





1 Senior Department of Cardiology, Sixth Medical Center of Chinese PLA General Hospital, 100853 Beijing, China
2 Medical School of Chinese PLA, 100853 Beijing, China
3 Emergency Department, First Medical Center of Chinese PLA General Hospital, 100853 Beijing, China
Abstract
With advances in therapies to reduce cardiovascular events and improvements in coronary imaging, an increasing number of clinical trials have demonstrated that treatments to reduce cardiovascular events in coronary artery disease are associated with favorable effects on atherosclerotic plaque size and characteristics. It has been observed that various drugs may induce plaque regression and enhance plaque stability after plaque formation. Numerous clinical trials have been conducted to verify the occurrence of plaque stabilization and regression and their beneficial effects on cardiovascular events. Using invasive imaging techniques such as intravascular ultrasound (IVUS) and optical coherence tomography (OCT), researchers have been able to gather evidence supporting the existence of coronary plaque stabilization and regression. In this review, we explore the possible mechanisms of plaque stabilization and regression, summarize the imaging features of plaque stabilization and regression, and assemble the evidence from clinical studies that have used different features as observational endpoints.
Keywords
- atherosclerotic
- plaque stabilization
- plaque regression
- lipid-lowing therapy
- intravascular ultrasound
- optical coherence tomography
Atherosclerosis is a chronic, progressive disease process with a long latency period, and clinical manifestations may not become apparent for two or three decades [1]. With increasing research into the pathogenesis of atherosclerosis, it is increasingly recognized that lipid deposition and macrophage infiltration at arterial wall lesions are reversible processes, and the hypothesis that the atherosclerotic process in humans can be reversed and regressed has persisted for decades [2]. After plaque formation, plaque volume and composition can be altered by a variety of therapies, including lipid lowering [3]. The use of invasive and non-invasive imaging techniques has made it possible to assess plaque burden and local plaque characteristics and to confirm that plaque stabilization and regression are real and reliable [4]. In recent years, study have confirmed the correlation between plaque stabilization and regression with reduced major adverse cardiovascular events (MACE) [5].
The aim of this review is to explore the possible mechanisms of plaque stabilization and regression, summarize the imaging features and clinical evidence, and predict the future research direction.
The researchers independently conducted a computerized literature search of three databases, PubMed, Embase, and Web of science, from 2018 to 2024. The search was conducted using the relevant search terms: “atherosclerotic plaque regression” or “plaque regression” and “plaque stabilization”, and more than 180 articles were retrieved. In addition, all the references of the retrieved articles were assessed for more information.
Plaque regression has traditionally been defined as an increase in lumen diameter as measured by coronary angiography [6]. Second, due to advances in intraplaque imaging, a reduction in atherosclerotic plaque volume as well as a reduction in markers related to plaque vulnerability can also be considered plaque regression [7, 8, 9]. However, there is no consensus about whether increased plaque stability is part of plaque regression.
Clinically, the development of therapies that result in regression and increased stability of atherosclerotic plaque is a desirable therapeutic goal for coronary heart disease, as most patients begin treatment after plaque formation. However, plaque regression is considered challenging due to the biological nature of advanced lesions, including necrotic cores, calcification, and fibrosis. Fortunately, many clinical studies have provided solid evidence of plaque stabilization and regression in humans following aggressive lipid-lowering therapy, giving researchers firm confidence to further explore the underlying mechanisms that regulate this complex process.
The mechanism of plaque stabilization and regression is not in itself equivalent to a reversal of the plaque development process, but rather consists of several important processes: the removal of lipids and necrotic components from the intima, the cessation of cell proliferation and undergoing phenotypic transformation, increase in the thickness of the fibrous cap, and increase in plaque calcification [10] (Fig. 1).
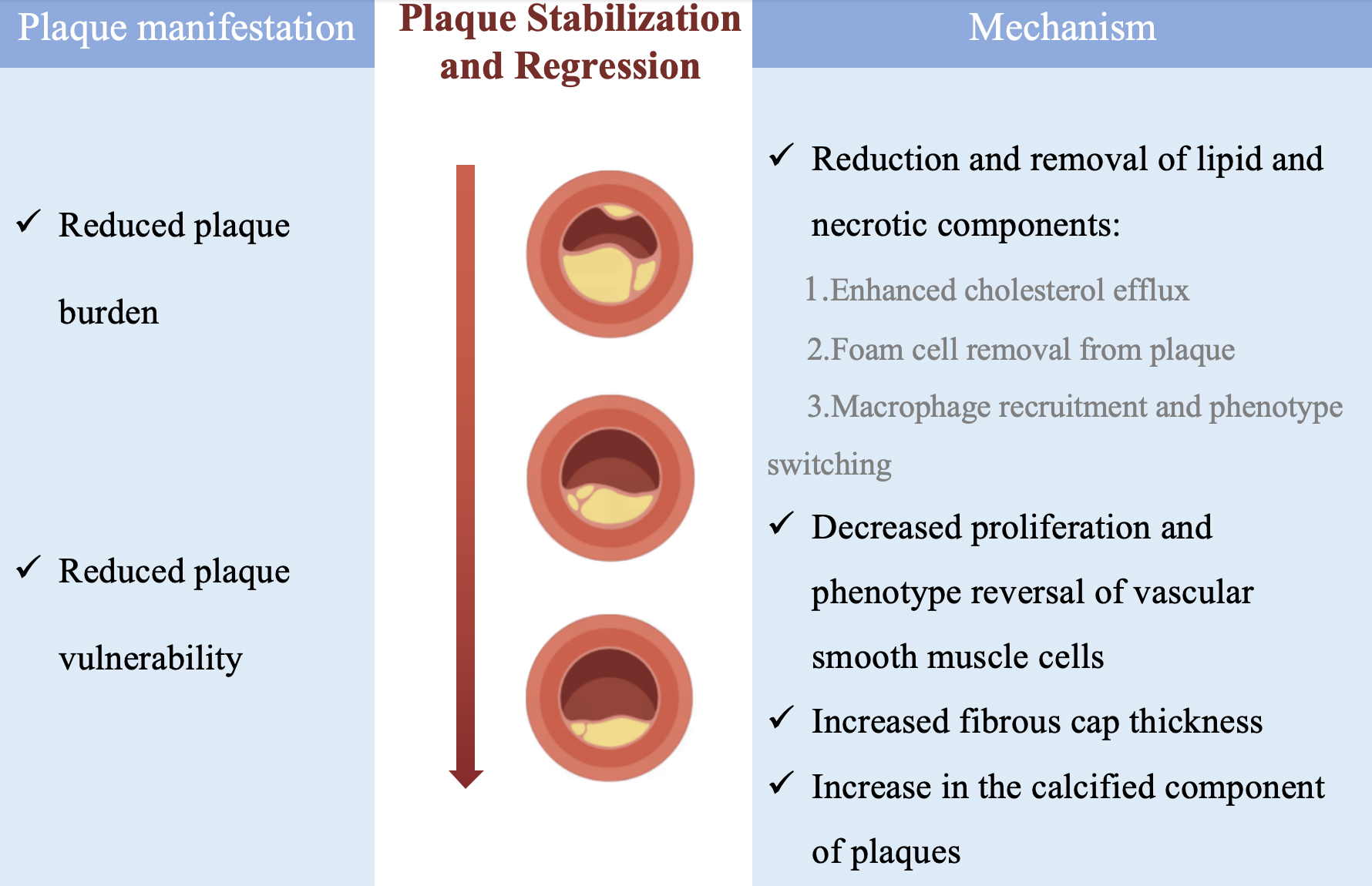 Fig. 1.
Fig. 1.
Possible mechanisms of plaque stabilization and regression. Manifestations and possible mechanisms of plaque stabilization and regression.
The most important step in plaque regression is the removal of lipids and necrotic cores from the plaque by various routes.
Animal study confirm that plaque regression is often accompanied by an increase in reverse cholesterol transport (RCT): the transfer of lipids from the periphery to the liver and their elimination from the body via the hepatobiliary pathway [11]. Promoting cholesterol efflux from macrophages is the first step in RCT. High-density lipoprotein (HDL) is essential to the RCT pathway as a cholesterol receptor. Several studies in large populations have shown that the ability of HDL to remove cholesterol is negatively correlated with cardiovascular disease [12, 13]. A preclinical evaluation showed that intravenous administration of autologous defatted HDL was able to result in a significant reduction in plaque volume by 6.9% [14].
One study silenced the expression of the macrophage surface protein Epsins, which enhances endocytosis of CD36 molecule to promote lipid uptake and impedes ATP-binding cassette subfamily G member 1 (ABCG1)-mediated cholesterol efflux, at the genetic level, and observed a reduction in necrotic core regions, a decrease in macrophage clusters, and a decrease in foam cell formation [15]. The results suggest that tilting the macrophage lipid balance toward cholesterol efflux may accelerate the achievement of plaque regression. In conclusion, enhanced transport of excess cholesterol from arteriolar lipid-rich lesions has become an important approach to anti-atherosclerotic drug development.
On the other hand, enhancing hepatic lipid uptake and clearance is also a feasible way to promote plaque regression. Ishigaki et al. [16] utilized viral transfection to ectopically express lectin-like oxLDL receptor-1 (LOX-1) in the liver to promote plasma oxidized low-density lipoprotein cholesterol (oxLDL-C) uptake and clearance. The results showed that the atherosclerotic plaques were almost completely regressed, strongly confirming the key role of lipid clearance in plaque regression.
Apoptosis and necrosis of foamy macrophages form the necrotic core. There is growing evidence that removal of foamy macrophages is associated with increased plaque stability and can occur concurrently with a decrease in plaque volume [17].
Some immune cells are involved in foam cell clearance. A mouse model of atherosclerosis suggests that enrichment of plaque regulatory T cells (Treg) is a common feature of plaque volume reduction, and that Treg are required for reduction of plaque burden, reduction of inflammation, and repair of arterial wall tissues. RNA sequencing of immune cells in plaques and flow cytometry suggest that Treg in plaque regression may originate from splenic induction of generation. Second, to further validate whether Treg is required for plaque stabilization and regression, removal of Treg using a CD25 monoclonal antibody in a mouse model showed that reduction of Treg prevented plaque stabilization and regression achieved by lipid-lowering therapy, possibly by inhibiting macrophage migration, preventing macrophage phenotypic switching, and hindering macrophage pro-resolving functions [18]. In addition, peripherally derived CD4+ T cells can be involved in regulating the onset of plaque stabilization and regression by altering the balance of effector T cells and M1:M2 macrophages, promoting the clearance of dead cells to reduce plaque necrosis, and stimulating the production of lipid mediators for inflammatory regression.
In addition, an aortic graft model showed that the route of destination for
foamy macrophages in plaques was the peripheral lymph nodes, and epigenomic
analysis of the transcriptome of plaque foamy macrophages revealed that the
Wnt-
However, macrophage recruitment is also thought to be responsible for plaque stabilization and regression, but this is not contradictory. Although macrophage recruitment and interactions with other cells directly induce plaque formation, it is also necessary for plaque stabilization and regression. Plaque stabilization and regression does not occur without newly recruited macrophages from the circulatory system.
A shift in macrophage polarization towards an M2 phenotype has also been
suggested to be a feature of plaque regression. Rahman et al. [17]
established an aortic graft model in which an enrichment of activated M2
macrophage markers was observed in plaque-incurring mice and confirmed that they
were derived from newly recruited Ly6Chi monocytes by single-cell RNA sequencing.
It was further found that it may induce M2 macrophage polarization by stimulating
the peroxisome proliferator-activated receptor-
In addition, study exploring the mechanisms of plaque regression has been conducted in humans. One study [23] achieved significant plaque regression by increasing the expression of proresolving lipid mediators in patients with stable angina pectoris (CAD) treated with statins, which was attributed to a decrease in the uptake of low-density lipoprotein cholesterol (LDL-C) by macrophages as well as enhanced phagocytosis by macrophages due to proresolving lipid mediators.
In conclusion, promotion of reverse cholesterol transport, promotion of macrophage-derived foam cell plaque efflux, and promotion of M2-type macrophage transformation are feasible ways to enhance plaque lipid efflux and increase stability (Fig. 2).
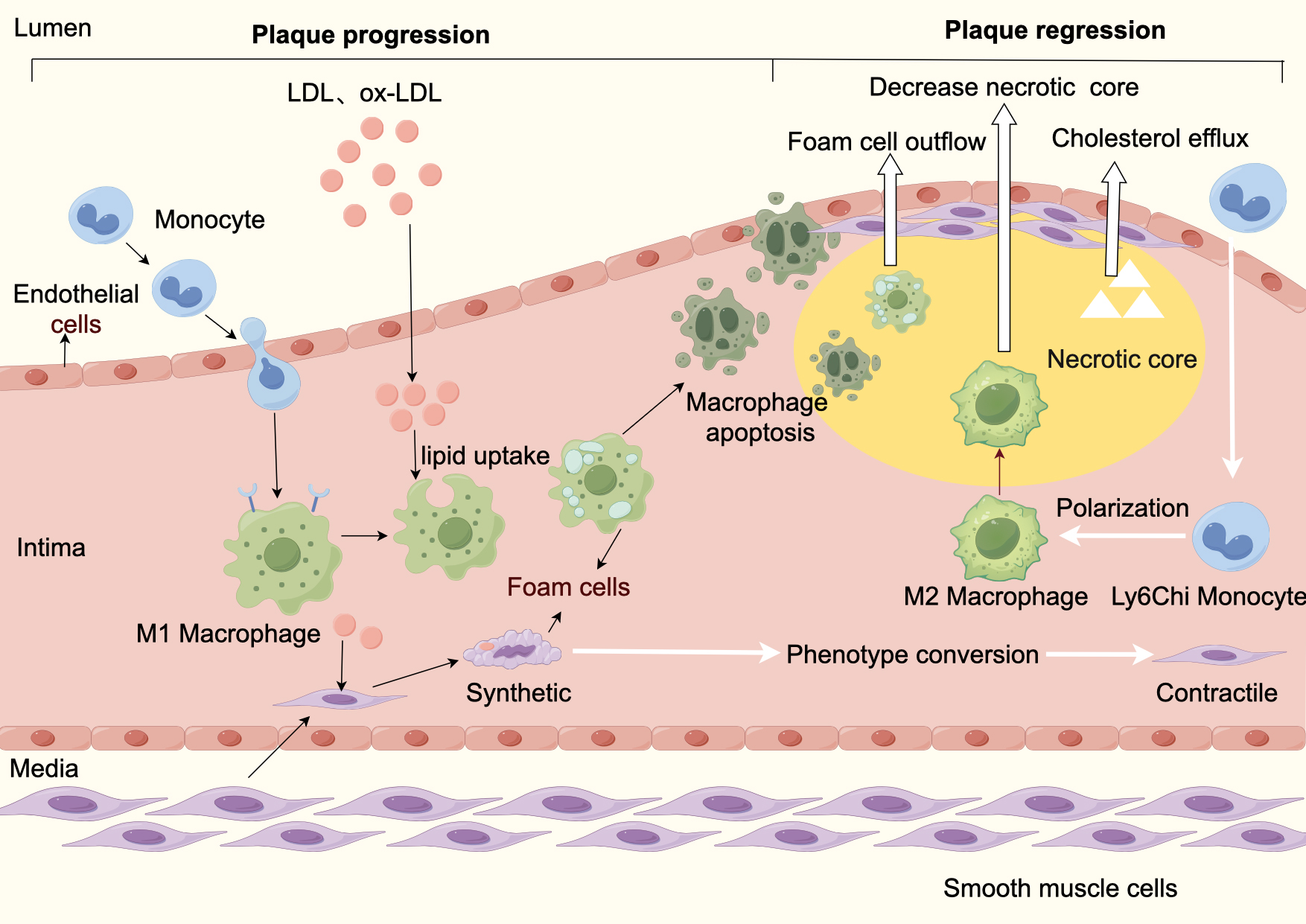 Fig. 2.
Fig. 2.
Atherosclerosis progression and regression. Plaque progression: Plaque formation begins with endothelial dysfunction caused by various pathogenic factors. Impaired endothelial function leads to increased permeability, which causes abnormal accumulation of lipid particles (mainly LDL) in the blood under the endothelium, which then attracts circulating monocytes to migrate to the sub endothelium and differentiate into macrophages, which phagocytose LDL-C to form foam cells. In addition, smooth muscle cells from the media shift from a contractile phenotype to synthetic phenotype phagocytizing lipids to form smooth muscle cell-derived foam cells. Foam cells apoptosis and secondary necrosis form a necrotic core, and synthetic-type smooth muscle cells proliferate and produce collagen, forming a fibrous cap and ultimately an atherosclerotic plaque. Plaque stabilization and regression: Lipid efflux and foam cell efflux cause a decrease in the necrotic core. LDL, low density lipoprotein cholesterol; Ox-LDL, oxidized low-density lipoprotein cholesterol; M1, pro-inflammatory macrophage phenotype; M2, anti-inflammatory macrophage phenotype.
Smooth muscle cells can serve as another important source of foam cells by mediating cholesterol influx via surface receptors. In addition, certain pro-inflammatory cytokines and growth factors triggered by the inflammatory process can promote the migratory proliferative phenotype of smooth muscle cells, which can lead to remodeling of the overall structure of the arterial wall and promote plaque progression [24]. However, the role of smooth muscle cells in plaque regression is poorly understood. One study attempted to remove lipids from smooth muscle cells and inhibit their proliferation and observed plaque regression accompanied by phenotype transformation. Possible mechanisms are that macrophage migration from the plaque area mediates a reduction in the inflammatory process within the plaque and a reduction in the lipid component that promotes phenotype switching of pathologic smooth muscle cells [10].
At present, no studies have detected changes in the phenotype and number of smooth muscle cells in animal models, and it is not clear whether this is the mechanism of plaque regression.
Thin-cap fibroatheroma (TCFA) is a plaque morphology often considered to be
prone to rupture and is usually defined as a plaque with necrotic nuclei and
macrophages near the arterial lumen with a thin fibrous cap measuring
Recent clinical studies have shown that statins can reduce plaque burden by decreasing the percentage of atherosclerotic plaque volume and total volume, while calcification volume increases [26]. Previous studies on the relationship between coronary calcification and plaque stability or instability have not been adequately investigated in clinical studies and remain controversial. However, recent pathologic and imaging study has shown that lamellar calcification is a marker of plaque stability, whereas minute, punctate, or fragmentary calcification is associated with early plaque or unstable plaque [26].
The ARCHITECT study characterized plaque throughout the coronary tree by
coronary computed tomography angiography (CCTA) in patients treated with a
combined lipid-lowering regimen and showed that the use of the combined
lipid-lowering regimen was observed at follow-up to be associated with a
significant reduction in plaque burden (–4.6%, p
Although imaging can assess metrics and estimate the incidence of plaque stabilization and regression, imaging tools do not capture the true complexity of the process, and the processes and mechanisms involved in the occurrence of plaque stabilization and regression remain to be discussed.
Analyzing the definition of plaque regression, the change in plaque volume is the most intuitive indicator of the occurrence of plaque regression. In addition, significant changes in plaque characteristics are also considered diagnostic criteria for increased plaque stability [28]. Multiple imaging strategies are available for comprehensive assessment of plaque. They can be categorized as invasive and noninvasive. Invasive imaging modalities include conventional coronary angiography, intravascular ultrasound (IVUS), optical coherence tomography (OCT), near-infrared spectroscopy (NIRS), and other intravascular imaging. Non-invasive imaging modalities include computed tomography angiography (CTA) and cardiac magnetic resonance imaging (MRI) (Fig. 3).
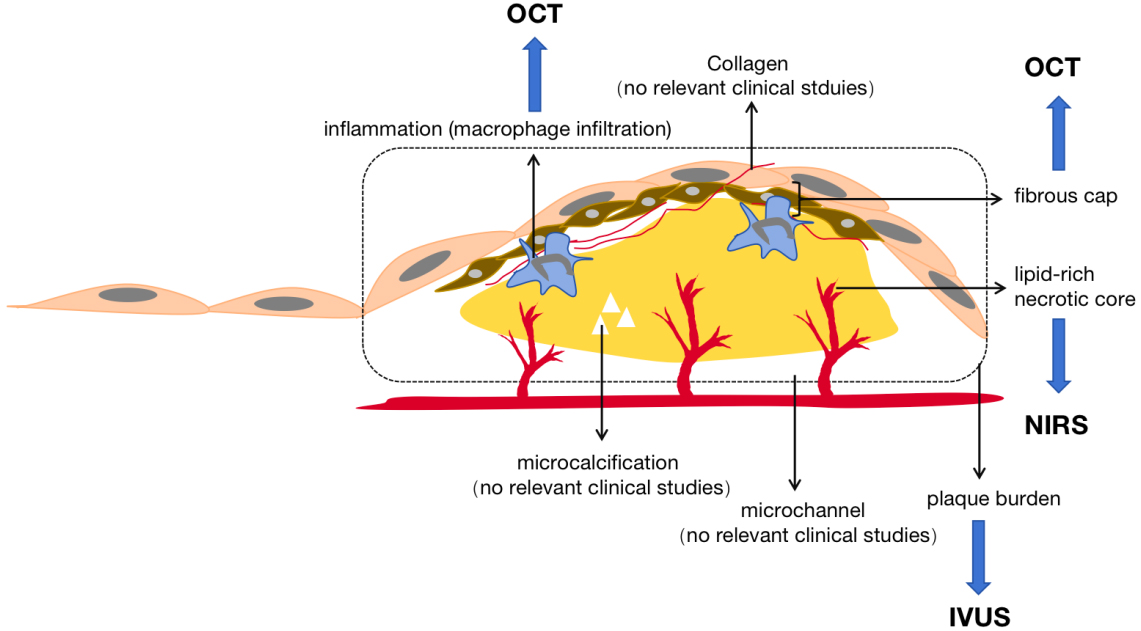 Fig. 3.
Fig. 3.
Plaque imaging modalities. Plaque imaging modalities. IVUS, intravascular ultrasound, used for plaque burden measurement; OCT, optical coherence tomography, used for measurement of plaque vulnerability characteristics; NIRS, near infrared spectroscopy, used for measurements on lipid-rich cores.
Conventional coronary angiography (CAG) provides information about the vessel lumen and can identify segments of vessels that visually cause significant narrowing of the lumen. However, because it is limited to the lumen and lacks information about plaque burden and composition, it is less commonly used to assess plaque stabilization and regression.
IVUS is the traditional standard for assessing plaque burden in clinical trials and has been used as a diagnostic imaging modality for plaque stabilization and regression imaging in many clinical studies. IVUS not only distinguishes plaques into fibrous, fibrofatty, calcified, and calcified necrotic subtypes, but also accurately measures plaque burden including atherosclerotic plaque area, total atherosclerotic plaque volume (TAV), and percent atherosclerotic plaque volume (PAV) [29]. These parameters can be measured over time to quantify plaque progression and regression [30].
OCT, a light-based modality for intravascular imaging of catheter delivery, has the advantage of high-resolution characterization of the surface elements of the vessel wall, allowing assessment of plaque stability by differentiating plaque morphology, estimating fibrous cap thickness, and showing the extent of macrophage infiltration. In recent years, many clinical trials have used OCT as an imaging modality to assess the effects of various drugs on plaque regression using plaque characteristics and composition as observational endpoints (including lipid content, presence of macrophage clusters, microcalcifications), with positive results. OCT is the gold standard for correctly measuring fibrous cap thickness in vivo, and there is a good correlation between the examined fibrous cap thickness and histology [31]. TCFA characterized by large necrotic cores and a thin fibrous cap overlying them are considered precursor lesions to plaque rupture leading to acute events. Radiofrequency (RF)-IVUS and OCT are the two intracoronary imaging techniques for detecting TCFA. Diagnosis of RF-TCFA is visually assessed based on the number and location of necrotic cores, regardless of cap thickness. Previous clinical studies have demonstrated that the presence of TCFA defined by RF-IVUS-defined presence of TCFA is associated with future MACE. A direct comparison regarding the diagnostic assessment of TCFA between RF-IVUS and OCT was confirmed by the IBIS-4 study [31] to be significantly inconsistent. This discrepancy may be due to the resolution of IVUS and the inherent limitations of assessing the necrotic core [31]. However, due to its poor penetration, it is often not used for plaque burden measurement [3]. The presence of macrophage clusters is one of the OCT predictors of future coronary events. The ability of plaque OCT to recognize macrophages has been validated, however different subtypes of macrophages in the plaque background could not be distinguished [32]. NIRS allows for quantitative analysis of lipid content in plaques and is used to assess the effect of lipid-modifying therapies on lipid content in plaques. The LRP and the PROSPECT II study reported that the lipid core burden index assessed by NIRS was significantly associated with future MACE [31]. While NIRS is the gold standard for in vivo lipid identification, it cannot distinguish between superficial and deep lipid pool. Thus, it is usually combined to IVUS catheter or more recently to OCT. Undeniably, the combination of NIRS and IVUS is still unable to accurately determine the location of plaque lipid as the lipid signal is characterized by a circumferential arc of its location around the IVUS image although not addressing the depth of the signal.
With advancements in technology, the current trend favors the use of noninvasive imaging for plaque assessment that can provide additional information beyond stenosis and plaque with minimal risk. Previous study has shown that plaque measurements using cardiac CTA are highly accurate compared to IVUS [33]. CTA has been shown to identify high-risk plaques associated with plaque vulnerability features. It can also show a strong correlation with macrophage infiltration observed on OCT by positive remodeling and low attenuation of plaque in the image [34]. Recent consensus has discussed the advantages and disadvantages of various imaging modalities for plaque and stenosis assessment, and CTA is considered the most suitable imaging modality for plaque assessment, making it the most valuable technique for practical clinical situations. In contrast, IVUS was considered suitable for assessing plaque composition and detecting culprit plaques [35].
Cardiac MRI has great advantages in assessing coronary artery wall thickness and positive remodeling, but it is not commonly used for coronary artery assessment because of the high level of expertise required for image recognition, the long examination time, and its applicability to large arteries [36].
Current clinical interventions for plaque stabilization and regression include pharmacologic as well as nonpharmacologic therapies. Pharmacological therapies are divided into two categories based on whether they target LDL-C.
Lowering LDL-C levels is the mainstay of plaque stabilization and regression. Most current clinical trials have focused on whether lipid-lowering therapies induce plaque regression or increase plaque stability (Table 1, Ref. [37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50]). High-quality clinical evidence exists to support the use of various lipid-lowering regimens to stabilize and regress coronary plaque.
| First author, year | Treatment | Imaging mode | Sample size | Follow-up weeks | Positive indicators related to plaque regression | Main findings |
| Nicholls, 2011 [38] | Atorvastatin 80 mg/d | IVUS | 1039 | 104 weeks | PAV | The high-intensity lipid-lowering treatment group had lower LDL-C levels, a greater reduction in PAV (–1.22% vs. 0.99%), and a more pronounced effect on TAV (–6.39 mm3 vs. –4.42 mm3). |
| Rosuvastatin 40 mg/d | ||||||
| Okazaki, 2004 [37] | Atorvastatin 20 mg/d | V-IVUS | 70 | 6 months | PAV | Plaque volume was significantly reduced in the atorvastatin group (13.1 |
| Percent change in plaque volume showed a significant positive correlation with follow-up LDL-C level (R = 0.456, p | ||||||
| Nissen, 2006 [49] | Rosuvastatin 40 mg/d | IVUS | 507 | 24 months | PAV, TAV | The mean (SD) change in PAV for the entire vessel was 0.98% (3.15%); Change in TAV showed a 6.8% median reduction. |
| Nissen, 2005 [50] | Atorvastatin 80 mg/d | IVUS | 502 | 18 months | PAV, TAV | Progression of coronary atherosclerosis occurred in the pravastatin group (2.7%; 95% CI: 0.2% to 4.7%; p = 0.001) compared with baseline. Progression did not occur in the atorvastatin group (−0.4%; 95% CI: −2.4% to 1.5%; p = 0.98) compared with baseline. |
| Pravastatin 40 mg/d | ||||||
| Tsujita, 2015 [39] | Atorvastatin 10 mg/d | IVUS | 202 | 9–12 months | PAV | The absolute change in PAV did show superiority for the dual lipid-lowering strategy (–1.4%; 95% CI: –3.4% to –0.1% vs. –0.3%; 95% CI: –1.9% to 0.9%, p = 0.001). |
| Atorvastatin + Ezetimibe 10 mg/d | ||||||
| Nicholls, 2016 [40] | Evolocumab 420 mg/L month | IVUS | 968 | 76 weeks | PAV, TAV | The primary efficacy parameter, PAV, increased 0.05% with placebo and decreased 0.95% with evolocumab (difference, −1.0% [95% CI: −1.8% to −0.64%], p |
| Yano, 2020 [42] | Rosuvastatin 5 mg/d | OCT | 58 | 4–12 weeks | Lipid content, Macrophage content | OCT analysis revealed that the increase in fibrous-cap thickness and decrease in macrophage grade were greater with a narrower lipid arc and shorter lipid length. |
| Rosuvastatin 5 mg/d + Evolocumab 140 mg/2 weeks | ||||||
| Sugizaki, 2020 [48] | Alirocumab 75 mg/2 weeks + Rosuvastatin 10 mg/dL | OCT | 24 | 36 weeks | Lipid content, Macrophage grade | Both the absolute increase (primary endpoint) and percentage increase in FCT were significantly greater in the alirocumab group than in the standard-of-care group (absolute change: 140 mm [78 to 163 mm] vs. 45 mm [10 to 78 mm] [p = 0.002]; percentage change: 273% [155% to 293%] vs. 100% [20% to 155%] [p = 0.004]). |
| Rosuvastatin 10 mg/dL | The macrophage grade decreased significantly in the alirocumab group but not in the standard-of-care group, and the percentage change in macrophage grade was significantly greater in the alirocumab group (–28.4% [–35.3% to –19.0%] vs. –10.2% [–25.3% to 4.3%], p = 0.033). | |||||
| Ota, 2022 [43] | Evolocumab 140 mg/2 weeks | NIRS-IVUS | 53 | 12 months | TAV, PAV, Lipid content | The percent reduction in normalized atheroma volume and absolute reduction in percent atheroma volume (PAV) were also significantly greater in the PCSK9i group (p |
| Furthermore, the PCSK9i group showed greater regression of maximal lipid core burden index for each of the 4-mm segments (maxLCBI4mm) than the control group (57.0 vs. 25.5, p = 0.010). | ||||||
| Hattori, 2012 [44] | Pitavastatin 4 mg | OCT | 42 | 9 months | FCT | Fibrous cap thickness over time between the pitavastatin and diet groups were highly significant. |
| Habara, 2014 [46] | Ezetimibe (10 mg/day) + Fluvastatin (30 mg/day) | OCT | 63 | 9 months | FCT | The change in the fibrous cap thickness was significantly greater in the ezetimibe + fluvastatin group (0.08 |
| Fluvastatin (30 mg/day) | ||||||
| Nicholls, 2022 [45] | Evolocumab 400 mg/L month | OCT | 161 | 52 weeks | FCT, PAV, Lipid content | The evolocumab group demonstrated a greater increase in minimum fibrous cap thickness (+42.7 vs. +21.5 mm, p = 0.015) and decrease in maximum lipid arc (–57.5° vs. –31.4°, p = 0.04) and macrophage index (–3.17 vs. –1.45 mm, p = 0.04) throughout the arterial segment. |
| Räber, 2022 [47] | Alirocumab 75 mg/2 weeks | IVUS, OCT, NIRS | 300 | 52 weeks | FCT, Lipid content | At 52 weeks, mean change in percent atheroma volume was −2.13% with alirocumab vs. −0.92% with placebo (difference, −1.21% [95% CI: −1.78% to −0.65%], p |
| Mean change in maximum lipid core burden index within 4 mm was −79.42 with alirocumab vs. −37.60 with placebo (difference, −41.24 [95% CI: −70.71 to −11.77], p = 0.006). | ||||||
| Mean change in minimal fibrous cap thickness was 62.67 µm with alirocumab vs. 33.19 µm with placebo (difference, 29.65 µm [95% CI, 11.75 to 47.55], p = 0.001). | ||||||
| Oh, 2021 [41] | Atorvastatin 10 mg + Ezetimibe 10 mg | NIRS-IVUS | 41 | 12 months | PAV, Lipid content | The combination of atorvastatin 10 mg and ezetimibe 10 mg showed comparable LDL-C lowering and regression of coronary atherosclerosis in the intermediate lesions, compared with atorvastatin 40 mg alone. |
| Atorvastatin 10 mg |
IVUS, intravascular ultrasound; V-IVUS, virtual intravascular ultrasound; OCT, optical coherence tomography; NIRS, near infrared spectroscopy; PAV, percentage of atherosclerotic plaque volume; TAV, total atherosclerotic plaque volume; FCT, fiber cap thickness; LDL-C, low-density lipoprotein cholesterol; PCSK9i, proprotein convertase subtilisin/kexin type 9 inhibitor.
Most of the current clinical studies on lipid-lowering therapy to induce plaque regression and promote plaque stabilization have used different imaging modalities, defined according to different plaque components and features. Overall, the observational metrics are categorized into the following 4 types.
TAV appears to be the logical definition of plaque regression [51]. Therefore, many clinical trials have used both PAV and TAV to reflect changes in plaque volume and to define whether plaque regression is occurring.
The idea that statins can significantly reduce plaque volume by lowering LDL-C has been recognized by researchers. Study has further confirmed the positive correlation between LDL-C reduction and plaque volume reduction [37]. Increasing the dose of statins is a means of achieving lower LDL-C levels. A study confirmed lower LDL-C levels in the high-intensity lipid-lowering therapy group, with a greater reduction in PAV (–1.22% vs. 0.99%). The effect on TAV was even more pronounced (–6.39 mm3 vs. –4.42 mm3) [38]. Although PAV and TAV are well-validated measures of IVUS-derived plaque volume, PAV incorporates the amount of plaque present in relation to the adaptive response of the vessel wall. As such, the concomitant arterial remodeling response of the vessel wall affects the calculation of PAV. PAV has thus become the primary efficacy endpoint for most trials using serial IVUS to assess changes in coronary atheromatous plaque volume [52]. More interestingly, in an analysis of clinical factors associated with changes in coronary artery volume after statin therapy, the researchers found that female patients on the same regimen were more likely to experience plaque regression and greater differences in PAV than male patients, and multivariate analyses also showed that women acted as independent predictors of PAV regression [52].
According to the latest global guidelines, the target LDL-C level for patients
at very high cardiovascular risk is
The ability of LDL-C lowering to induce atherosclerotic plaque regression has been recognized by researchers. However, the relationship between lipid therapy-induced plaque regression and the occurrence of MACE remains controversial. Researchers now believe that plaque regression achieved by therapeutic regimens targeting LDL-C reduction does correlate with a favorable prognosis. Previous studies have confirmed that intensive lipid-lowering regimens can control lipids below recommended levels and reduce the incidence of MACE by 2.2% in absolute terms and 22% in relative terms [58, 59]. Similarly, a meta-analysis of 17 lipid therapy trials showed that a 1% reduction in mean PAV induced by treatment of dyslipidemia was associated with a 20% reduction in the incidence of MACE [5]. A systematic review and meta-analysis published in the journal of the American Medical Association (JAMA) in 2020 also used PAV as a surrogate for plaque regression, pooled 23 studies related to lipid-lowering therapy, and concluded that 1% plaque regression was associated with a 14%–25% reduction in the incidence of MACE, providing strong support for changes in PAV as a surrogate marker for MACE [56]. However, we should not oversimplify the correlation between PAV and MACE. Future studies will need to demonstrate this causal relationship by temporally sequencing changes in PAV and MACE events in individuals. What remains constant, however, is that lowering LDL cholesterol levels always seems to benefit patients who already have atherosclerotic disease.
However, imaging studies using serial IVUS have demonstrated only modest
reductions in atherosclerotic plaque burden despite low (
Radiofrequency IVUS analysis has shown that statins induce favorable changes in plaque composition, leading to a decrease in lipid body mass and an increase in fiber content, and are more effective in reducing lipid composition when combined with PCSK9i. For example, the Yano study [42] from 2020 used OCT as an imaging method and observed a significant reduction in lipid length and maximal lipid arc in the lipid-lowering treatment group (–40° vs. –24°). In addition, the Ota et al. [43] used NIRS-IVUS as an imaging tool for semi-quantitative detection of plaque lipids and similarly observed that a lipid-lowering regimen significantly reduced lipid composition, decreased formation of lipid necrotic cores, and enhanced plaque stability.
Some researchers believe that statins may contribute to plaque stability by making these macrocalcifications more integrated and denser [60]. The SATURN study [61] performed serial IVUS measurements in patients treated with statins and showed that plaque regression was accompanied by an increase in dense calcium volume with no change in fiber or necrotic core tissue volume. The first trial using CTA to assess plaque in 2013 showed that statins significantly reduced low-attenuation and noncalcified plaque, which is one of the criteria for high-risk plaque [62].
Calcified plaques are considered the most stable form of plaque [63]. Paradoxically, some investigators have also suggested that plaque calcification is characteristic of progressive plaques and that the pattern and distribution of calcification correlates with the severity of CAD, with microcalcification more likely to be associated with vulnerable plaques with clinical events [64]. Therefore, the potential mechanisms and types of statin-mediated calcification deserve further investigation.
Fibrous cap thickness as assessed by OCT was an important determinant of plaque vulnerability. Increased fiber cap thickness is a pathologic manifestation of increased plaque stability. Statin therapy after acute myocardial infarction has been reported to cause an increase in fibrous cap thickness [65]. Kousuke et al. [44] performed prospective OCT in patients with stable angina pectoris and showed a significant increase in fibrous cap thickness after statin treatment compared to baseline (140 µm vs. 189 µm), which was not significant in the control group. In addition, serial OCT analyses in patients with stable angina have been reported to show that the percentage reduction in LDL-C with statin therapy correlates with the percentage increase in fibrous cap thickness [45]. A clinical study conducted by Habara et al. [46] used OCT as an imaging modality to analyze fibrous cap thickness, a metric associated with plaque vulnerability, and the results suggested that the statin combined with ezetimibe group had a more pronounced increase in fibrous cap thickness (0.08 mm vs. 0.04 mm), a significant reduction in lipid plaque angle, and significantly better plaque regression. In addition, combination with PCSK9i also increased fibrous cap thickness in lipid-rich plaques compared with statin monotherapy, as confirmed by the HUYGENS [45] study.
From the perspective of imaging methods to observe plaque characteristics, some researchers believe that OCT is not the best way to observe atherosclerotic plaques with thin fibrous caps and may lead to false-positive results due to the presence of microcalcifications, foam cells, and thrombi [66]. Therefore, researchers hope to improve the resolution and sensitivity of plaque characterization using multimodal imaging. The PACMAN-AMI study [47] was the first to evaluate the effect of high-intensity statins in combination with PCSK9 inhibitors on plaque using simultaneous IVUS, OCT, and NIRS imaging and showed that the combination therapy resulted in satisfactory plaque regression. Surprisingly, the study also confirmed the benefits of PCSK9 inhibitors in improving plaque stability even with the use of high-intensity statins. Therefore, current guidelines recommend the use of PCSK9 inhibitors in combination with statins and ezetimibe in patients at very high risk of atherosclerotic cardiovascular disease when maximum tolerability of statins and ezetimibe remains unsatisfactory [67].
Decreased macrophage content may also serve as a surrogate marker for increased plaque stability [68]. The 2020 ALTAIR study [48] also observed a significant reduction in macrophage grade and a greater percentage change in the statin plus PCSK9i group (–28.4% vs. 10.2%). The likely reason for this is the lower LDL-C levels induced by the combination therapy. The results provide possible mechanistic insights into the efficacy of adding alirocumab to standard-dose statins to improve clinical outcomes.
However, despite the substantial reduction in LDL cholesterol because of lipid-lowering therapy, residual cardiovascular risk may remain. Mean on-treatment LDL cholesterol levels in SATURN [61] were the lowest achieved in any previously conducted atherosclerosis imaging study (62 mg/dL in patients taking Rosuvastatin and 70 mg/dL in patients taking Atorvastatin), but plaque progression was still present in one-third of patients. This suggests that alternative therapeutic strategies should still be actively sought, to reduce the burden of atherosclerosis.
Increasing blood HDL levels is also a hot topic of research in plaque stabilization and regression [69]. Steven et al. [70] investigated the role of elevated HDL on plaque volume and observed that patients given different doses of complex HDL had a mean decrease in plaque volume of 4.2% after 5 weeks of treatment and that HDL levels were positively correlated with plaque regression. However, there is also study in which plaque regression was not observed with HDL-mimicking drugs, so the ability of elevated HDL to promote plaque regression needs to be confirmed in further large randomized controlled trials [29].
In addition, residual inflammatory risk may be an important cause of cardiovascular events beyond cholesterol [19]. Schuett et al. [71] observed significant plaque regression in mice using inhibitors of IL-6 signaling. However, there are fewer clinical trials examining changes in plaque composition and volume after anti-inflammatory therapy. The exact relationship between anti-inflammatory therapy and plaque regression deserves further exploration.
In addition to inflammation, the risk of cardiovascular disease that remains after well-controlled LDL levels may be due to elevated triglyceride (TG)-rich lipoproteins, which are common dyslipidemias in patients with diabetes and metabolic diseases. A large, randomized trial using icosapent ethyl (IPE) showed that lowering triglycerides resulted in a significant reduction in adverse cardiovascular events in statin-treated patients and was proportional to the blood concentration of eicosapentaenoic acid (EPA) [72]. However, EPA/docosahexaenoic acid (DHA) blends, which possess similar triglyceride-lowering effects, did not show the same benefits in the trial. The inconsistency of the results has prompted further investigation into the possible mechanisms. Researchers believe that EPA in combination with statins maintains normal membrane cholesterol distribution, enhances endothelial function, and improves features associated with plaque stability. In addition, researchers believe that the apparent benefits of IPE in multiple trials may stem from multiple effects associated with therapeutic levels of EPA, not just triglyceride lowering. These effects include alterations in platelet function, inflammation, cholesterol distribution, and endothelial dysfunction [72].
In addition to medication, lifestyle changes to control other diseases can affect the degree of plaque regression. Examples include quitting smoking, controlling weight, avoiding comorbid diabetes, lowering blood pressure, and increasing exercise.
It is well acknowledged that atherosclerosis is a major cardiovascular complication of diseases associated with chronic inflammatory status, increased oxidative stress and disorders of lipid metabolism. Systemic oxidative stress states predispose LDL to oxidation, forming oxidized LDL, which displays pro-atherosclerotic activity through a complex mechanism. Oxidized LDL cholesterol-rich oxidation products, also known as oxysterols, can exert various biological effects on vascular cells such as promoting apoptosis, inducing oxidative stress and cytotoxicity involved in plaque formation and destabilization, which have been well documented. It is now well demonstrated that oxidative stress is a determinant for the formation of oxysterols as well as signaling pathways evoked by deleterious oxysterols [73]. Therefore, the protective effect of reducing the production of oxysterols through antioxidant therapy is valuable and deserves further investigation. Cigarette smoking, a major health hazard, contributes to atherosclerosis, thrombosis, and inflammation through multiple mechanisms, including endothelial dysfunction and increased oxidative stress. Oxygen radical-mediated oxidative stress plays a central mechanism in smoking-mediated atherosclerotic disease. These free radicals may come directly from cigarette smoke or indirectly from endogenous substances [74]. And increased oxidative stress is largely associated with prothrombotic effects (increased platelet reactivity, decreased endogenous fibrinolysis, and lipid peroxidation) and inflammatory responses to the vessel wall. Furthermore, antioxidants or drugs that reduce oxidative stress have been shown to ameliorate or reverse the prothrombotic and proinflammatory features associated with smoking [74].
In a very small, randomized study published in 1990, 28 patients with coronary atherosclerosis were randomly assigned to receive intensive lifestyle changes, including smoking cessation, or usual care. After 1 year, percent diameter coronary stenosis assessed by coronary angiography was reduced from a mean of 40.0% to 37.8% in the intensive life-style group and increased from a mean of 42.7% to 46.1% in the control group [75]. The findings tentatively suggest that smoking cessation has an effect on volume regression after coronary plaque formation. In addition, much of the literature strongly suggests that smoking adversely affects all stages of atherosclerosis, for example, by promoting thrombosis or activating MMPs, which promote the formation and rupture of vulnerable plaques, thereby triggering acute events [76]. Zhang et al. [77] divided patients with acute coronary syndromes (ACS) after percutaneous coronary intervention (PCI) into smoking cessation group, persistent smoking group, and nonsmoking group. All three groups were treated with statins after surgery, and OCT was used to focus on the morphology of non-culprit plaques. It was found that the persistent smoking group had a smaller fibrous cap thickness and a higher incidence of TCFA compared with the other two groups. It was concluded that persistent smoking attenuated the effect of statin therapy on plaque stabilization in ACS patients. The results of this study suggest that smoking cessation may have a stabilizing effect on plaques by increasing fibrous cap thickness and improving plaque morphology [77].
Despite the paucity of data on plaque stabilization and regression after smoking cessation, it is undeniable that smoking is an important risk factor for patients with thrombotic coronary events (especially plaque erosion), and smoking cessation significantly reduces acute events by decreasing coronary thrombosis and attenuating the inflammatory response [78].
Previous study has found that the higher the patient’s body mass index, the greater the offset of plaque regression [79]. In addition, diabetes mellitus has a negative impact on plaque regression. The TRUTH study [80] confirmed that diabetes mellitus significantly impairs the plaque stabilizing effects of statins through complex mechanisms, including activation of hematopoiesis, increased inflammatory cell infiltration, and impeded macrophage polarization. On the other hand, the PESA study confirmed that factors such as non-smoking and being female may promote the occurrence of plaque regression [81]. In addition, the study has confirmed the positive effect of exercise intensity on plaque regression [82]. However, because the available data are low in quality, whether lifestyle modification has clinically significant effects on coronary plaque stabilization and regression remains uncertain.
In conclusion, pharmacologic therapy is the cornerstone of reducing plaque volume and enhancing plaque stability, on top of which we should also actively control other factors that affect the plaque volume and stability in order to minimize the occurrence of clinical events.
Favorable results from clinical studies in recent years have underscored the importance of achieving very low LDL-C levels in patients at high cardiovascular risk and have emphasized that combining multiple types of lipid-lowering agents provides clinical benefit in achieving these effects in most patients. Combination lipid-lowering therapy has become a common strategy to achieve plaque regression through greater reductions in LDL-C levels. However, it has not been established whether combination therapy with ezetimibe and statins is more effective than statins when LDL cholesterol levels are comparable. Although available data suggest that the lower the LDL cholesterol level, the greater the degree of coronary plaque regression. However, the long-term safety of very low LDL cholesterol levels remains to be investigated and confirmed by clinical trials.
Through the initial elucidation of the mechanisms underlying the onset of plaque regression and plaque stabilization, increasing attention has been paid to the role of inflammatory cells and inflammatory factors in this context, and more basic studies targeting the reduction of inflammation levels and the promotion of cellular phenotypic shifts may be needed in the future to confirm the onset of plaque regression and enhanced plaque stability. In addition, the role of neovascularization in promoting plaque formation and progression has been emphasized by researchers. The local hypoxic environment of plaques can upregulate proangiogenic factors to trigger neovascularization. Pathological, morphological, and functional characteristics of neovascularization such as morphologic disturbances, loss of basement membrane and pericytes, and abnormal increase in permeability allow for the delivery of inflammatory cells and lipoproteins to the lesion site, thereby exacerbating the lipid and inflammatory microenvironment within the plaque and promoting plaque formation and destabilization [83]. Animal study has demonstrated that inhibition of pathologic neovascularization facilitates increased plaque stability, as evidenced by a decrease in lipid content and macrophage accumulation. The likely mechanism is that protocatechuic aldehyde increases pericyte proliferation, migration, and adhesion, which serves to increase pericyte coverage of plaques and reduce vascular endothelial growth factor-A production, inhibiting plaque neovascularization. In addition, it can alleviate oxidized LDL-induced pericyte dysfunction and maintain capillary structure and stability [84].
Furthermore, the occurrence of progression of different plaque phenotypes and plaque regression has not been studied. It has been suggested that lipid-rich plaques have the most therapeutic value with the possibility of reversal with intensive LDL-lowering therapy and control of risk factors, whereas calcified plaques, even when LDL is lowered to very low levels, are less likely to be reversible in such plaques. What is certain, however, is that the plaque phenotype may change over time as a result of drug use and episodes of subclinical events [32].
There is increasing clinical evidence that patients with coronary atherosclerosis benefit from lipid-lowering therapy and that plaque stabilization and regression improve patient survival. However, various imaging techniques remain a surrogate endpoint, and plaque volume reduction and composition changes should not be interpreted as equivalent to clinical benefit in the prevention of cardiovascular events [38]. Despite these limitations, we believe that the effectiveness of currently used drugs in reducing plaque volume and increasing plaque stability through certain mechanisms is noteworthy. In addition, although clinical studies evaluating short-term plaque volume changes suggest that plaque regression is possible with intensive lipid lowering and intravascular imaging, these changes are small compared with control populations. In conclusion, we do not believe that plaque regression is the only therapeutic goal. We always believe that risk factors should be tightly controlled and treated early to prevent or minimize plaque progression and enhance its stability, regardless of whether plaque regression occurs. In addition, low-cost, low-risk circulating biomarkers have been independently associated with prognosis and may serve as an adjunct to identify patients more likely to benefit from lipid-lowering therapy.
XZ contributed to the design and implementation of the article, data collection and article writing. HHF, YH, XHY, and MTJ contributed to the literature search, screening and figure production. HHF and MTJ provide help and advice on writing articles. YH and XHY revised the manuscript. WW and LG provided guidance on the design and implementation of the article, data collection and article writing, and financial support. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This study was supported by the Beijing Natural Science Foundation (7242138), the Capital Development Health Research Special Project (2024-2-5072), and the Innovation Cultivation Fund of the Sixth Medical Center of the General Hospital of the Chinese People’s Liberation Army (No. CXPY202202).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.