF. Furthermore, Abe et al. [30] noted that
HIF-1 is capable of triggering inflammatory and fibrotic changes within
epicardial adipose tissue by upregulating adipose angiopoietin-like protein 2
(ANGPTL2) expression, further contributing to AF progression. HIF-1 may
thus function via multiple pathways to shape the atrial structural remodeling
observed in AF.
Reparative fibrosis occurs after extensive loss of cardiomyocytes, and its role
in the initiation and progression of atrial fibrillation remains controversial.
Generally, scarring, primarily composed of fibroblasts and extracellular matrix,
is generally considered to be non-conductive [31]. These collagen-based scars
directly interfere with conduction, reducing the occurrence of atrial
fibrillation [32]. However, at the infarct border zone, fibroblasts can couple
with cardiomyocytes via connexin 43 (Cx43). Since fibroblasts have a lower
membrane potential than the resting potential of the atria, they decrease the
resting potential of the surrounding cardiomyocytes, thereby reducing the
conduction velocity of action potentials and inducing AF. Among these mechanisms,
HIF-1 may influence the expression of Cx43 and may thus facilitate the
development and progression of AF [33].
4.3 HIF-1 Induces Atrial Fibrillation-Related Myocardial
Metabolic Remodeling
In the absence of pathological changes, cardiomyocytes primarily rely on adenosine triphosphate (ATP)
generated by mitochondrial oxidative phosphorylation as their main source of
energy, with only a minor contribution from glycolysis [34]. Most glycolytic
enzyme-encoding genes have been established as direct HIF-1 targets
that can be induced under inflammatory or hypoxic conditions [35]. In hypoxic
settings, HIF-1 is directly involved in the transitioning of cells
between oxidative phosphorylation and glycolysis. Initially, HIF-1 can
promote the upregulation of pyruvate dehydrogenase kinase 1 (PDK1), leading to
the dephosphorylation of pyruvate dehydrogenase (PDH) involved in the
tricarboxylic acid cycle and the blockade of pyruvate conversion into acetyl-coenzyme A (CoA).
Furthermore, HIF-1 can induce the upregulation of glycolytic enzymes
and the glucose transporter 1 (GLUT1) and glucose transporter 3 (GLUT3), enhancing glucose uptake
within cells to help ensure an adequate supply of ATP [36].
HIF-1 can also drive lactate dehydrogenase A (LDHA) expression,
resulting in the conversion of pyruvate to lactate and the regeneration of
nicotinamide adenine dinucleotide (NAD)+ for further glycolytic cycling mediated by glyceraldehyde 3-phosphate dehydrogenase (GAPDH) [35]. Additionally,
monocarboxylic acid transporter protein 4 (MCT4) plays a crucial role in the
transport of lactate out of cells [37]. A shift away from oxidative
phosphorylation in favor of glycolytic dependence results in a reduction in the
consumption of oxygen necessary to produce ATP, leading to a drop in
mitochondrial reactive oxygen species (ROS) biogenesis, shielding cells against
oxidative injury [38, 39]. As glycolytic intermediates, lactate and pyruvate can
also directly prevent the release of Ca2+ from the sarcoplasmic reticulum
through a reduction in ryanodine receptor (RyR) activity [40]. Enhanced phosphofructokinase (PFK)
activity can induce pathologic cardiac hypertrophy and influence the expression
of key cardiac metabolism- and remodeling-related genes [41]. The enhanced
production of lactate and associated lactate signaling activity have been
established as a key regulator of atrial structural remodeling linked to
oxidative stress-related damage and mitochondrial apoptosis [42].
Cardiac tissue primarily relies on fatty acid oxidation to generate 60–90% of
its total ATP, with pyruvate oxidation contributing the remaining 10–40% [35].
In individuals with permanent AF, this metabolic balance is disrupted.
Transcriptomic analyses have shown a downregulation of key enzymes involved in
fatty acid oxidation [43]. Krishnan et al. [44] demonstrated that
activation of the HIF1-peroxisome proliferators-activated receptors
(PPAR) pathway leads to disrupted myocardial metabolism.
This activation results in HIF-1 upregulating glycolytic genes, while
PPAR enhances glycolytic flux and the expression of fatty acid uptake
genes, particularly affecting the glycerol-phosphate pathway. Concurrently, there
is a decline in the expression of critical enzymes for fatty acid metabolism,
such as carnitine palmitoyltransferase-1 (CPT-1), and a reduction in triglyceride
oxidative utilization, leading to triglyceride accumulation and cardiac
steatosis. Furthermore, HIF-1 can activate caspase-3 through the
PPAR/octamer-binding transcription factor 1 (Oct1)/growth arrest and DNA damage-inducible alpha (GADD45A) axis, triggering cardiomyocyte apoptosis. This
cascade of events prompts compensatory responses, including myocardial
hypertrophy and fibrosis, ultimately contributing to the development of AF.
5. Clinical Prospects and Challenges
Recent studies [16, 17] underscore the correlation between HIF-1 expression and
AF incidence, suggesting that the HIF signaling axis may serve as a target for
novel therapeutic interventions. Specifically, pharmacological modulation of
HIF-1 activity can improve atrial structural and electrical remodeling,
reducing the burden of AF. Metformin, for instance, is a commonly prescribed
hypoglycemic drug that reportedly exerts cardioprotective activity. In an animal
study, metformin administration improved cardiomyocyte lipid metabolism, a
protective effect linked to the inhibition of HIF-1 expression and a
subsequent reduction in downstream PPAR levels mediated by activation
of adenosine monophosphate activated protein kinase (AMPK) [45]. Furthermore, Bi
et al. [46] determined that LDN-57444 can reduce LV remodeling,
inflammation, and abrogating oxidative stress induced by angiotensin-2 by
ubiquitin C-terminal hydrolase L1 (UCHL1). This ultimately curtailed AF
incidence and duration by inhibiting the activation of atrial HIF-1,
TGF-, and Smad 2/3 signaling. Several antitumor drugs have also been
designed to target HIF-1, including 32-134D, PX-478, and acriflavine.
The safety and efficacy of these drugs in AF patients, however, has yet to be
established [8, 47].
AF has a complex pathogenesis, and the specific contributions of HIF-1
warrant further study. While HIF-1 can reduce oxidative
phosphorylation-mediated ROS biogenesis, under conditions of intermittent hypoxia
it can also upregulate NADPH oxidase 2 (NOX2) and inhibit mitochondrial electron transport chain
complexes I and III, resulting in higher levels of ROS production [48]. The
degree to which HIF-1 can promote AF development, through changes in
ion channel concentrations and autonomic nervous function, will also require
further study. Moreover, comprehensive efforts are needed to characterize the
links between common therapeutic agents, including metformin or antiarrhythmic
drugs, and HIF-1. These studies will be critical for the production of
new drugs that can aid in the prevention and treatment of AF.
6. Conclusions
In conclusion, HIF-1 is an essential regulator in AF pathophysiology.
At the cellular level, HIF-1 can contribute to the exacerbation of
atrial structural, electrical, and metabolic remodeling which disrupt normal
electrophysiological activities, cellular structures, and cardiomyocyte energy
metabolism. These adverse modifications perpetuate a deleterious feedforward
cycle and a worsening of AF (Fig. 1). Interventional strategies focused on
targeting HIF-1 hold promise as a means of managing patients suffering
from this form of arrhythmia. The modulation of HIF-1 activity may help
disrupt the progressive electrophysiological and metabolic deterioration that
characterizes AF progression within the atria, thereby preventing disease
progression and potentially reversing the course of the disease. However, further
research is essential to determine the safety and efficacy of these treatments,
aiming to provide AF patients with more efficacious and precise pharmacological
options for disease management.
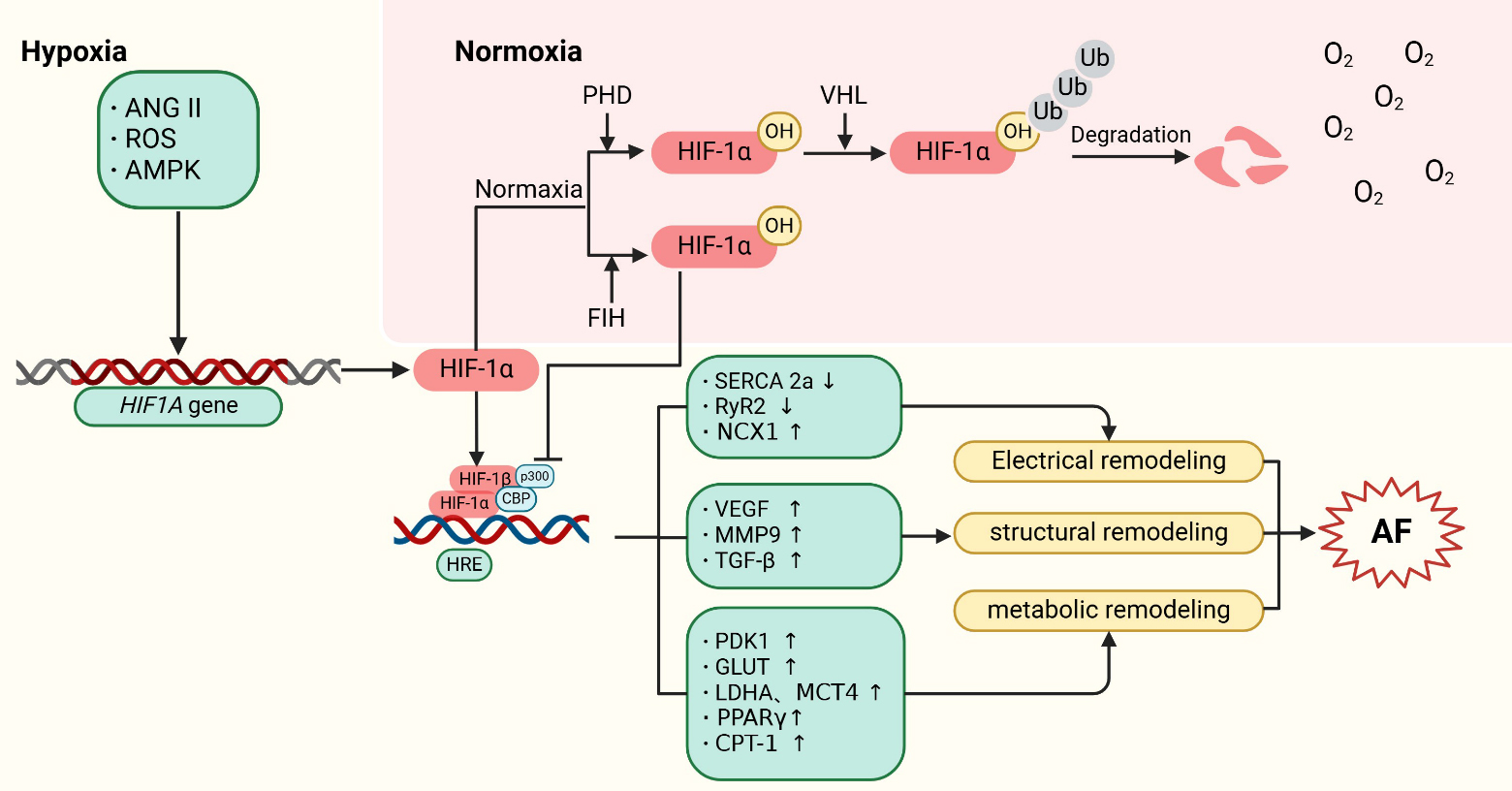 Fig. 1.
Fig. 1.
Mechanisms of HIF-1 regulation in atrial
fibrillation. HIF-1 is encoded by the HIF1A gene. Under
normoxic conditions, HIF-1 is hydroxylated by PHDs, leading to its
subsequent proteasomal degradation following ubiquitination by pVHL.
Additionally, factor inhibiting HIF (FIH) hydroxylates an asparagine residue on
HIF-1, reducing its interaction with CBP/p300 and thus inhibiting its
transcriptional activity. Under hypoxic conditions, upstream factors such as
ANGII, ROS, and AMPK stimulate the expression of the HIF1A gene.
Concurrently, the activities of PHDs and FIH are reduced, allowing
HIF-1 to translocate to the nucleus. There, it forms a complex with
HIF-1 and CBP/p300, and binds to hypoxia-response elements (HREs) in the
promoter regions of HIF target genes. This interaction activates the expression
of downstream proteins, contributing to the development of AF. Specifically,
HIF-1 promotes atrial structural remodeling by inducing the expression
of target genes such as VEGF, MMP9, and TGF-. It also drives atrial
metabolic remodeling through the upregulation of PDK1, GLUT, LDHA, MCT4,
PPAR, and CPT-1. Furthermore, HIF-1’s binding to HRE
sequences competitively inhibits the expression of SERCA 2a and RyR2, thereby
promoting atrial electrical remodeling.
ANGII, angiotensin II; ROS, reactive oxygen species; AMPK, adenosine
monophosphate activated protein kinase; HIF, hypoxia-inducible factor; PHD,
prolyl-4-hydroxylases; pVHL, von hippel-lindau proteins; HRE, hypoxia response
elements; FIH, factor inhibiting HIF; SERCA, sarcoplasmic reticulum Ca2+ ATPase; RyR2, ryanodine
receptor 2; NCX1, Na+/Ca2+-exchanger 1; VEGF, vascular endothelial
growth factor; MMP9, matrix metalloproteinase 9; TGF-, transforming
growth factor ; PDK1, pyruvate dehydrogenase kinase 1; GLUT, glucose
transporters; LDHA, lactate dehydrogenase A; MCT4, monocarboxylate transporter 4;
PPAR, peroxisome proliferators-activated receptors ; CPT-1,
carnitine palmitoyl transferase-1; AF, atrial fibrillation; VHL, von hippel-lindau; Ub, ubiquitin; CBP, CREB-bindingprotein.
Author Contributions
JZ, TW and RW selected the topic, prepared the initial manuscript draft and searched the literature. DW and FZ assisted in reviewing the literature, generated all figures and revised the manuscript critically for important intellectual content. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Ethics Approval and Consent to Participate
Not applicable.
Acknowledgment
Not applicable.
Funding
This project was supported in part by the National Natural Science Foundation of China (82370342) and Natural Science Foundation of Jiangsu Province (BK20231145).
Conflict of Interest
The authors declare no conflict of interest.



