
 , Yinghui Huang 1,*
, Yinghui Huang 1,*1 Department of Nephrology, The Key Laboratory for the Prevention and Treatment of Chronic Kidney Disease of Chongqing, Chongqing Clinical Research Center of Kidney and Urology Diseases, Xinqiao Hospital, Army Medical University (Third Military Medical University), 400037 Chongqing, China
2 Medical Division, Xinqiao Hospital, Army Medical University, 400037 Chongqing, China
3 Department of Oncology, Southwest Cancer Center, Southwest Hospital, Army Medical University, 400038 Chongqing, China
†These authors contributed equally.
Abstract
Cardiorenal syndrome type 3 (CRS3) is defined as acute kidney injury (AKI)-induced acute cardiac dysfunction, characterized by high morbidity and mortality. CRS3 often occurs in elderly patients with AKI who need intensive care. Approximately 70% of AKI patients develop into CRS3. CRS3 may also progress towards chronic kidney disease (CKD) and chronic cardiovascular disease (CVD). However, there is currently no effective treatment. Although the major intermediate factors that can mediate cardiac dysfunction remain elusive, recent studies have summarized the AKI biomarkers, identified direct mechanisms, including mitochondrial dysfunction, inflammation, oxidative stress, apoptosis and activation of the sympathetic nervous system (SNS) and renin-angiotensin-aldosterone system (RAAS), inflammasome, as well as indirect mechanisms such as fluid overload, electrolyte imbalances, acidemia and uremic toxins, which are involved in the pathophysiological changes of CRS3. This study reviews the main pathological characteristics, underlying molecular mechanisms, and potential therapeutic strategies of CRS3. Mitochondrial dysfunction and inflammatory factors have been identified as the key initiators and abnormal links between the impaired heart and kidney, which contribute to the formation of a vicious circle, ultimately accelerating the progression of CRS3. Therefore, targeting mitochondrial dysfunction, antioxidants, Klotho, melatonin, gene therapy, stem cells, exosomes, nanodrugs, intestinal microbiota and Traditional Chinese Medicine may serve as promising therapeutic approaches against CRS3.
Keywords
- CRS3
- mitochondrial dysfunction
- crosstalk
- molecular mechanisms
- therapeutic strategies
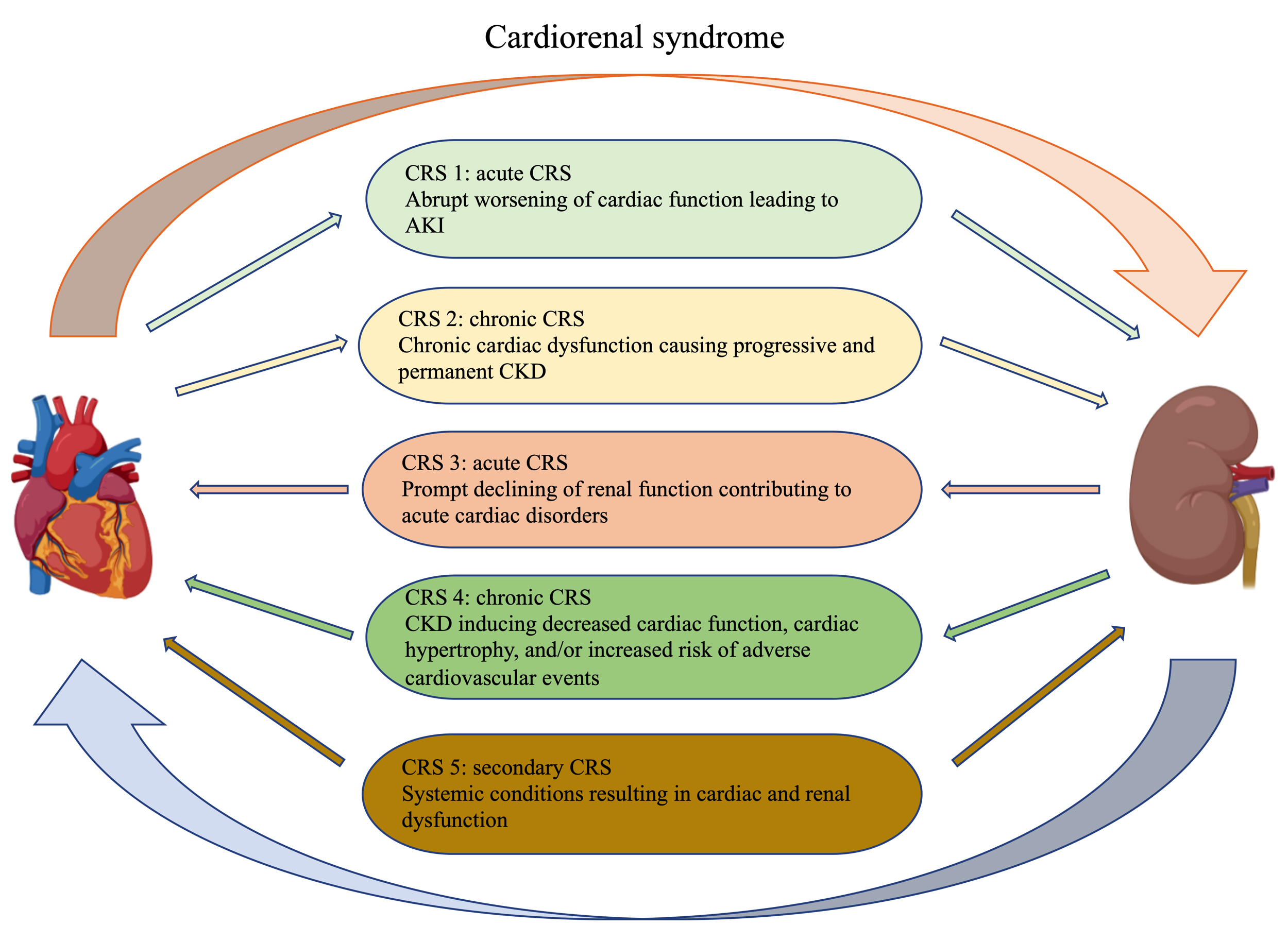
Physiological communication between the heart and kidney is essential to preserve metabolic waste removal, hemodynamic stability and bodily function [1]. However, in pathological states, an impaired organ often leads to the dysfunction of another organ. Cardiorenal syndrome (CRS) was used to describe this complex pathological interaction between the heart and kidney [2, 3, 4]. According to the primary or secondary organic dysfunction, CRS is divided into cardiorenal syndrome (type 1 and 2) and renal-cardiac syndrome (type 3 and 4). Depending on whether the primary organ dysfunction is acute (type 1 and 3) or chronic (type 2 and 4) at the time of onset [5]. In addition, CRS type 5 describes a systemic disease such as diabetes or sepsis that causes both cardiac and renal dysfunction [6] (Fig. 1). In fact, many patients may develop or transform between different CRS subtypes during their disease progression.
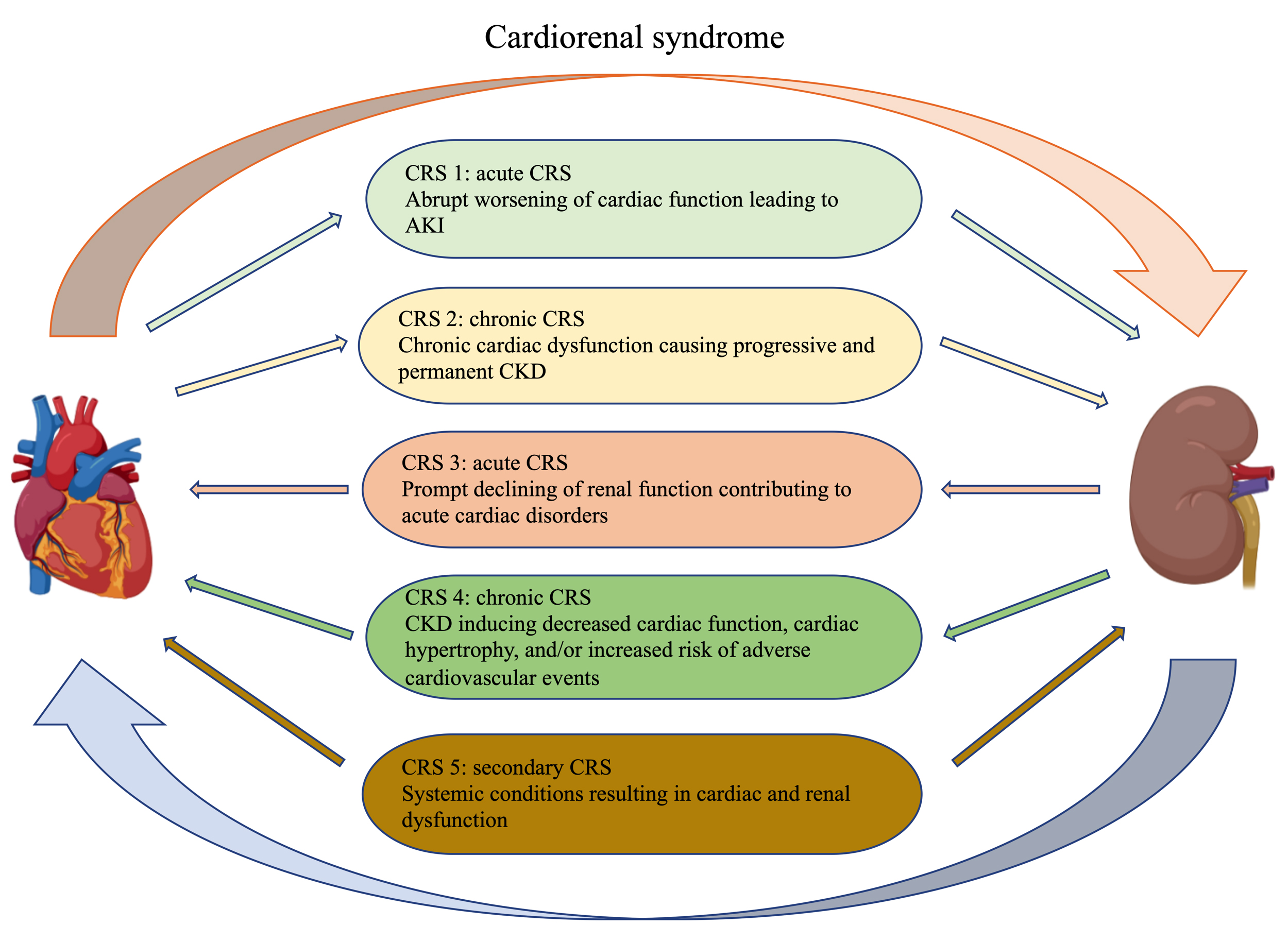 Fig. 1.
Fig. 1.Classification of cardiorenal syndrome. The Figures in this review are constructed by the online website (https://app.biorender.com).
Cardiorenal syndrome type 3 (CRS3), also known as acute renal-cardiac syndrome, is an episode of acute cardiac dysfunction caused by acute kidney injury (AKI) [7]. The main clinical manifestations of CRS3 include acute heart failure, acute myocardial infarction, tachyarrhythmia and acute cardiogenic shock [4, 8]. It is generally accepted that AKI is the pathogenic factor and initiator of CRS3, and approximately 70% of AKI patients develop into CRS3 [9, 10]. AKI is also the result of a rapid deterioration of cardiac function in CRS type 1, as well as the cause of acute cardiac injury in CRS type 3. Therefore, the interplay between the damaged kidney and heart may form a vicious cycle, which further aggravates the development CRS3 [5]. Thus, timely and effective intervention is necessary to impede the progression of this disease. Although CRS3 has attracted increased attention in recent years, the pathophysiological and molecular mechanisms of CRS3 remain largely unknown.
In this review, the literature search strategy and search terms include AKI, Cardiorenal syndrome type 3, molecular mechanisms of CRS3, therapeutic strategies of CRS3 as well as targets of CRS3. We summarized the pathophysiological changes, pathogenesis, and underlying mechanisms of CRS3, and discussed the potential therapeutic targets for CRS3. We have summarized and classified the novel biomarkers of AKI caused by different injuries. These biomarkers will be beneficial for accurate diagnostic and prognostic evaluation of progressive AKI, which can guide the adoption of therapeutic management strategies. Moreover, these AKI biomarkers may also be key molecules in the interplay between the kidney and heart. Timely monitoring and interventions of these AKI biomarkers may also delay the progression of CRS3 (Table 1, Ref. [11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36]).
| Biomarker | Sample | Origin | References |
| TIMP-2 | Urine | Distal tubule | [11, 12] |
| IGFBP7 | Urine | Proximal tubule | [11, 13] |
| NGAL | Urine or Plasma | Distal tubule, Epithelial cells, Neutrophils | [14] |
| KIM-1 | Urine | Proximal tubule | [15] |
| Collectrin↓ | Urine | Proximal tubule | [16] |
| OLFM4 | Urine | Neutrophils and Epithelial cells | [17] |
| L-FABP | Urine | Proximal tubule | [18] |
| CCL14 | Urine | Tubular epithelial cells | [19] |
| Dickkopf-3 | Urine | Tubular epithelial cells | [20] |
| IL-18 | Urine | Multiple cell types | [21, 22] |
| Cd, Cu and Zn | Urine | — | [23] |
| MMP-9 | Urine | Proximal tubule | [24] |
| Urine | Hepatocytes | [25] | |
| Urine | Hepatocytes | [26] | |
| Albumin | Urine or Serum | Hepatocytes | [27] |
| N-acetyl- |
Urine | Proximal tubule | [28] |
| Calprotectin | Urine or Plasma | Neutrophils | [29, 30] |
| Cystatin C | Plasma or Serum | Nucleated cells | [31] |
| Lnc-HILPDA, Lnc-PRND | Serum | Kidney | [32] |
| suPAR | Plasma | Immune cells, Endothelial cells | [33, 34] |
| Proenkephalin A | Plasma | Multiple cell types | [35] |
| GDF15 | Plasma | Proximal tubule | [36] |
| TIMP-2, tissue inhibitor of metalloproteinases 2; IGFBP7, insulin-like growth factor-binding protein 7; NGAL, neutrophil gelatinase-associated lipocalin; KIM-1, kidney injury molecule 1; OLFM4, Olfactomedin-4; L-FABP, liver-type fatty acid-binding protein; CCL14, C-C motif chemokine ligand 14; Dickkopf-3, Dickkopf-related protein 3; IL-18, interleukin-18; MMP-9, matrix metalloproteinase-9; Lnc-HILPDA, long non-coding RNA HILPDA; Lnc-PRND, long non-coding RNA PRND; suPAR, soluble urokinase plasminogen activator receptor; GDF-15, growth/differentiation factor 15; ↓ represents a reduced level. | |||
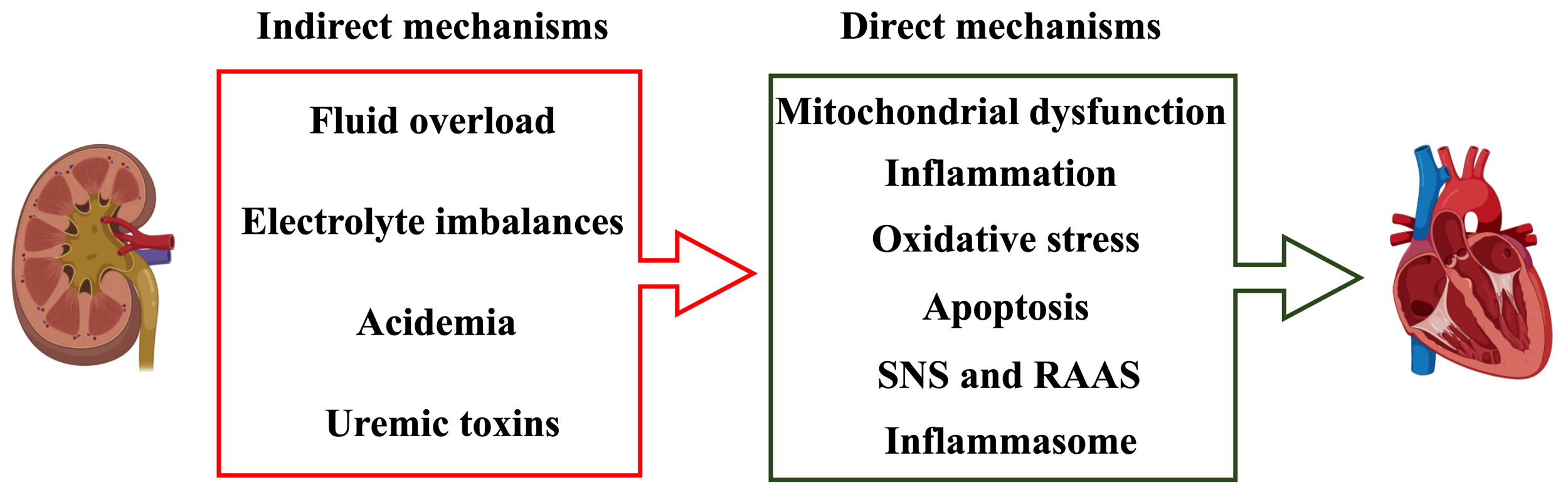
Although the precise pathophysiological mechanisms of CRS3 remain unclear, AKI is considered to possess direct and indirect mechanisms on cardiac structure and function. Direct mechanisms can be attributed to mitochondrial dysfunction, inflammation, oxidative stress, apoptosis, activation of the sympathetic nervous system (SNS), renin-angiotensin-aldosterone system (RAAS) and inflammasome. Indirect mechanisms include fluid overload, electrolyte imbalances, acidemia and uremic toxins [4, 37].
Recent studies have demonstrated that mitochondrial dysfunction as an important
contributor to myocardial injury in CRS3 [7]. Therefore, maintaining
mitochondrial homeostasis is a promising strategy for the treatment of CRS3 [7, 38]. Both mitochondrial unfolded protein response (UPR
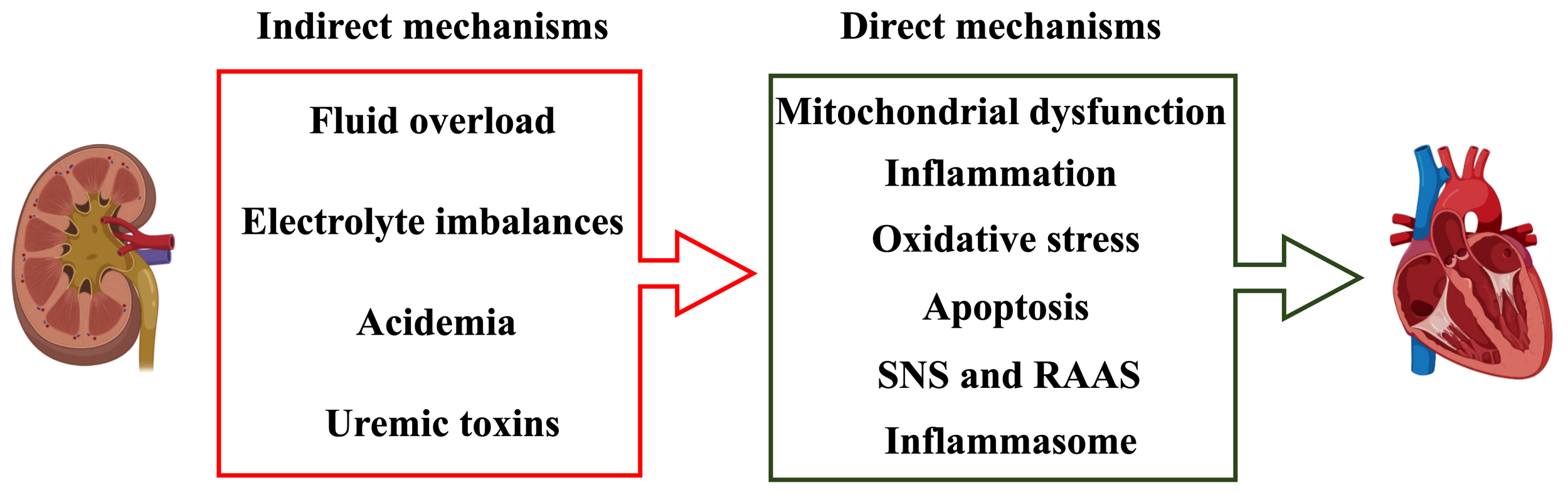 Fig. 2.
Fig. 2.Direct and indirect mechanisms contributing to CRS3.
Inflammatory response is an important pathological process and therapeutic
target of CRS [49, 50]. It has been observed that the expressions of several
inflammatory factors are significantly altered after IRI in the unilateral
kidney, including tumor necrosis factor-a (TNF-
Accumulating evidence has demonstrated that oxidative stress is closely related to the dysfunction of cellular components that can lead to organ disorders, and is recognized as one of the major causes of IRI-induced distant organ injury [59, 60, 61]. Oxidative stress is also considered to be a direct by-product of mitochondrial damage in myocardial cells involved in the pathogenesis of CRS3 [62]. Oxidative stress increases the channels of calcium release, such as the inositol 1,4,5-triphosphate receptor, and inhibits the activity of calcium reuptake proteins, including sarcoplasmic/endoplasmic reticulum calcium ATPase 2a [63]. This results in a systolic calcium deficit and/or diastolic calcium leak, which increases the possibility of arrhythmogenic events and diminishes cardiac contractile capacity [64]. Eventually, impaired mitochondria provoke cell death by activating caspase 3/7/9 in myocardial cells during CRS3 [65]. The death of myocardial cells then prompts the inflammatory response and the release of inflammatory factors, further aggravating the damage of myocardial cells in the process of CRS3 [10]. Caio-Silva et al. [62] used a unilateral renal IRI mouse model to evaluate oxidative stress and antioxidant parameters of the kidney and heart. The results showed that the activities of antioxidant enzymes superoxide dismutase (SOD) in kidney tissues significantly increased, and the bioavailability of nitric oxide (NO) was also increased [62]. The activities of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) and nitric oxide synthase (NOS) in myocardial tissues were enhanced, accompanied by the aggravation of cell damage, 8 days after renal IRI. Cai et al. [65] reported in a CRS3 mouse model that CRS3 resulted in lower heart function, increased inflammatory responses and exacerbated myocardial oxidative stress than in sham mice. Mitochondrial oxidative stress and the inflammation response have been proposed to reduce cardiomyocyte viability and function during CRS3. Neres-Santos et al. [7] reported that vitamin C, an antioxidant, exerted renocardiac-protective effects by reducing NO levels and inducible nitric oxide synthase (iNOS) expression in the kidney and heart. These studies suggest that oxidative stress is a direct mechanism involved in the pathogenesis of CRS3.
Various types of cell death have been reported to be involved in acute renal and
cardiac injury, including apoptosis, necrosis, pyroptosis, ferroptosis and
autophagy [66, 67, 68, 69, 70, 71, 72, 73]. Apoptosis is one of the most common modes of cell death, which
is activated by death receptors [74]. Kelly et al. [57] first reported
that AKI leads to an upregulated expression of intercellular cell adhesion
molecule-1 (ICAM-1) and a significant increase in peroxidase activity and
apoptosis in cardiomyocytes, accompanied by a decrease in left ventricular
dilatation and short-axis shortening of the left ventricle. This study found that
the inflammatory factors, TNF-
AKI is accompanied by continuous activation of the sympathetic nervous system (SNS) and the renin-angiotensin-aldosterone system (RAAS) [56, 77, 78, 79]. The activated SNS will induce the release of neuropeptide Y, which is responsible for the formation of neointima, vasoconstriction and impairment of the immune system [80]. SNS activation has multiple direct negative effects on the heart, including increasing myocardial oxygen demand, destroying calcium homeostasis, promoting cardiomyocyte apoptosis and hypertrophy [81, 82, 83]. In addition, SNS can further activate RAAS. Studies have shown that RAAS activation has become a major risk factor for AKI. RAAS plays a vital role in regulating renal hemodynamics, function and pathophysiology during kidney diseases [84, 85]. Recent studies have demonstrated that urinary renin, the rate limiting enzyme of RAAS, is an indicator of renal RAAS activity, and the increase of renin is related to the severity of AKI [78, 86]. RAAS activation leads to an increase in angiotensin II, which results in systemic vasoconstriction and extracellular volume expansion by increasing sodium retention [87], leading to cardiac remodeling and ventricular hypertrophy [88]. In vitro studies have shown that angiotensin II can induce hypertrophy, cell reprogramming and necrosis of cardiomyocytes, and cardiac fibrosis [89, 90, 91]. Although the role of RAAS and SNS in CRS3 still lacks solid evidence, further in-depth experimental studies may help to elucidate the precise effects and underlying mechanisms.
Inflammasome, a multiprotein complex which can trigger the cleavage and
activation of proinflammatory cytokine including IL-1
| Inflammasome component | Samples | Outcomes | References |
| NLRP3 | Serum samples | NLRP3 in the septic shock group was significantly higher than that in the healthy control group. | Huang et al., 2022 [104] |
| IL-1 |
Serum samples | IL-1 |
Zheng et al., 2021 [105] |
| Shi et al., 2021 [106] | |||
| IL-1 |
Serum samples | IL-1 |
Yang et al., 2021 [107] |
| IL-1 |
Plasma samples | IL-1 |
Brocca et al., 2015 [108] |
| IL-18 | Plasma samples | IL-18 were significantly elevated in CRS. | Pastori et al., 2015 [109] |
| IL-18 | Urine samples | Urine IL-18 were independently associated with AKI stage. | Duff et al., 2021 [110] |
| Caspase1, IL-18 | Urine samples | IL-18 and caspase-1 were increased in patients undergoing coronary angiography. | Lau et al., 2018 [111] |
| NLRP6 | Renal tubules | NLRP6 was reduced during human kidney injury. | Valiño-Rivas et al., 2020 [112] |
| ASC, Active-Caspase1 | Renal interstitium | ASC and Active-Caspase1 were significantly increased in the RIAKI case compared to a healthy control. | Grivei et al., 2020 [113] |
| NLRP3, Nod-like receptor pyrin domain-containing protein 3; IL-1 | |||
Fluid overload, electrolyte imbalances, acidemia, and uremic toxins contribute to CRS3 under pathophysiological conditions. Fluid overload will lead to physiological abnormalities in multiple organs, especially in AKI patients [114, 115]. There is a time correlation between volume overload and the development of ventricular arrhythmias [116], since fluid overload increases the work of the heart, contributing to arrhythmias [117]. Therefore, complex arrhythmias have become a serious complication of AKI-induced cardiac injury and myocardial dysfunction. In turn, arrhythmias also increase the risk of renal failure [118].
Hyperkalemia is a common complication of severe AKI. Hyponatremia can cause premature atrial and ventricular contractions, while severe hypokalemia can cause prolongation of the Q-T interval, leading to ventricular tachycardia, ventricular fibrillation and cardiac arrest [119]. Hypernatremia will affect the heart during severe dehydration, and can result in tachycardia, decreased blood pressure, intracranial hemorrhage, and edema.
Metabolic acidosis is a common complication of AKI and a common indication for initiation of renal replacement therapy (RRT) [120], since elevated plasma hydrogen ion concentrations in patients with metabolic acidosis can seriously affect cardiac function [121, 122, 123]. Severe acidosis results in a marked decrease in cardiac contractility, which can be significantly improved by correcting acidosis [124, 125].
AKI causes an acute uremic state, as evidenced by electrolyte disturbances, disrupted volume stability, as well as the accumulation of metabolic toxins, including small water-soluble compounds, large intermediate molecules, and protein-bound uremic toxins (PBUTs) [126], which have been extensively investigated in chronic kidney disease (CKD); particularly indoxyl sulfate (IS), P-cresol sulfate (PCS), and indole-3-acetic acid (IAA) [127, 128, 129, 130]. Multiple studies have shown that IS promotes cardiac fibrosis and hypertrophy by inducing oxidative stress and inflammation. Similar to IS, PCS is toxic to blood vessels and the heart [131]. Huang et al. [132] demonstrated the toxic effect of PCS on cardiomyocytes by reducing cardiomyocyte proliferation and inducing mitochondrial damage. Lekawanvijit et al. [133] confirmed its deleterious effect on vascular reactivity in an in vitro model of an aortic ring exposed to PCS.
These accumulated metabolic toxins can result in cellular and tissue damage to the kidney and heart, while renal dysfunction increases the accumulation of uremic toxins, ultimately leading to the progression of CRS3. Although acute uremia in AKI may contribute to the cardiotoxicity of CRS3, further studies are required to determine their complex roles and underlying mechanisms in cardiotoxicity after AKI [134] (Fig. 2). Several studies, including our own, indicate that uremic toxins are closely related to CVDs [135, 136].
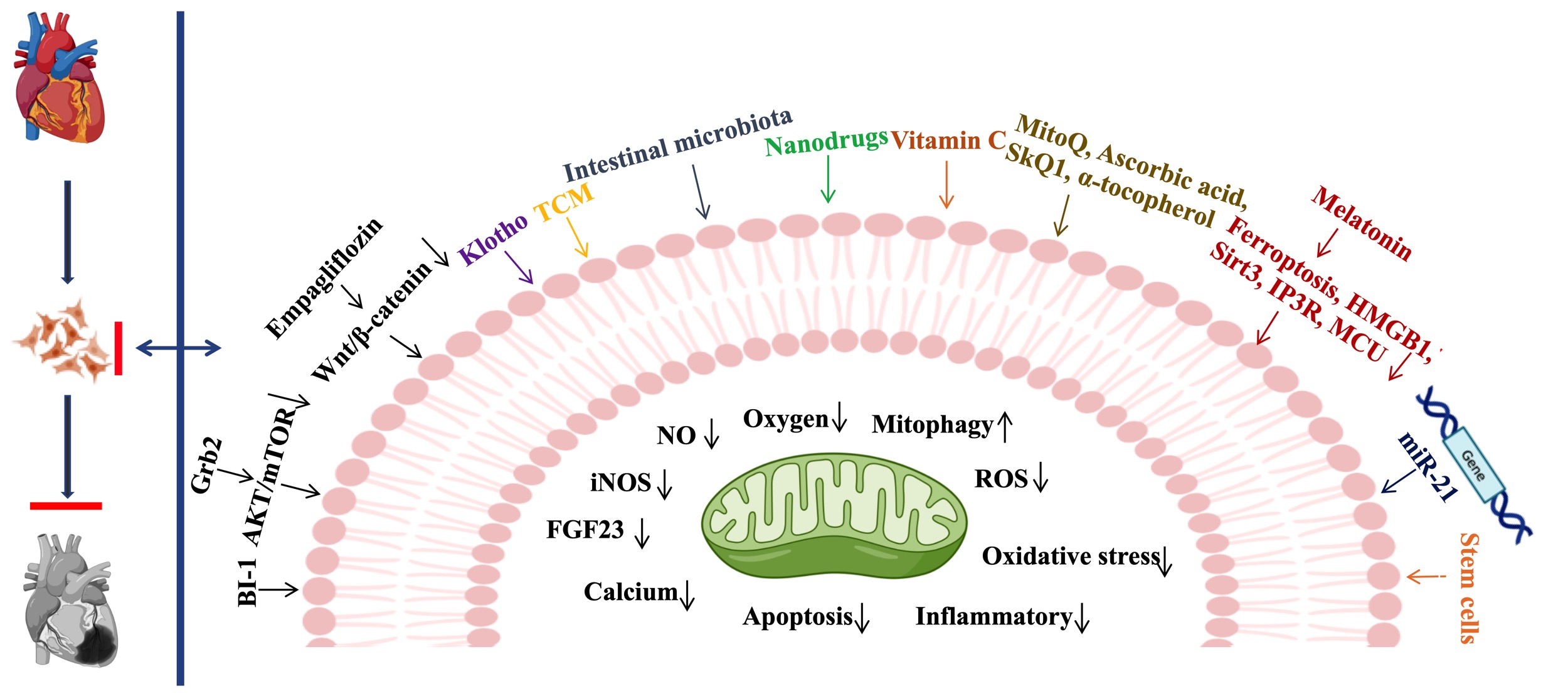
The treatment of CRS3 is a difficult clinical challenge, since drugs used to treat CVD may possess potential nephrotoxicity, while treatment for AKI usually provokes myocardial damage. Therefore, targeted therapies to ameliorate AKI-related cardiac dysfunction are urgently needed. Since the pathophysiologic mechanism of CRS3 remains largely unknown, it is necessary to comprehensively study its molecular mechanisms and develop novel therapeutic targets for CRS3. In Fig. 3 we summarize the recently reported potential treatments targeting CRS3 .
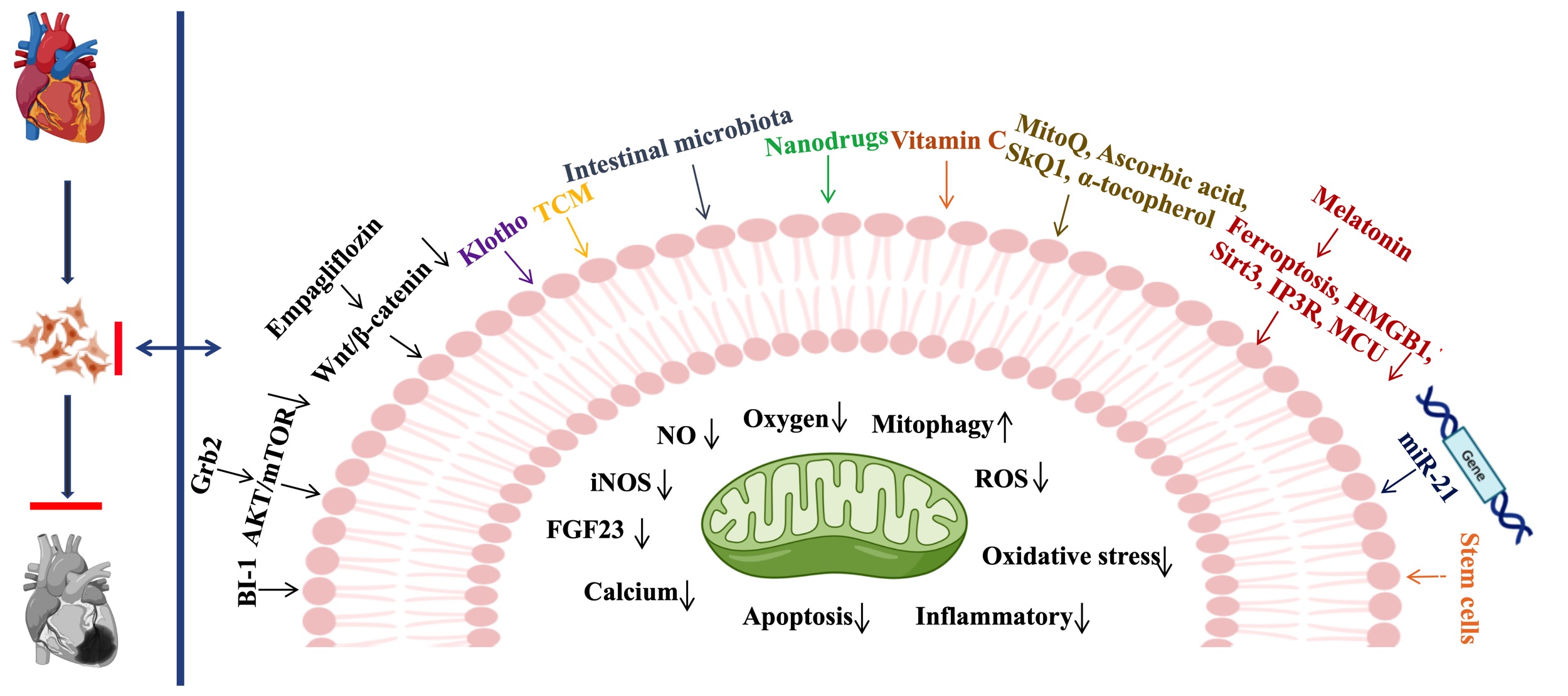 Fig. 3.
Fig. 3.Therapeutic strategies for CRS3. BI- 1, Bax inhibitor-1; Grb2, Growth factor receptor-binding protein 2; AKT, AKT Kinase; mTOR, Mammalian target of rapamycin; TCM, Traditional Chinese Medicine; Wnt, Wingless/Integrated; MitoQ, Mitoquinone; SKQ1, 10-(6′-plastoquinonyl) decyltriphenyl phosphonium; HMGB1, high mobility group box 1; Sirt3, sirtuin-3; IP3R, Inositol 1,4,5-trisphosphate receptor; MCU, Mitochondrial calcium uniporter; NO, nitric oxide; iNOS, inducible nitric oxide synthase; FGF23, Fibroblast growth factor 23; ROS, reactive oxygen species.
Emerging evidence confirms that Bax inhibitor-1 (BI-1) could ameliorate
myocardial injury in patients with CRS3 by activating mitochondrial UPR and
FUN14 domain-containing protein 1 (FUNDC1) -mediated mitophagy, suggesting that BI-1 plays a crucial role in CRS3
[39]. Wang et al. [10] performed a proteomic analysis of CRS3 and
identified Grb2 as an important regulator involved in AKI-related myocardial
injury. Elevated Grb2 contributed to
diastolic dysfunction and mitochondrial bioenergetics impairment, while the
application of Grb2-specific inhibitor reversed these pathological changes during
AKI. Abnormally elevated levels of Grb2 promotes mitochondrial metabolic disorder
of myocardial cells by inhibiting the Akt/mTOR signaling pathway, which can lead
to cardiac dysfunction [10]. Cai et al. [65] reported that empagliflozin
can preserve mitochondrial structure, stabilize cardiomyocyte structure, maintain
cardiac systolic and diastolic function, and reduce myocardial inflammation via
activating the Wingless/integrated (Wnt)/
Collectively, these findings suggest that mitochondrial dysfunction is a common pathological feature and molecular mechanism of AKI and its related to cardiac injury. Targeting mitochondrial dysfunction and maintenance of mitochondrial homeostasis are promising strategies for the treatment of CRS3. However, the upstream mechanisms regulating mitophagy in CRS3 remain incompletely defined and further mechanistic studies are required.
Accumulating evidence suggests that vitamin C
confers renal and cardioprotective roles in CRS3. Recent studies have shown that
vitamin C can not only improve AKI, but also protect the heart after AKI [7, 137].
It has been reported that vitamin C treatment can preserve kidney weight, restore
renal function, reduce NO levels and iNOS expression, and improve oxygen
consumption. After vitamin C treatment, oxygen consumption and NO levels were
improved, oxidative stress was attenuated, mitochondrial damage was ameliorated,
and myocardial cell damage was reduced. This study also showed that when the
kidney was injured, vitamin C should be given as soon as possible to protect the
kidney and heart from IRI [7]. However, this study also has some limitations. It
does not clarify the mechanism by which ROS derived from the kidney affects
cardiac damage and the role of vitamin C in the crosstalk between the kidney and
the heart. In addition, several potential therapeutic approaches, such as
Mitoquinone (MitoQ), ascorbic acid, 10-(6′-plastoquinonyl) decyltriphenyl phosphonium (SkQ1), and
Klotho is an anti-aging protein, predominantly expressed in the kidney. Previous
studies have confirmed that Klotho exerts a protective role in AKI and CVD [139, 140]. Klotho deficiency not only aggravates AKI, promotes the transition of AKI
to CKD, but also is closely related to CVD, suggesting that regulating endogenous
or exogenous Klotho can provide renal and cardiac protection [141]. Another study
has confirmed that Klotho has cardioprotective effect on CRS3 induced by renal
IRI, mainly by preventing cardiac hypertrophy and Ca
Previous studies have confirmed that melatonin plays a protective role in AKI. Melatonin reduces AKI by inhibition of nuclear factor erythroid 2-related factor 2/solute carrier family 7 member 11 (Nrf2/Slc7a11) axis-mediated ferroptosis [143]. Melatonin significantly decreases folic acid (FA) induced AKI injury by inhibiting the nuclear translocation of high mobility group protein B1 (HMGB1) in renal tubular epithelial cells [144]. In addition, melatonin alleviates contrast-induced kidney injury by activating Sirt3 [145]. Melatonin also has cardioprotective effects, in cardiac IRI [146], septic cardiomyopathy [147], and drug-induced cardiotoxicity [148]. These results strongly suggest that melatonin may serve as a potential therapeutic treatment for CRS3. Wang et al. [9] confirmed that melatonin protects cardiac function from CRS3 by inhibiting inositol 1,4,5-trisphosphate receptor-mitochondrial calcium uniporter (IP3R-MCU) signaling. Melatonin preconditioning attenuates renal IRI-induced cardiac injury by maintaining myocardial diastolic function and reducing cardiomyocyte death. Melatonin can also enhance the effects of other treatments or medications for CRS. For example, the combination of melatonin and Exendin-4 has a protective effect on the heart and kidney of rats with CRS [149]. It was also reported that melatonin enhanced the therapeutic effect of mesenchymal stem cell-derived exosomes on renal IRI in rats [150].
microRNAs (miRNAs) are associated with the
development and progression of various injuries, including renal and cardiac
diseases, and CRS [151, 152]. Therefore, miRNAs are commonly used as disease
biomarkers and potential therapeutic targets. Recent studies have shown that
multiple miRNAs are also closely related to CRS [153]. It was reported that
miR-21 is highly expressed in both the heart and kidneys and circulating miR-21
can serve as a diagnostic and prognostic marker in CRS2. miR-21 has been
associated with poor prognosis in most primary organ failures, which suggests
that inhibition of miR-21 may be a potential therapeutic target for CRS3. Using
bioinformatics analysis, Romana Ishrat et al. [154] determined that some
miRNAs, such as miR-122-5p, miR-222-3p, miR-21-5p, miR-5p, miR-3p, miR-24-3p and
miR-143-3p as well as some related target genes including transforming growth factor-β1, X-linked inhibitor of apoptosis protein, Lamin-B2, N-alpha-acetyltransferase 50, Nucleoside diphosphate-linked moiety X motif 3, YME1-like protein 1, Insulin-like growth factor 1 receptor, DEAD box protein 6, Protein argonaute-2, Myc proto-oncogene protein, and Protein Hook homolog 3 (TGF-
Stem cells have the potential to treat many diseases in regenerative medicine
due to their self-renewal and multi-directional differentiation potential. Stem
cells function through paracrine mechanisms, modulating apoptosis, reducing
oxidative stress and inflammatory mediators, improving damaged tissue and
inducing a favorable remodeling environment for organs. These favorable qualities
has been demonstrated in experimental models of acute and chronic kidney injury
[155, 156, 157]. A recent study reported that adipose-derived stem cells (ADSCs) can
alleviate the pathophysiological changes of CRS3 [158]. The results of this study
demonstrated that the levels of inflammatory factors, such as serum
Interferon-
In comparison with bone marrow-derived mesenchymal stem cells (MSCs), which must be obtained through minimally invasive surgery, ADSCs have many advantages. For example, ADSCs can be obtained from adipose tissues, which are more abundant than bone marrow stem cells, easier to culture, and faster to grow. Although stem cells have favorable therapeutic prospects in the treatment of CRS3, there are still many limitations in their clinical usage, such as the complexity of stem cell sources, the safety and effectiveness of stem cell therapy due to the uncontrolled preparation process and quality control of stem cells. In addition, stem cells may be potentially carcinogenic, which greatly limits their applications in clinical medicine. There is emerging evidence that MSC-exosomes can serve as natural carriers for targeted drug delivery. Therefore, therapeutic drugs can be efficiently incorporated into exosomes and then delivered to damaged tissues. In addition, MSC exosomes also comprise bioactive substances such as proteins, mRNAs and miRNAs. Studies have reported MSC-exosomes in AKI [159], and it is also a promising method for cell-free treatment of AKI and CRS3. These studies have demonstrated that MSC-exosomes can improve kidney and heart damage, implying that MSC-exosomes are a promising cell-free therapy for CRS3.
There is evidence that the burst of active oxygen and reactive nitrogen (RONS) are major contributors to the progression of AKI [160, 161]. Because of the complex and unique physiological structure of the kidney, most anti-oxidation and anti-inflammatory small molecule drugs are ineffective due to the lack of specificity to kidney tissue and their side effects [162]. Recent studies, including our own, show that nano drugs can target the kidney to solve the limitations of current AKI treatment by controlling the size, shape and surface characteristics of nano drugs [163]. Nano drugs for AKI mainly include nano-RONS-sacrificial agent, antioxidant nano enzyme, and the nano carrier of antioxidant anti-inflammatory drugs. These nano drugs have demonstrated important therapeutic effects, such as reducing oxidative stress damage, restoring kidney function, and are associated with low adverse effects [162]. Ni et al. [164] found that molybdenum-based nanoclusters can be utilized as antioxidants to improve AKI. Zhao et al. [165] studied the redox mediated artificial non enzyme antioxidant MXene nano platform to alleviate AKI. Zhang et al. [166] developed biodegradable self-assembled ultra-small nano dots as active oxygen/nitrogen species scavenger, which can significantly improve AKI. Yu et al. [167] found that cerium dioxide nanoparticles targeting mitochondria with atorvastatin combined with the ROS responsive nano drug delivery system has favorable effects in the treatment of sepsis-induced AKI. Wang et al. [163] found that selenium nanoparticles can alleviate AKI via regulating the GPx-1/NLRP3/Caspase-1 pathway. In addition, nano drugs may play a role in treating heart disease. Haley et al. [168] discussed the clinical feasibility of the therapeutic strategy of delivering anti-inflammatory drugs to the heart muscle through biodegradable polymers, liposomes, hydrogels and nanoparticles-based drug delivery models (NDDM). NDDM is a promising method to successfully treat ischemic HF by delivering anti-inflammatory agents to the myocardium. Tang et al. [169] found that platelet nanobubbles fused with stem cells can target repair of cardiac injury. Collectively, these studies suggest that nano drugs not only can be used to treat AKI, but also to improve CRS.
Compared with traditional drugs, nano drugs possess several advantages in the treatment of AKI. Nanodrugs have a variety of materials, flexible sizes and shapes [162]. In addition, nano drugs can be targeted by molecular modification to locate the lesion site, to better control targeting of specific tissues and organs. Furthermore, nanomaterials can also be used as a drug delivery platform to improve the biocompatibility and stability of drugs, and the controlled release of drugs. However, the difference between species is the most important challenge for the clinical transformation of nano drugs. Most studies on nano drugs are only based on rodent models, and not human AKI disease models [170]. Compared with humans, the capillary density and mitochondrial density of mouse kidney tissue are much higher because the metabolic rate of mice is almost seven times than that of humans. AKI patients have diverse genetic and disease backgrounds, such as diabetes, liver and other diseases [171, 172], which may affect the metabolism and efficacy of nano drugs. Therefore, different animal models are necessary to establish multi-dimensional validation, such as the use of the AKI model of zebrafish for validation [173, 174]. Finally, the biocompatibility and long-term safety of nano drugs are unknown and need to be validated in human studies prior to their use in clinical medicine [175].
There is now evidence that intestinal dysbiosis is closely linked to AKI, shedding light on kidney–intestine crosstalk in AKI [176, 177, 178]. Zhu et al. [179] demonstrated that the supplementation of Lactobacillus casei Zhang could prevent AKI and impede the progression of CKD by improving intestinal flora, increasing the levels of short chain fatty acids (SCFAs) and nicotinamide in the serum and kidney. Yang et al. [176] reported that the increase of Enterobacteriaceae and the decrease of Lactobacillus and Ruminococcus were hallmarks of dysbiosis induced by IRI and were related to the decreased levels of SCFAs, intestinal inflammation, and the leaky gut. They also confirmed that the intestinal microbiota controls the severity of AKI through regulation of the immune system. This renal protective effect is related to the reduction of Th17 and Th1 responses as well as the expansion of regulatory T cells and M2 macrophages [176]. A study by Andrade Oliveira et al. [180] also showed that SCFAs derived from intestinal microbiota prevents IRI-AKI, suggesting a crosstalk between kidney and intestine. Lee et al. [181] found that Lactobacillus salivarius BP121 prevented cisplatin-induced AKI by inhibiting uremic toxins and alleviating dysbiosis. Metabolites derived from gut microbiota also play an important role in AKI. For example, D-serine derived from gut microbiota can mitigate AKI [182]. Thus, targeting intestinal microbiota may provide a new therapeutic strategy for AKI and CRS3.
A number of studies, including our own, have also confirmed that TCM and active
monomers can also improve kidney function in AKI though different mechanisms,
including inhibiting inflammation, cell apoptosis, necroptosis, ferroptosis, and
decreasing oxidative stress [183, 184]. Our recent study found that Oroxylin A,
the main active component of Scutellaria baicalensis, prevented AKI and
progression to CKD by inducing PPAR
Mitochondrial dysfunction, one of the molecular links between the kidneys and heart, plays a crucial role in CRS3. Targeting mitochondrial dysfunction may serve as a therapeutic target to treat kidney and heart disease in CRS3. However, the mechanism for these benefits has not been fully clarified. The underlying mechanisms of inflammatory factors and biomolecules in damaged kidney tissues and their effects on heart tissues after AKI remain elusive. Although several mechanisms are involved in maintaining mitochondrial function, determining which regulatory mechanism possesses better therapeutic effects in CRS3 still require further investigation. Myocardial damage can also be caused by mechanical stress such as calcium metabolism disorders, intracellular acidosis and fluid overload. The relationship between these factors and mitochondrial dysfunction will also need to be clarified.
In addition to mitochondrial dysfunction, the crosstalk between the kidney and
heart is also key in the treatment of CRS3. Mechanistically, the crosstalk
between organs after tissue injury may involve soluble mediators and their target
receptors, cellular mediators, especially immune cells, as well as newly
discovered neural immune connections. Khamissi et al. [191] identified
kidney-released circulating osteopontin (OPN) as a novel AKI-acute lung injury
(ALI) mediator. OPN released from renal tubule cells triggered lung endothelial
leakage, inflammation, and respiratory failure [191]. In CRS3, the mechanism of
kidney derived inflammatory factors, including IL-1
Although different modes of cell death have been observed in renal tubular epithelial cells and cardiomyocytes during the progression of AKI and cardiac injury, whether there is a common mechanism of cell death in the kidney and heart remains unknown. Thus, developing convenient, accurate and noninvasive methods to predict the severity of tissue injury and the types of cell death in CRS3 will be necessary to develop therapeutic treatments for CRS3.
The contribution of different cell types to the progression of CRS3 still needs to be investigated. Targeting distinct cell types in kidney or heart tissues may generate different outcomes. For example, promoting mitophagy in vascular smooth muscle cells (VSMCs) advances the progression of atherosclerosis (AS), while in endothelial cells and macrophages, promoting mitophagy is atheroprotective. These controversial results provide further evidence that different cell types playing different roles may determine the progression and fate of certain diseases. Therefore, comprehensive studies of the roles and regulatory mechanisms of different cells in CRS3 will be beneficial for developing new therapeutic targets for treating CRS3.
These studies have shown that melatonin combined with other treatments can overcome the shortcomings of a single drug and enhance its efficacy. For example, melatonin combined with mitochondria-targeting drugs or stem cells can achieve an enhanced therapeutic effect. Innovative drug delivery systems such as nanoparticle-loaded drug delivery, extracellular vesicles, and molecular structure optimization to increase bioavailability and tissue targeting will also be required [149, 150].
The occurrence of AKI is attributed to a rapid deterioration of cardiac function in CRS1, and is responsible for acute cardiac damage in CRS3. Thus, the interaction between the kidney and heart is a bidirectional regulation pattern, which forms a vicious cycle and ultimately contributes to the progression of CRS3. Emerging evidence suggests that mitochondrial and inflammatory factors may be the central link for developing therapeutic targets in CRS3. Targeting mitochondrial dysfunction or inflammatory mediators may serve as a promising therapeutic strategy. We reviewed the existing strategies for CRS3 therapy, including targeting mitochondrial dysfunction, antioxidant, Klotho, melatonin, gene therapy, stem cell therapy, nanodrugs, intestinal microbiota and TCM. In addition, according to the different pathological characteristics of heart and kidney injuries, we propose a combined treatment scheme to overcome the shortcomings of a single factor treatment and enhance the therapeutic efficacy. We also suggest developing more sensitive and accurate non-invasive biomarkers of CRS3 to grade and judge the degree of damage. We also recommend timely and targeted treatment according to the degree of injury and disease progression. However, current studies are mainly based on cell and animal models. Further validation in clinical trials to understand the efficiency and safety of these potential therapeutic strategies are urgently needed.
CRS3, Cardiorenal syndrome type 3; AKI, acute renal injury; CKD, Chronic kidney
disease; CVD, chronic cardiovascular disease; ROS, reactive oxygen species; SNS,
sympathetic nervous system; RAAS, renin-angiotensin-aldosterone system;
UPR
YH and JZ proposed the concept and revised the manuscript. YL, XG and YS wrote the manuscript and designed the figures. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
We would like to express our gratitude to all those who helped us during the writing of this manuscript.
This study was supported by research grants from Key program of the Natural Science Foundation of China (No. 82030023), the Natural Science Foundation of China (Nos. 81800621, 81802783, 81873605, 82170705), Chongqing Science and Technology Talent Program (cstc2021ycjh-bgzxm0145), Natural Science Foundation of Chongqing Science & Technology Commission (cstc2021jcyj-msxmX0672, CSTB2022NSCQ-MSX0220), Frontier specific projects of Xinqiao Hospital (No. 2018YQYLY004), and Personal training Program for Clinical Medicine Research of Army Medical University (No. 2018XLC1007).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.