





1 Cardiovascular Surgery, Xijing Hospital, Fourth Military Medical University, 710032 Xi'an, Shaanxi, China
†These authors contributed equally.
Academic Editors: Daniel I. Simon and Teruo Inoue
Abstract
Warfarin is clinically used as the first choice for long-term anticoagulant
therapy, and for the prevention of thromboembolic events. However, when used at
low doses in the long term or high doses in the short term, warfarin treatment
may result in tissue calcifications—such as calcifications in the coronary
arteries, peripheral vascular system, blood vessels of patients with atrial
fibrillation and chronic kidney disease, and vascular valves—and
atherosclerotic plaque calcification. These warfarin-induced calcifications may
affect cardiovascular function and exacerbate diseases such as diabetes and
hypertension. Studies have shown that quercetin, osteoprotegerin, sclerosin, and
sodium thiosulfate may alleviate these effects by interfering in the
Wnt/
Keywords
- warfarin
- anticoagulation
- calcification
- prevention
Oral warfarin anticoagulation (OAC) administration is the main strategy for clinical anticoagulation, and effectively prevents various thromboembolic diseases. It is considered the first choice among long-term anticoagulant drugs for prevention of diseases, such as pulmonary embolism and deep vein thrombosis after mechanical heart valve replacement [1, 2]. However, one of the lesser-known long-term side effects of warfarin use is an increase in systemic arterial calcification [3]. Clinical and animal experimental data have demonstrated that long-term use of warfarin can lead to calcification of multiple tissues throughout the body [4, 5], leading to increased vascular wall stiffness and reduced compliance. These pathological side-effects may lead to serious complications, such as atherosclerosis, valvular calcification, and coronary artery calcification.
While the anticoagulant effect of warfarin is used extensively in clinical
practice, treatment strategies addressing warfarin-induced calcification are
still lacking. Previous studies have shown that quercetin, osteoprotegerin,
sclerostin, and sodium thiosulfate can alleviate warfarin-induced calcification,
mainly through the activity of Wnt/
Calcification of coronary arteries is a well-known risk factor for mortality in
ischemic heart disease. Poterucha TJ et al. [14] demonstrated that the
use of warfarin was associated with increased systemic calcification, including
calcification of the coronary arteries and the surrounding vasculature. Andrews J
et al. [15] evaluated the effects of warfarin on coronary percent
atheroma volume (PAV) and calcium index (CaI), in patients with coronary heart
disease. The results revealed that warfarin had no significant effect on PAV, but
was independently correlated with increased CaI in a multivariate model. Namba
et al. [16] assessed 42 patients with atrial fibrillation who had a high
risk of developing atherosclerosis. The results revealed that long-term warfarin
treatment may be related to osteoporosis and VC in hypertensive patients 60–80
years old. Villines et al. [17] conducted a cross-sectional analysis on
the severity of coronary artery calcification (CAC) in patients without coronary
heart disease treated with warfarin and found that the severity of CAC was
positively correlated with the duration of warfarin use. Wei et al. [18]
investigated the correlation between age and VC induced by warfarin. The data
revealed that there was a dose-time-response for warfarin that was positively
correlated with the distribution of the aortic calcification (AC) score and
plasma IL-6 levels in patients less than 65 years old, but this correlation was
not observed in patients
Animal experiments have also highlighted the effects of warfarin on AC. Uto
et al. [19] investigated the role of collagen metabolism in AC. Male
Sprague-Dawley rats (5 weeks old) were fed a diet containing warfarin and vitamin
K1 (WVK) to establish a VC model;
Han et al. [20] assessed the incidence of peripheral AC in 430 patients treated with warfarin and found that warfarin was correlated with lower limb AC, but not with age, sex, diabetes status, or other characteristics. Using in vivo experiments, Mackay et al. [21] showed that mutations in the zebrafish (Danio rerio) lineal homologue Abcc6a, led to extensive and high mineralization of the axial skeleton, while warfarin aggravated its calcification phenotype, and vitamin K reduced ectopic calcification to normal levels.
Tantisattamo et al. [22] found that warfarin administration increased the incidence of calcification of breast arteries in women. In a multivariate logistic model, warfarin was an important determinant of AC in women, and the severity of calcification was related to the age and duration of warfarin utilization, but not to the length of time after stopping warfarin treatment, indicating that warfarin-induced calcification of the breast arteries is cumulative and might be irreversible.
Breast fat necrosis (BFN) is usually considered a benign inflammatory response to breast trauma. AlQattan et al. [23] reported the case of a 65-year-old woman with atrial fibrillation who took warfarin. Examination of the histopathology revealed fat necrosis caused by calcification. Considering the background of the patient, the diagnosis was secondary BFN due to calcification induced by warfarin. Alappan et al. [24] investigated whether oral warfarin-induced VC could be reversed after renal transplantation and assessed the progression of calcification in the breast artery before and after renal transplantation. The data showed that VC is irreversible after renal transplantation, which highlighted the importance of prevention.
In fact, cardiovascular events are one of the major causes of deaths among patients affected with kidney disease and diabetes [25]. VC is a common complication in elderly patients with diabetes or renal insufficiency [26]. Warfarin was reported to cause vascular calcification, and renal arteries calcification with a decline in kidney function. As a result of kidney insufficient, most of the drugs used for cardiovascular risk reduction become unavailable [27]. Some patients with diabetes and hypertension need to take warfarin for an extended period of time, and some individuals need higher doses of warfarin to maintain a normal international normalized ratio (INR). However, long-term use of warfarin may lead to the calcification of small and medium-sized blood vessels and aggravate underlying diseases.
Zhang YT et al. [26] found that long-term warfarin treatment in patients with mechanical heart valve replacement, atrial fibrillation, hemodialysis, and chronic kidney disease could induce and accelerate VC, which not only leads to serious complications, such as atherosclerosis, valvular calcification, and CAC, but also aggravates diseases, such as diabetes and hypertension. Siltari et al. [28] revealed that warfarin increased the risk of further VC in patients with atherosclerosis. Bell DSH et al. [29] indicated that the incidence of non-valvular atrial fibrillation in patients with type II diabetes increased by 40%, and the risk of thromboembolism associated with atrial fibrillation increased by 79% compared with patients with atrial fibrillation without diabetes. Moreover, the use of warfarin in these patients improved thromboembolism, but decreased the level of matrix Gla protein, which may promote the calcification of the coronary and renal arteries, thus increasing the risk of cardiovascular disease and accelerating the decline of renal function. It has been reported that warfarin may accelerate hypertension in high-risk patients, especially in those with diabetes or uncontrolled hypertension [30].
Atrial fibrillation (AF) is a common complication in dialysis patients. Lee et al. [31] investigated the relationship between warfarin and congestive heart failure and peripheral arterial occlusive disease in AF patients on hemodialysis. The results revealed that warfarin-induced VC increased the risk of congestive heart failure and peripheral arterial occlusive disease in AF patients. Yamagishi et al. [32] evaluated the clinical efficacy and safety of warfarin use in patients with diabetes mellitus complicated with AF. Changes in blood glucose levels of diabetic patients may affect the pharmacokinetics and anticoagulant activity of warfarin, therefore the risk-benefit balance of warfarin may easily become impaired in these patients. Additionally, due to the vitamin K-dependent gamma-glutamyl carboxylation of warfarin inhibitors (Gla protein), the use of warfarin may increase the risk of osteoporotic fracture and VC, which are the main reasons for diminished quality of life in patients with diabetes complicated with AF. Brimble et al. [33] explored the relationship between end-stage renal disease (ESRD), AF, and the use of anticoagulants to prevent ischemic stroke. The data suggested that warfarin may not only increase the risk of bleeding, but also promote VC in this patient population.
Increased VC is “one of the main underlying mechanisms for cardiovascular death in patients with chronic kidney disease (CKD) mediated by cardiovascular disease (CVD)” [34, 35]. Clinical data has shown that warfarin is related to renal VC and the deterioration of renal function [36, 37]. Warfarin leads to the calcification of small and medium-sized arteries in patients with renal transplantation. Hristova et al. [38] reported that treatment with warfarin accelerated VC in patients who underwent renal transplantation, and that this was mainly noted in small and medium-sized arteries. On the contrary, there was almost no calcification in the aorta. Interestingly, calcification mainly occurred in the intima, indicating that the response to warfarin is different between the intima and media, and between the different vascular beds. In contrast to highly calcified renal vessels, renal allografts were not calcified.
Warfarin is one of the main factors associated with VC in patients with CKD and
hemodialysis (HD) [39, 40]. Fusaro et al. [5] conducted epidemiological
studies evaluating 387 hemodialysis patients that were followed for three years,
to analyze the changes in mortality and the incidence of vertebral fracture and
VC. In a multivariate logistic regression analysis, it was found that the use of
warfarin was associated with an increased risk of aortic (OR 2.58, p
Experimental data suggest that long-term use of warfarin can lead to valvular
calcification. Levy et al. [48] observed the effects of vitamin K
deficiency on mineral and bone metabolism. In vitro studies have shown
that human aortic valve interstitial cells were calcified in the presence of high
phosphate and a vitamin K antagonist. Using a rat model of calcific aortic valve
disease (CAVD) induced by warfarin administration and vitamin K, Fang et
al. [49] explored the role of miR-29b and TGF-
Warfarin treatment has been shown to increase the volume of atherosclerotic
plaques [50]. Van Gorp et al. [51] found that short-term treatment with
warfarin promoted the formation of atherosclerotic plaques with a
pro-inflammatory phenotype. Additionally, they found that warfarin aggravated the
progression of plaque calcification and atherosclerotic disease. Long-term
treatment with warfarin significantly accelerated the calcification of
atherosclerotic plaques. Florea et al. [52] established a murine model
of atherosclerosis using ApoE
Warfarin can induce fetal chondrodysplasia punctata (CDP), which is the abnormal calcification of skeletal cartilage during fetal development. CDP is usually inherited, but maternal vitamin K deficiency also leads to this particular pathology. Since warfarin is an oral anticoagulant that acts on vitamin K-dependent coagulation factors, the use of warfarin in pregnant women may facilitate the development of CDP. Therefore, warfarin is considered to be one of the non-genetic etiological factors associated with developing CDP [53]. Songmen et al. [54] reported that a 27-year-old woman who had taken warfarin after artificial heart valve surgery produced a fetus that developed warfarin syndrome. The baby was born with a sunken nasal bridge and narrow nostrils. X-ray images showed spotted osteophytes of the vertebrae, femur, and humerus, which supported the diagnosis of fetal warfarin syndrome. Between 1991 and 2007, Wainwright et al. [55] performed autopsies on 13 fetuses with warfarin embryonic disease, with a gestation period of 17 to 37 weeks, and among these fetuses, 11 cases had nasal dysplasia.
These studies suggest that warfarin may induce calcification of small and medium-sized arteries, breast arteries, and fetal skeletal cartilage (Fig. 1), while also promoting VC in patients with chronic diseases. These considerations should be highlighted in clinical practice when initiating warfarin treatment (Fig. 1).
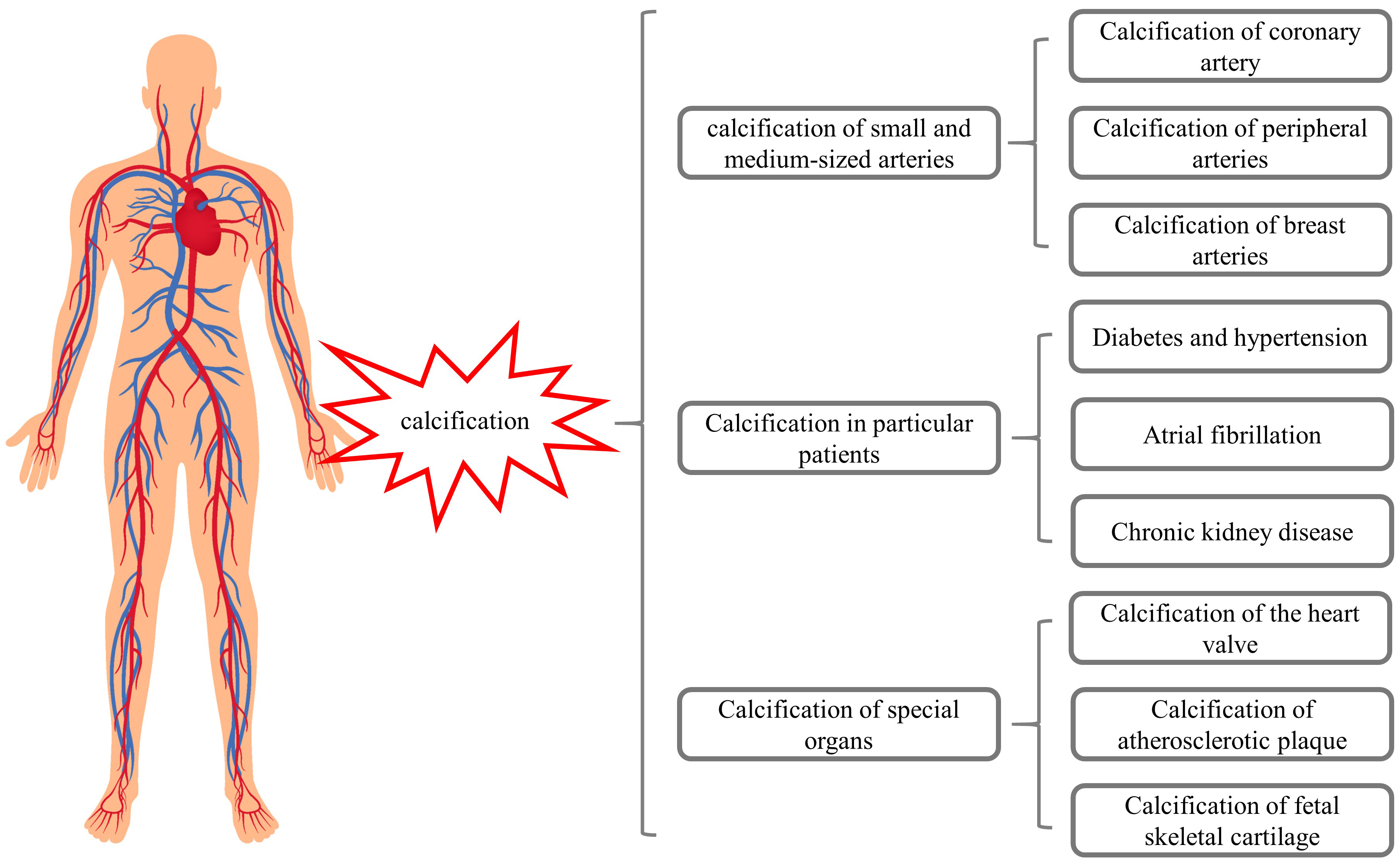 Fig. 1.
Fig. 1.Warfarin may induce calcifications of different organs.
Warfarin-induced calcification is mainly due to the decreased synthesis and
activity of matrix Gla protein (MGP). Gla is a vitamin K-dependent (VKD) amino
acid that binds to calcium. It is mainly formed by post-translational
modifications of glutamate induced by VKD
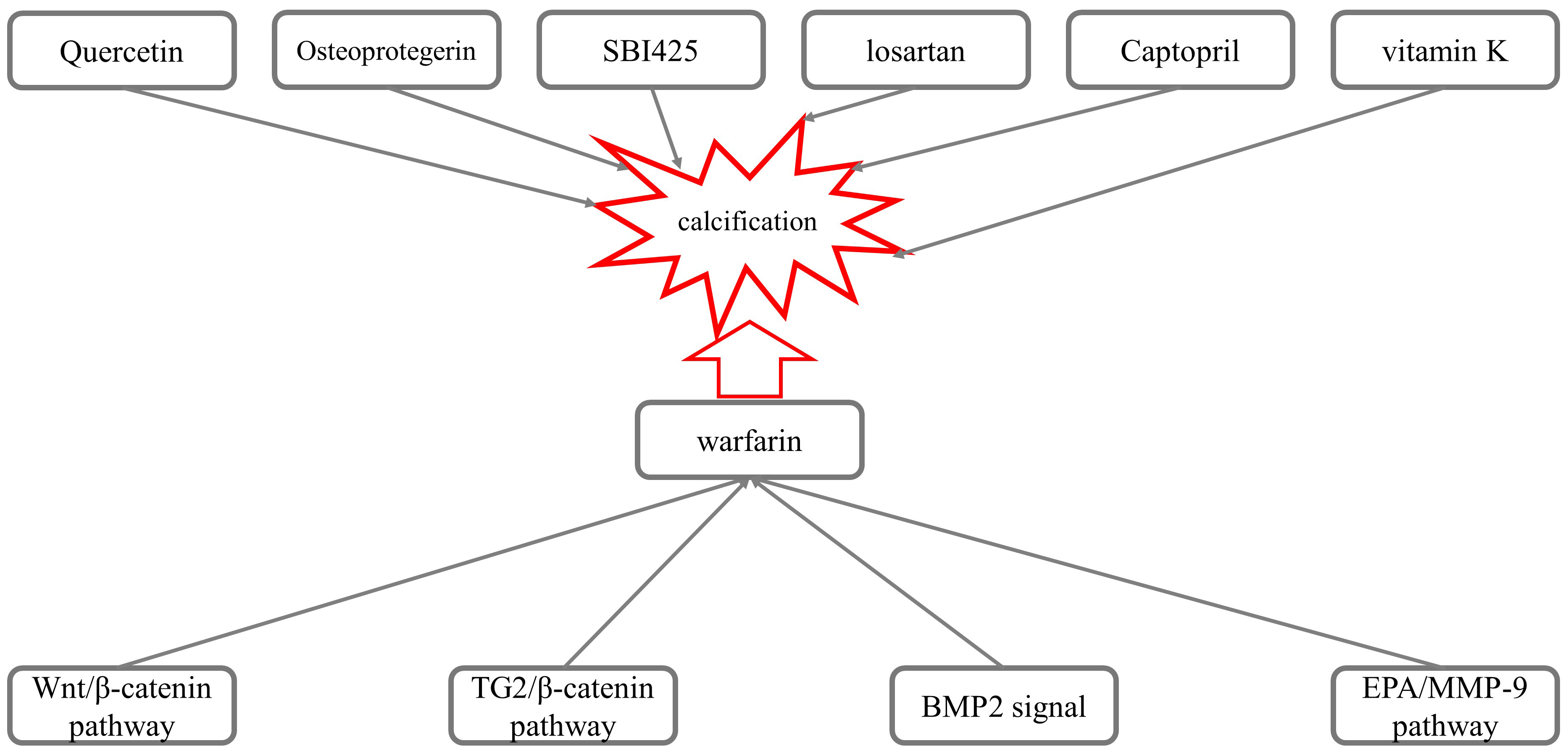 Fig. 2.
Fig. 2.Mechanisms underlying the calcification induced by warfarin.
Nie et al. [7] investigated the role of the Wnt/
Several experimental results have suggested that the Transglutaminase 2
(TG2)/
BMP2 also participates in warfarin-mediated VC. Li et al. [11] observed the effect of losartan on warfarin-induced VC in rats. Compared with the control group, administration of losartan (100 ng/kg/day) for two weeks inhibited the expression of mRNA and the BMP2 and Runx2 proteins, as well as reduced the apoptosis of VSMCs and calcification induced by warfarin, suggesting that losartan inhibited VC via inhibiting the expression of Runx2 and BMP2. The results presented by Yu Z et al. [12] showed that warfarin accelerated the calcification of human aortic valve interstitial cells (HAVIC) in patients with aortic stenosis (AS) through the PXR-BMP2-ALP pathway.
The use of eicosapentaenoic acid (EPA) reduced the arterial calcification
induced by warfarin in rats [13]. Sprague-Dawley rats were treated with warfarin
(3 mg/g in food) and vitamin K1 (1.5 mg/g in food) for two weeks to induce medial
arterial calcification, and then treated with EPA (1 g/kg/day).
Immunohistochemical and RT-PCR analysis showed that EPA decreased the expression
of osteopontin and osteogenic markers, such as alkaline phosphatase and core
binding factor-
Price et al. [66] revealed that osteoprotegerin effectively inhibited
arterial calcification induced by warfarin. Compared to rats treated with
warfarin alone, VC was significantly decreased in rats treated with both warfarin
and osteoprotegerin. Osteoprotegerin completely prevented CAC induced by warfarin
and reduced the levels of calcium and phosphate in the abdominal aorta
(p
Furmanik et al. [67] investigated the possibility that warfarin-induced aortic calcification and VC are primarily caused when endoplasmic reticulum stress increases the expression of Grp78 and ATF4 in rat aortas and VSMCs, increasing the release of extracellular vesicles through the PERK-ATF4 pathway and thus promoting VC. Opdebeeck et al. [68] found that administration of the tissue-nonspecific alkaline phosphatase (TNAP) inhibitor SBI-425 significantly reduced aortic and peripheral arterial calcification in a warfarin-induced calcification rat model. De Maré A et al. [69] used a diet containing warfarin to induce VC in rats to investigate the role of the bone formation inhibitor sclerosin (Sclerostin) in VC. The results showed that the severity of warfarin-induced VC was time-dependent, and the levels of serum sclerosin gradually increased.
Warfarin is widely used in patients who require long-term anticoagulation because of its effective anticoagulant properties, specific antagonism, and low cost. Presently, there is no ideal alternative drug [70, 71, 72]. More attention should be paid to keeping INR within a certain range to avoid bleeding or embolism when using warfarin. The dosage of warfarin is affected by many factors, such as sex, age, diet, and medication, and each patient should be dosed according to their individual needs and therapeutic goals [73]. However, the calcification induced by warfarin has not been highlighted by clinicians, as they prefer to focus on its anticoagulant effect. As such, there is no ideal strategy to prevent or treat warfarin-induced calcification. Therefore, reducing warfarin-induced calcification while ensuring the anticoagulant effect is an urgent clinical problem that needs to be resolved. It would be of great clinical significance to establish a scheme to reduce the calcification effects of warfarin.
OAC is a double-edged sword; on the one hand, warfarin exerts its anticoagulant
effect by antagonizing vitamin K, on the other hand, it also induces VC by
reducing the synthesis and activity of VKD MGP. Therefore, warfarin could not
achieve a balance between anticoagulation and VC reduction by interfering with
MGP. It has been confirmed that QU attenuates warfarin-induced VC via
Gla/
Early clinical diagnosis, early intervention, and multidisciplinary approaches act in the prevention and treatment of VC induced by warfarin. Emamy et al. [75] reported that the direct factor Xa inhibitor slightly reversed calcification in coronary arteries and heart valves, which was induced by warfarin and other vitamin K inhibitors. Yang et al. [76] cultured HAVIC from patients with warfarin-induced aortic valve stenosis in a high inorganic phosphate medium, and showed that menaquinone-4 accelerated warfarin-induced calcification in HAVIC.
Some studies reported that warfarin-treated patients with supratherapeutic INRs had a much higher risk of adverse renal outcomes [77, 78, 79]. Clinicians need to make a trade-off between warfarin and new anticoagulant drugs, such as apixaban, for patients with AF on HD. On the one hand, warfarin has some shortcomings, such as inducing VC; on the other hand, anticoagulants such as apixaban or rivaroxaban have disadvantages, such as short half-life, lack of effective antagonism, and high cost, which limits their usability. Coleman CI et al. [34] compared the effects of rivaroxaban and warfarin on renal failure in patients with non-valvular atrial fibrillation (NVAF). Compared with warfarin, rivaroxaban reduced the incidence of renal failure. Reilly et al. [80] believed that apixaban is an ideal drug to replace warfarin in the treatment of HD complicated with NVAF. However, although both apixaban and rivaroxaban show good pharmacokinetic characteristics in ESRD, due to the potential risk of dialysis drug accumulation and the lack of adequate understanding of their mechanism, Brancaccio et al. [81] believed that neither of these two drugs could be used safely in dialysis patients. Saito et al. [82] reported five HD patients, four with medial arterial calcium deposits and one with skin calcium deposits; four patients were treated with sodium thiosulfate and three patients with low calcium dialysate. The average follow-up period was 7.4 months; however, four patients were cured and one died of infection. These data suggested that multidisciplinary, early management, and strict detection of minerals and bone markers may improve the process of warfarin-induced VC.
Warfarin-induced calcification is gradually becoming a concern for clinicians and some have tried to use newly developed drugs [3]. Still, all of these drugs have cumulative effects or lack definite antagonists, so they are unable to totally replace warfarin. Therefore, reducing calcification while ensuring the anticoagulant effect of warfarin is an urgent clinical problem that needs to be solved. The appropriate use of warfarin will be important for reducing calcification.
In this review, we summarize the clinical phenomenon of warfarin-induced calcification, its possible molecular mechanisms, and the current prevention and treatment strategies. We hope this review can provide a theoretical reference for further improvement of warfarin anticoagulation therapy, promotion of the rational use of warfarin anticoagulation, and minimization of its calcification effects.
Conceptualization—JL; original draft preparation—XW, LP; review and editing— JM, JL; figure preparation—LZ; supervision—JL; funding acquisition—XW, JL. All authors have read and agreed to the published version of the manuscript.
All subjects gave their informed consent for inclusion before they participated in the study. The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of Xijing Hospital, Fourth Military Medical University (approval number: KY20192087).
Not applicable.
This study was supported by grants from the National Natural Science Foundation of China (82070264, 82070503), the Key R&D Program of Shaanxi Province (2022ZDLSF01-09).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.