

 , Samuel S. Lee 1,*
, Samuel S. Lee 1,*
1 Liver Unit, University of Calgary Cumming School of Medicine, Calgary, AB T2N 4N1, Canada
2 Division of Gastroenterology, Yangsan Hospital, Pusan National University Faculty of Medicine, 50612 Pusan, Republic of Korea
3 Telemedicine Center, Second Hospital of Hebei Medical University, 050004 Shijiazhuang, Hebei, China
†These authors contributed equally.
Abstract
Cirrhotic cardiomyopathy is defined as systolic and diastolic dysfunction in patients with cirrhosis, in the absence of any primary heart disease. These changes are mainly due to the malfunction or abnormalities of cardiomyocytes. Similar to non-cirrhotic heart failure, cardiomyocytes in cirrhotic cardiomyopathy demonstrate a variety of abnormalities: from the cell membrane to the cytosol and nucleus. At the cell membrane level, biophysical plasma membrane fluidity, and membrane-bound receptors such as the beta-adrenergic, muscarinic and cannabinoid receptors are abnormal either functionally or structurally. Other changes include ion channels such as L-type calcium channels, potassium channels, and sodium transporters. In the cytosol, calcium release and uptake processes are dysfunctional and the myofilaments such as myosin heavy chain and titin, are either functionally abnormal or have structural alterations. Like the fibrotic liver, the heart in cirrhosis also shows fibrotic changes such as a collagen isoform switch from more compliant collagen III to stiffer collagen I which also impacts diastolic function. Other abnormalities include the secondary messenger cyclic adenosine monophosphate, cyclic guanosine monophosphate, and their downstream effectors such as protein kinase A and G-proteins. Finally, other changes such as excessive apoptosis of cardiomyocytes also play a critical role in the pathogenesis of cirrhotic cardiomyopathy. The present review aims to summarize these changes and review their critical role in the pathogenesis of cirrhotic cardiomyopathy.
Keywords
- cirrhotic cardiomyopathy
- pathogenic mechanisms
- heart failure
- ventricular dysfunction
- adrenergic receptor
- nitric oxide
- endocannabinoid receptor
- bile acids
- myofilaments
- ion channel
- myosin heavy chain
Cirrhotic cardiomyopathy (CCM) is generally agreed to be a combination of systolic dysfunction, impaired diastolic relaxation and altered morphology such as left atrial enlargement, in the absence of prior heart disease or another identifiable cause in patients with cirrhosis. The cardiac dysfunction is usually not obvious at rest. However, when challenged such as by exercise, drugs, and surgery, cardiac dysfunction is manifested as a blunted ventricular inotropic and chronotropic response to these stimuli [1, 2].
Cardiomyocytes are the main functional cells of ventricular contraction and are essential for maintaining the normal pumping function of the heart. Our previous study demonstrated that the contractile and relaxation velocities of cardiomyocytes isolated from cirrhotic animals are significantly attenuated [3]. The mechanisms (Fig. 1) are multifaced [4, 5, 6, 7, 8]. Among many factors, alterations in cytoplasmic membrane receptors (Table 1, Ref. [3, 9, 10, 11, 12, 13, 14, 15, 16]), ion channels, biophysical membrane fluidity and myofilaments, and excessive cardiomyocyte apoptosis play important roles. Although there are many studies on the cellular pathogenic mechanisms responsible for CCM, they have not yet been fully clarified.
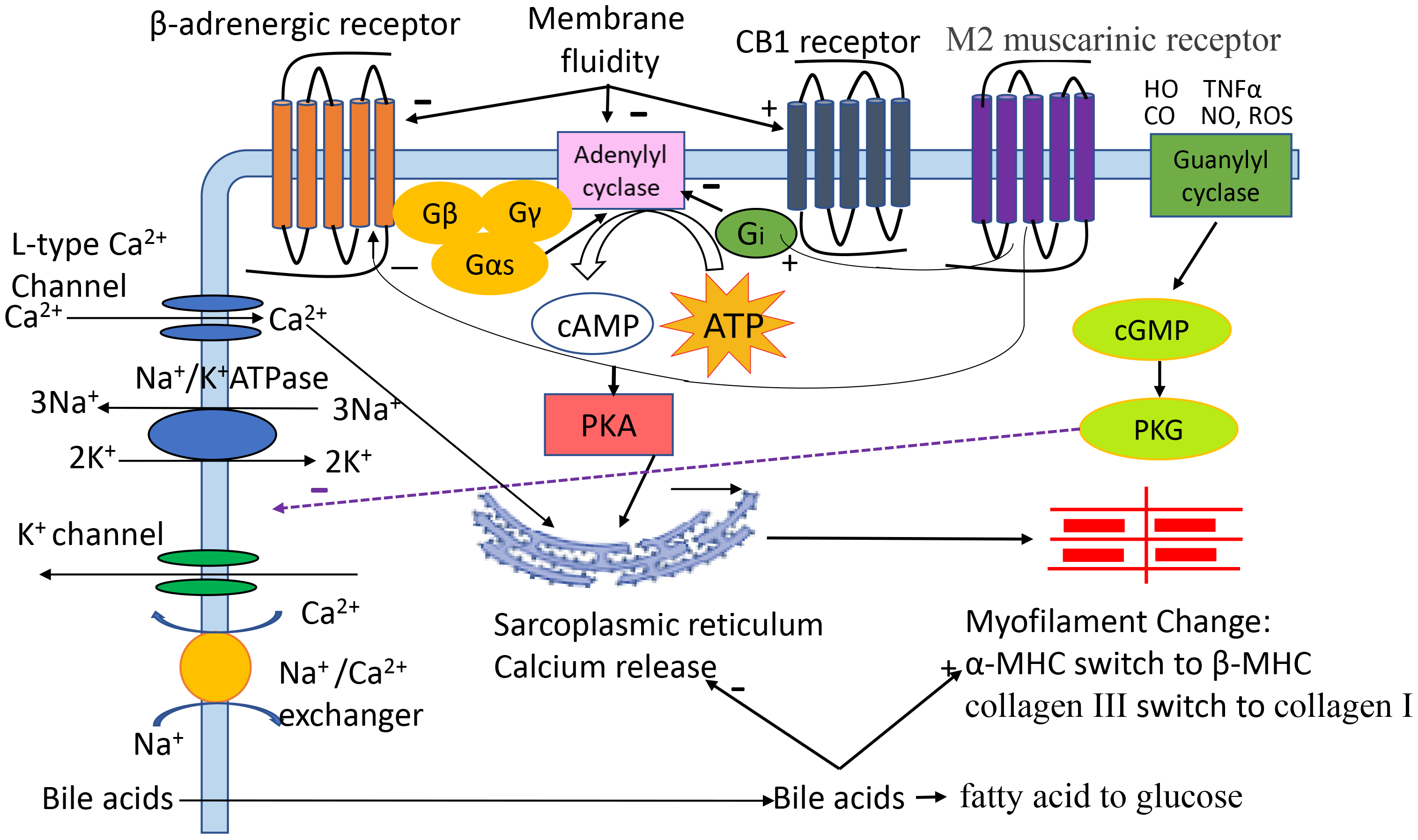 Fig. 1.
Fig. 1.
The pathogenic role of different factors in cirrhotic
cardiomyopathy (CCM). Cardiac contractility and relaxation are complex
processes. Activation of
| Receptor | Structural changes | Mechanism | Impact on cardiac function |
| Downregulated [9] | Overdrive [10] | Blunted response to | |
| Anti- | |||
| CB1 | No change [12, 13] | Local increased CB1 agonist | Blunted response to |
| M2 | Downregulated [14] | Compensatory role | Blunted response to carbachol |
| No change [15] | |||
| cGMP systems | Upregulated [3] | Nitric oxide upregulation | Downregulate L-type Ca2+ channel [16] |
In patients or animal models with heart failure, sympathetic nervous system
activity is increased and the density of
We demonstrated a similar phenomenon of
The mechanisms underlying the decrease of
The anti-
Since anti-
There are 5 subtypes of muscarinic receptors: M1, M2, M3, M4 and M5. M2 is the
main subtype in cardiomyocytes [25, 26]. Using an enzyme-linked immunosorbent
assay (ELISA), Duan et al. [27] demonstrated that not only are anti-
In a cirrhotic rat model, we did not find a significant decrease in M2 receptor density on the cytoplasmic membrane of cirrhotic cardiomyocytes [15]. However, the magnitude of the inotropic response to carbachol was blunted in cirrhotic hearts, suggesting that the attenuated muscarinic responsiveness is due to post-receptor factors [15]. We speculate that the blunted muscarinic function represents a compensatory response to the numerous factors inhibiting ventricular contractility in cirrhosis.
In addition to sympathetic (
Mărieş and Maniţiu reviewed the role of endocannabinoids in
cirrhotic CCM [12], it is not the changes of CB1 receptors on cardiomyocytes from
these hearts, consistent with the results of Bátkai and coworkers [13] in
CCl4-cirrhotic rats. It is the increase of endocannabinoids in the cirrhotic
heart. The dose-response curve of cardiac contractility to the
The cardiac action potential (AP) is a rapid sequence of changes in the voltage across the plasma membrane of cardiomyocytes. The pathophysiological consequences of voltage channel changes (Table 2, Ref. [31, 32, 33, 34, 35]) impair electro-mechanical coupling. The abnormalities of ventricular contractile and relaxation velocities in CCM may be at least in part due to abnormalities of ion channels. Our studies have revealed abnormalities of two ion transients, calcium [31, 32] and potassium [33], in rat cirrhotic ventricular myocytes.
| Protein/Ion | Structural or functional change | Impact on cardiac function |
| Potassium [33] | Downregulation of I(t), Isus | Q-T interval prolongation |
| Calcium [32] | Downregulation of L-type calcium channels | Ca2+ dynamic abnormalities, impaired contractility |
| Na⁺/K⁺-ATPase [34] | Downregulated | Impaired contractility |
| SR [32] | No change [35] | Ca2+ dynamic abnormalities |
| SERCA [32] | No change [35] | Unclear |
| NCX | Downregulated | Ca2+ dynamic abnormalities |
| Ca2+ leakage [31] | Increased | Decreases contractility and relaxation |
I(t), Ca2+-independent transient outward K+ current; I𝑠𝑢𝑠, delayed rectifier K+ current; SERCA, sarcoplasmic/endoplasmic-reticulum Ca2+-ATPase; NCX, sodium-calcium exchanger; SR, sarcoplasmic reticulum.
Potassium channels are widely distributed in virtually all organisms [36], and control a wide variety of cell functions [37]. Potassium currents, such as Ca2+-independent transient outward K+ current (I(t)), delayed rectifier K+ current (I𝑠𝑢𝑠), and inwardly rectifying potassium current (I(K1)), are generated via these channels. It is clear that potassium currents play an essential role in the action potential. I(t) is a crucial determinant of excitation-contraction (EC) coupling: in the early phase of repolarization of the cardiac action potential and in setting the plateau voltage level of the action potential. Therefore, it extensively affects membrane current flow in the plateau window. It has been demonstrated that I(t) and its molecular constituents are reduced in cardiac hypertrophy and heart failure [38, 39, 40].
I(t) reduction prolongs action potential duration, and the waveform and duration of the action potential intensely affect the Ca2+ transient and thus mechanical shortening (contractility) [38]. I(t) reduction also causes cardiomyocyte hypertrophy [41], and is a consistent finding in non-cirrhotic heart failure [42, 43] and CCM [33].
Our lab tested the status of potassium channels in isolated cirrhotic cardiomyocytes. We first used bile duct ligation to create a cirrhotic model in rats; sham-operated rats served as controls. Single myocytes were current- and voltage-clamped using standard whole-cell methods. Under the blockade of L-type Ca2+ currents by cadmium chloride (CdC) 12, we measured three different K+ currents in isolated single myocytes from the atria and ventricles of sham-operated and cirrhotic rats: Ca2+-independent transient outward K+ current (I(t)), delayed rectifier K+ current (I𝑠𝑢𝑠), and inwardly rectifying potassium current (I(K1)). We showed that the potassium currents were unchanged in isolated atrial cardiomyocytes between cirrhotic and sham-control rats. In ventricular myocytes from cirrhotic animals, the only significant functional changes were decreases of I(t) and I𝑠𝑢𝑠. Further analysis revealed that the observed changes are due to a decrease in current density, i.e., fewer functional K+ channels.
Although many factors can prolong action potentials, activation of the K+ channels is essential for both early and final repolarization and therefore the decreases of I(t) and I𝑠𝑢𝑠 [24] largely explain the prolonged electrocardiographic Q-T interval, which afflicts about 30–70% of patients with cirrhosis [44]. Whether these K+ channel abnormalities also contribute to the higher rates of arrhythmias such as atrial fibrillation [45] in patients with cirrhosis, remains unclear at present.
Like potassium channels, voltage-gated Ca2+ channels are key transducers of membrane potential changes which play a pivotal role in the cardiac action potential. There are ten members of the voltage-gated Ca2+ channel family in mammals, comprising low-voltage activated (or T-type) and high-voltage activated Ca2+ (L-, N-, P/Q- and R-type) channels. Among them, N-, P-, Q-, and R-type Ca2+ currents are most prominent in neurons [46]. In the heart, Ca2+ influx is mainly carried out by L-type Ca2+ channels [47]. L-type Ca2+ channels transport Ca2+ from outside the cell to the cytosol and therefore these channels are fundamental for the initiation and regulation of EC coupling in cardiomyocytes.
In EC coupling, Ca2+ enters the cytoplasm via the L-type Ca2+ channels where Ca2+ combines with the Ca2+- release channels (ryanodine receptor), triggering Ca2+ release from the sarcoplasmic reticulum (SR). The cytosolic Ca2+ released from the SR combines with the troponin complex and generates actin-myosin cross-bridge linking which results in cell contraction [48]. This process is called excitation-contraction coupling. The cytosolic Ca2+ concentration in cardiomyocytes is the unique determinant of contractile function.
After contraction, both Ca2+ channels on the cytoplasmic membrane and Ca2+ release channels in the cytosol are closed, and the Ca2+ is removed from the cytosol via two main systems: sarcoplasmic-endoplasmic reticulum calcium-ATPase (SERCA) and the sodium-calcium exchanger (NCX). The SERCA system pumps back the Ca2+ from the cytosol to SR and the NCX extrudes the Ca2+ from the cytosol to the extracellular space [49]. A well-maintained Ca2+ balance between the Ca2+ entering the cytosol before cardiac contraction and that removed from the cytosol after contraction is a prerequisite for normal cardiac systolic and diastolic function. If the amount of Ca2+ entering the cell is not equal to that extruded in each cardiac cycle, the cardiomyocytes would either gain or lose Ca2+ [50] which would seriously impair contractility over a few cycles and be completely untenable over a longer term. Pertinent studies from our lab demonstrated that Ca2+ transport is abnormal in cirrhotic cardiomyocytes [31, 32].
We showed that L-type Ca2+ channels are decreased in cirrhotic cardiomyocytes [32]. Ca2+ entry from outside the cardiomyocyte is essential for triggering EC-coupling: removal of Ca2+ from the perfusion buffer discontinued cardiac contraction of the frog heart [51] which confirms that external Ca2+ is required for cardiac systole. The decrease of L-type Ca2+ channels theoretically impacts the amount of cytosolic Ca2+ before contraction. Indeed, the current densities of Ca2+ influx via L-type Ca2+ channels were significantly lower in cardiomyocytes measured from cirrhotic cardiomyocytes compared with that from sham controls [23]. The decrease of L-type Ca2+ channels may therefore play a significant role in decreased contractility of cardiomyocytes in CCM.
Another abnormality in the Ca2+ handling system lies in the SR. The root mean square value of sarcomere length fluctuations (RMSSL) quantitates the amount of spontaneous sarcomere length fluctuation during diastole, which is believed to be an index of calcium leakage from the SR. We found that RMSSL is significantly higher in ventricular trabeculae from cirrhotic rat hearts at all stimulus rates, especially with relatively higher stimulus rates, compared with that from sham-control rats [31]. Accordingly, this indicates that the leakage of Ca2+ from the SR in cirrhotic cardiomyocytes is higher than that from sham controls. Such leakage may cause insufficiency of Ca2+ storage in SR and consequently reduce its Ca2+ release when Ca2+ enters the cytosol via L-type Ca2+ channels. The resulting outcome will be a decreased contractility of cirrhotic cardiomyocytes.
Besides the abnormalities of the Ca2+ handling system, the sensitivities of
myofilament to Ca2+ are also reduced in cirrhotic cardiomyocytes.
Metzger et al. [52] chemically induced hypothyroidism in adult
rats, and showed that this was associated with a myosin heavy chain (MHC) shift
from the predominant stronger
Na+/K+-ATPase is an essential enzyme found in the plasma membrane of all animal cells [54]. The Na+/K+-ATPase consists of alpha- and beta-subunits and actively transports 3 Na+ out and 2 K+ ions into the myocyte and thus removes one positive charge carrier from the intracellular space per pump cycle [55]. Na+/K+-ATPase is the main structure that maintains the sodium (140 mM vs 10–30mM) and potassium (3.5–5 mM vs 130–140mM) concentration gradient across the membrane of the cell. In cardiomyocytes, regular activity of the Na+/K+-ATPase and its Na+/K+ pump activity is essential for maintaining ion gradients, cell excitability, propagation of action potentials, and electro-mechanical coupling. Schwinger et al. [56] showed that total Na+/K+-ATPase concentration is decreased by approximately 40% in patients with cardiac dysfunction and this decrease is correlated with cardiac function. Our preliminary data indicated that Na+/K+-ATPase is decreased in CCM (unpublished data). Therefore, the decrease of Na+/K+-ATPase in the cirrhotic heart may also be involved in the pathogenesis of CCM.
Another sodium transporter is the NCX, a Ca2+ and Na+ transport protein, that couples the transport of three Na+ and one Ca2+ ion across the cell membrane. Interestingly, the transport direction depends on ionic concentrations and membrane potential, either Ca2+ extrusion/Na+ entry (forward mode) or Ca2+ entry/Na+ extrusion (reverse mode) [57]. There are three isoforms of NCX: NCX1, NCX2, and NCX3. Only NCX1 is expressed in cardiac myocytes. NCX1 on the membrane of cardiomyocytes usually operates in a “forward” direction and plays a role in cardiac relaxation. However, when the intracellular Na+ is increased, such as during the early phase of an action potential, NCX1 also operates in “reverse” mode. NCX protein expression is increased in human heart failure [58]. Our preliminary data indicated that NCX expression was decreased in cirrhotic cardiomyocytes (unpublished observations). The discrepancy between the non-cirrhotic heart failure and CCM may be due to an increase of bile acids in our BDL-induced cirrhotic rat model [59] because bile acids have an inhibitory effect on NCX [60].
Our lab compared the cardiac sarcolemmal plasma membrane differences between
cirrhotic rats and controls, examining both structural and functional changes. We
demonstrated that the membrane cholesterol content of the cirrhotic myocyte was
significantly increased (178.1
Many years ago, we demonstrated how decreased fluidity impairs
Hyperdynamic circulation, including peripheral vasodilatation and increased cardiac output, is a feature in cirrhosis which can lead to hypertrophy of cardiomyocytes due to the increased cardiac workload. Inserte and coworkers [63] demonstrated that compared with controls, cirrhotic rats showed 30% increase in heart weight, 30% increase in cross-sectional area of the left ventricular wall, and 12% increase in the width of cardiomyocytes from left ventricles. Whether there are myofilament changes in cirrhotic cardiomyocytes needs to be investigated.
Myofilaments (Table 3, Ref. [31, 64]) include myosin, actin, and titin [65, 66, 67]. They play critical roles in cardiac contraction. We investigated titin [64] and MHC [31]. We did not demonstrate any structural changes in titin either the whole protein or isoforms.
| Myofilaments | Structural or functional change | Impact on cardiac function |
| MHC [31] | Switch from |
Reduces contractile force and velocity |
| Collagen [64] | Switch from type III to type I | Increases diastolic stiffness |
MHC, myosin heavy chain.
There are two isoforms of MHC,
The other filament-related proteins that may be worthwhile investigating in CCM
is the troponin complex. Troponin is a component of thin filaments. There are
three isoforms, troponin C, troponin I, and troponin T. Among them, troponins I
and T are cardiac-specific. In the process of excitation–contraction coupling,
calcium first combines with troponin and triggers cardiac contraction [69].
However, to date, there is no pathogenic study on the role of troponin in CCM.
The pertinent studies are on the role of troponin in the diagnosis of cardiac
dysfunction. Coss et al. [70] found that a troponin I level
Cardiac collagens are produced by fibroblasts. In subjects with cirrhosis, the increased pro-inflammatory cytokines stimulate fibroblasts in the heart to produce collagens, leading to cardiac fibrosis [73]. Our study also found a switch of collagen from the compliant subtype III to stiffer type I in cirrhotic rat hearts, which likely impairs diastolic relaxation [64].
Cardiomyocytes are the unique functional cells of cardiac contraction. Cell death plays an essential role in cardiac dysfunction. Cell death can occur by necrosis or programmed cell death. Necrosis is a passive, accidental cell death due to uncontrolled environmental perturbations, such as inflammation. In comparison, programmed cell death, including apoptosis, pyroptosis, and ferroptosis, is an active, programmed process with a series of molecular steps that lead to cell death. Bacteria/viral infections can cause pyroptosis; the cell death is initiated with cellular membrane rupture. Ferroptosis is caused by iron overload and characterized by the accumulation of lipid peroxides, and cell death begins with mitochondria. To date, there are no studies on cardiomyocyte necrosis, pyroptosis, and ferroptosis in CCM. However, there is extensive previous work on apoptosis in noncirrhotic cardiac conditions, and a few studies in CCM pathogenesis, described below.
Apoptosis of cardiomyocytes occurs in most cardiovascular diseases [74, 75]. It was demonstrated that only 0.023% of cardiomyocyte apoptosis is sufficient to cause a lethal, dilated cardiomyopathy [76]. There are two pathways that lead to apoptosis, the intrinsic pathway and the extrinsic pathway [77, 78]. The extrinsic pathway is initiated via death receptors on the surface of plasmic membrane [79], the intrinsic pathway, also called mitochondrial pathway, begins when an injury occurs within the cell. Intrinsic stresses cause mitochondrial dysfunction which releases cytochrome c. The later combines with apoptotic protease activating factor-1 (APAF1), and forms the apoptosome, which activates caspase-9 and caspase-3 [80]. Caspase-3 is the major executor of apoptosis [81], both extrinsic and intrinsic pathways execute apoptotic effects via caspase-3. In CCM, both extrinsic and intrinsic pathways are involved in cardiomyocyte apoptosis.
We tested intrinsic and extrinsic pathways in the cirrhotic model induced by BDL in mice, and showed that the extrinsic pathway plays a major role in the apoptosis of cirrhotic cardiomyocytes, whereas the intrinsic pathway actually appeared to exert a compensatory protective role. Our immunohistochemistry demonstrated a significant increase of PARP (poly-ADP ribose polymerase) staining of cardiomyocytes from cirrhotic hearts. As it is known that PARP represents direct evidence of ongoing apoptosis [82, 83], these results therefore indicated that apoptosis is indeed occurring in the cardiomyocytes of cirrhotic hearts [84]. Another study also found that apoptosis plays an important role in CCM [85].
Bile acids are increased in the serum of cirrhotic patients [86] and exert
inhibitory effects on cardiac contractility [87]. Therefore, bile acids may play
a role in the decreased cardiac contractility in patients with CCM. The possible
mechanisms include facilitation of
Nitric oxide (NO) is overproduced in cirrhotic patients and experimental
cirrhotic animals [91, 92]. The elevated NO exerts an inhibitory role on cardiac
contraction in patients with cirrhosis. The mechanism of the negative contractile
effect of NO on cardiac function is via cGMP signaling. cGMP further decreases
calcium sensitivity of myofilaments [93] and blunts
Carbon monoxide (CO) is another evanescent gas that acts as a cardiac contractile inhibitor. CO is generated by heme oxygenase (HO). Like NO, CO levels are also significantly increased in the cirrhotic heart [95]. The mechanism of cardiac inhibition by CO is via cGMP stimulation. The HO inhibitor, zinc protoporphyrin IX, reduced the elevated cGMP levels and restored the inhibited cardiac contractility in a BDL-rat cirrhotic heart. These findings implicate the involvement of an HO-CO-cGMP pathway in the pathogenesis of CCM.
The most investigated cytokine in CCM is TNF
The pathogenesis of CCM is multifaceted: from the cytoplasmic membrane to the cytosol and nucleus. Among these, membrane receptors, voltage channels, plasma membrane biochemical and biophysical changes, contractile myofilaments, cardiomyocyte apoptosis and direct contractility inhibitors have been demonstrated to play essential roles.
SSL: conception of the review idea. DR, FY, KY, HL: literature review. DR, FY, HL wrote the first draft. All authors contributed intellectual content and extensive revisions of the draft, read and approved the final version. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.