

 , Nemanja Muric 6,7, Vladislava Stojic 8, Stefani Bolevich 9, Sergey Bolevich 10, Vladimir Jakovljevic 1,10
, Nemanja Muric 6,7, Vladislava Stojic 8, Stefani Bolevich 9, Sergey Bolevich 10, Vladimir Jakovljevic 1,101 Department of Physiology, Faculty of Medical Sciences, University of Kragujevac, 34000 Kragujevac, Serbia
2 Department of Pharmacology, Faculty of Medical Sciences, University of Kragujevac, 34000 Kragujevac, Serbia
3 Department of Internal Medicine, Faculty of Medical Sciences, University of Kragujevac, 34000 Kragujevac, Serbia
4 Clinic of Pulmology, University Clinical Center Kragujevac, 34000 Kragujevac, Serbia
5 Clinic of Cardiology, University Clinical Center Kragujevac, 34000 Kragujevac, Serbia
6 Department of Psychiatry, Faculty of Medical Sciences, University of Kragujevac, 34000 Kragujevac, Serbia
7 Clinic of Psychiatry, University Clinical Center Kragujevac, 34000 Kragujevac, Serbia
8 Department of Medical Statistics and Informatics, Faculty of Medical Sciences, University of Kragujevac, 34000 Kragujevac, Serbia
9 Department of Pathophysiology, 1st Moscow State Medical University IM Sechenov, 119991 Moscow, Russia
10 Department of Human Pathology, 1st Moscow State Medical University IM Sechenov, 119991 Moscow, Russia
Academic Editors: Filippos Triposkiadis and Davide Bolignano
Abstract
As the ultimate pathophysiological event, heart failure (HF) may arise from various cardiovascular (CV) conditions, including sustained pressure/volume overload of the left ventricle, myocardial infarction or ischemia, and cardiomyopathies. Sacubitril/valsartan (S/V; formerly termed as LCZ696), a first-in-class angiotensin receptor/neprilysin inhibitor, brought a significant shift in the management of HF with reduced ejection fraction by modulating both renin-angiotensin-aldosterone system (angiotensin II type I receptor blockage by valsartan) and natriuretic peptide system (neprilysin inhibition by sacubitril) pathways. Besides, the efficacy of S/V has been also investigated in the setting of other CV pathologies which are during their pathophysiological course and progression deeply interrelated with HF. However, its mechanism of action is not entirely clarified, suggesting other off-target benefits contributing to its cardioprotection. In this review article our goal was to highlight up-to-date clinical and experimental evidence on S/V cardioprotective effects, as well as most discussed molecular mechanisms achieved by this dual-acting compound. Although S/V was extensively investigated in HF patients, additional large studies are needed to elucidate its effects in the setting of other CV conditions. Furthermore, with its antiinflamatory potential, this agent should be investigated in animal models of inflammatory heart diseases, such as myocarditis, while it may possibly improve cardiac dysfunction as well as inflammatory response in this pathophysiological setting. Also, discovering other signalling pathways affected by S/V should be of particular interest for basic researches, while it can provide additional understanding of its cardioprotective mechanisms.
Keywords
- sacubitril/valsartan
- heart failure
- cardiovascular diseases
- cardioprotective mechanisms
Cardiovascular diseases (CVDs) remain global mortality contributors with nearly doubled prevalence in the last three decades [1]. As the ultimate pathophysiological event, heart failure (HF) may arise from various cardiovascular (CV) conditions, including sustained pressure/volume overload of the left ventricle (LV), myocardial infarction (MI) or ischemia, and cardiomyopathies [2]. The devastating prevalence of HF, which affects more than 64 million people worldwide [3], underscores the great importance of implementing novel therapeutic strategies in this population of patients.
According to the European Society of Cardiology (ESC), currently accepted HF
categorization, based on LV ejection fraction (EF) (LVEF), divides HF patients
into following subgroups: HF with reduced EF (HFrEF, EF
The knowledge of the natriuretic peptide system (NPS) paved the way for continuous research into therapeutic options for HF. The possibility of augmenting NPS by neprilysin (NEP) inhibition, and therefore amplifying the desired physiological actions of natriuretic peptides (NPs) has gained considerable scientific interest. Sacubitril/valsartan (S/V; formerly termed as LCZ696), a first-in-class angiotensin receptor/neprilysin inhibitor (ARNI), brought a significant shift in the management of patients with HFrEF by modulating both NPS (NEP inhibition by sacubitril) and RAAS (angiotensin II type I receptor blockage by valsartan) pathways. The pivotal evidence on its overwhelming benefits arrived in 2014 from a landmark, the PARADIGM-HF (Prospective Comparison of ARNI with angiotensin-converting enzyme inhibitor (ACEI) to Determine Impact on Global Mortality and Morbidity in Heart Failure) trial [8], which subsequently changed the recommendations for HFrEF therapeutic approach [5, 9].
It is worth mentioning that sodium-glucose co-transporter 2 inhibitors (SGLT2i) have recently also strengthened the therapeutic artillery for the management of HFrEF [4, 10]. Although initially developed as antidiabetics, these agents (empagliflozin and dapagliflozin) were proved effective in reducing CV death and HF hospitalization in HFrEF patients, irrespectively of their diabetes status [11, 12]. The landmark trial DAPA-HF (Dapagliflozin and Prevention of Adverse Outcomes in Heart Failure) demonstrated that in comparison to the placebo, dapagliflozin significantly reduced the incidence of CV death and HF worsening in HFrEF patients regardless of the presence of diabetes [11]. Similar results arrived from the EMPEROR-REDUCED (Empagliflozin Outcome Trial in Patients with Chronic Heart Failure and a Reduced Ejection Fraction) trial, where empagliflozin proved its superiority against placebo in HFrEF patients by reducing the relative risk of CV mortality and HF hospitalization, irrespectively of their diabetes status [12]. Furthermore, SGLT2i were equally effective in HFrEF patients with or without S/V treatment, suggesting that the use of both agents could achieve the most prominent cardioprotective effect in this population of patients [13, 14].
In recent years, the efficacy of S/V has been also investigated in the setting of other CV pathologies which are during their pathophysiological course and progression deeply interrelated with HF. However, its mechanism of action is still not entirely clarified, suggesting other off-target effects contributing to its cardioprotection.
Herein, we aimed to highlight up-to date clinical and experimental evidence on S/V cardioprotective effects, as well as the most discussed molecular mechanisms.
NPs are a family of structurally similar but genetically distinct bioactive
peptides involving atrial natriuretic peptide (ANP), B-type (or brain)
natriuretic peptide (BNP), and C-type natriuretic peptide (CNP) [15]. A surge of
scientific interest for NPs began in 1981 after Bold and colleagues [16] observed
increased diuresis (
Two known pathways can accomplish NPs breakdown: (1) NRP-C-mediated internalization and lysosomal degradation [28, 29], (2) enzymatic degradation performed by NEP, a zinc-dependent metalloprotease that is widely distributed in various tissues, such as endothelial and epithelial tissue, smooth muscle cells, cardiac myocytes, adipocytes, and pancreatic islets [30]. It is considered a principal enzyme for the degradation of numerous vasoactive peptides with different physiological roles (with vasodilatator or vasoconstrictive effects) in the CV system. NEP exhibits the greatest affinity for ANP, CNP, angiotensin I and II, while the lowest for BNP, endothelin-1, and bradykinin [23]. Unlike NRP-C, NEP metabolism has a minor contribution in NPs clearance under normal conditions, whereas, in pathological states with an increased level of circulating NPs, it is the dominant mode of NPs breakdown. On the contrary, NPR-C may become saturated [31].
Except in maintaining BP homeostasis, NPs also exhibit a diverse myriad of physiological effects, including antifibrotic, antihypertrophic, anti-inflammatory, and lusitropic effects, as well as sympathoinhibition and RAAS suppression [32, 33]. In most of their physiological aspects, NPs act antagonistically to RAAS. In HF, NPs also has a significant diagnostic role. In particular, BNP and its inactive terminal fragment, N-terminal pro b-type natriuretic peptide (NT-proBNP) plasma levels rise in response to increased ventricular stress and are considered a gold standard for HF diagnosis, prediction of its severity and prognosis [5, 9]. However, despite the significant elevation of NPs in congestive HF, its protective effects diminish as the disease progress, with a dominance of RAAS and SNS in disease deterioration [34]. In his recently published paper, Diez comprehensively described a variety of potential mechanisms considered to decrease the beneficial effects of NPs in the state of chronic HF [34]. However, we will not discuss it here, while it was not the focus of our present review.
Given the adopted knowledge on favourable actions of NPs, the goal of numerous studies that followed was to develop an appropriate therapeutic weapon that would enhance its effects and facilitate HF management.
An urgent need for manufacturing novel agents for HF treatment led to an explosion of investigations in recent years. Initial studies related to maximizing the NPs physiological actions aimed to examine the efficacy of exogenous NPs administration in HFrEF patients, however, the results were not promising. Nesiritide, a recombinant human BNP, beneficially influenced hemodynamics in the setting of acute decompensation of chronic HF [35], however it failed to reduce mortality and HF rehospitalization in comparison to placebo [36]. On the other hand, carperitide, a recombinant human ANP, is widely used in patients with acute HF in Japan [37], although no robust evidence proved its efficacy in improving clinical end-points [38]. On contrary, one recent cohort even reported that the use of carperitide was associated with worse outcomes in patients with acute HF than those assigned to nitrates [39]. Therefore, another strategy for endogenous NP reinforcement rushed into scientific focus concerning inhibition of NEP-mediated NPs’ breakdown.
In 1980, Roques and colleagues [40] reported the earliest preclinical findings on NEP inhibition effects. In healthy individuals, administration of NEP inhibitors as monotherapy improved natriuresis and diuresis and increase plasma ANP levels [41, 42]. However, despite an increase of ANP due to chronic use of candoxatril, an oral NEP inhibitor, sustained lowering of BP in hypertensive patients has not been achieved, so further research on its usage was discontinued [43]. As discussed earlier, there are various NEP substrates, including those with vasoconstrictive effects (angiotensin or endothelin). Therefore, suppression of its physiological actions not only increases the plasma levels of NPs, but vasoconstrictor levels too [6], leading to annulation of its preferred effects. Nevertheless, to overcome the shortcomings of lone NEP inhibition, manufacturing a dual-acting compound that would simultaneously activate NPS and inhibit RAAS seemed like a convenient solution.
After exceptional results from extensive clinical studies, CONSENSUS and SOLVD-Treatment trials [44, 45], ACEi have strengthened their place in the treatment of HFrEF for more than 30 years. Therefore, these agents were first tested in synergy with NEP inhibitors. However, in the OVERTURE trial, omapratilat, a first combined ACE and NEP inhibitor, was not superior over enalapril in reduction of the primary clinical event in patients with chronic HF [46]. Furthermore, this drug was associated with an unacceptable risk of angioedema [46], which was attributed to its dual mechanism of action; inhibiting both ACE and NEP, which are responsible for the breakdown of bradykinin, led to its excessive accumulation and consequent angioedema [47]. From this moment, the most promising drug class for combination with NEP inhibitor became ARBs.
A first-in-class ARNI is comprised of two molecular moieties in a 1:1 M ratio, sacubitril, which is rapidly metabolized after oral ingestion into sacubitrilat, NEP inhibitor, and valsartan, a well-known ARB with established efficacy in treating CVDs [48]. S/V primarily targets two neurohormonal pathways critical for the pathophysiology of HF and thus increase plasma cGMP concentration as a consequence of enhanced NPS in one side and plasma renin and angiotensin II mediated by ARB on the other [49, 50].
With increasing evidence on its protective effects in HFrEF [8], S/V has also been evaluated in patients with HFpEF [51], the most sensitive patient population regarding available treatment options. Even more, its effectiveness was also assessed in terms of other CV conditions, including MI, cardiac arrhythmias, and recently in cardiac dysfunction related to cancer therapy (Fig. 1). The summary of main clinical studies with S/V in different clinical settings is presented in Table 1 (Ref. [8, 52, 53, 54, 55, 56, 57]).
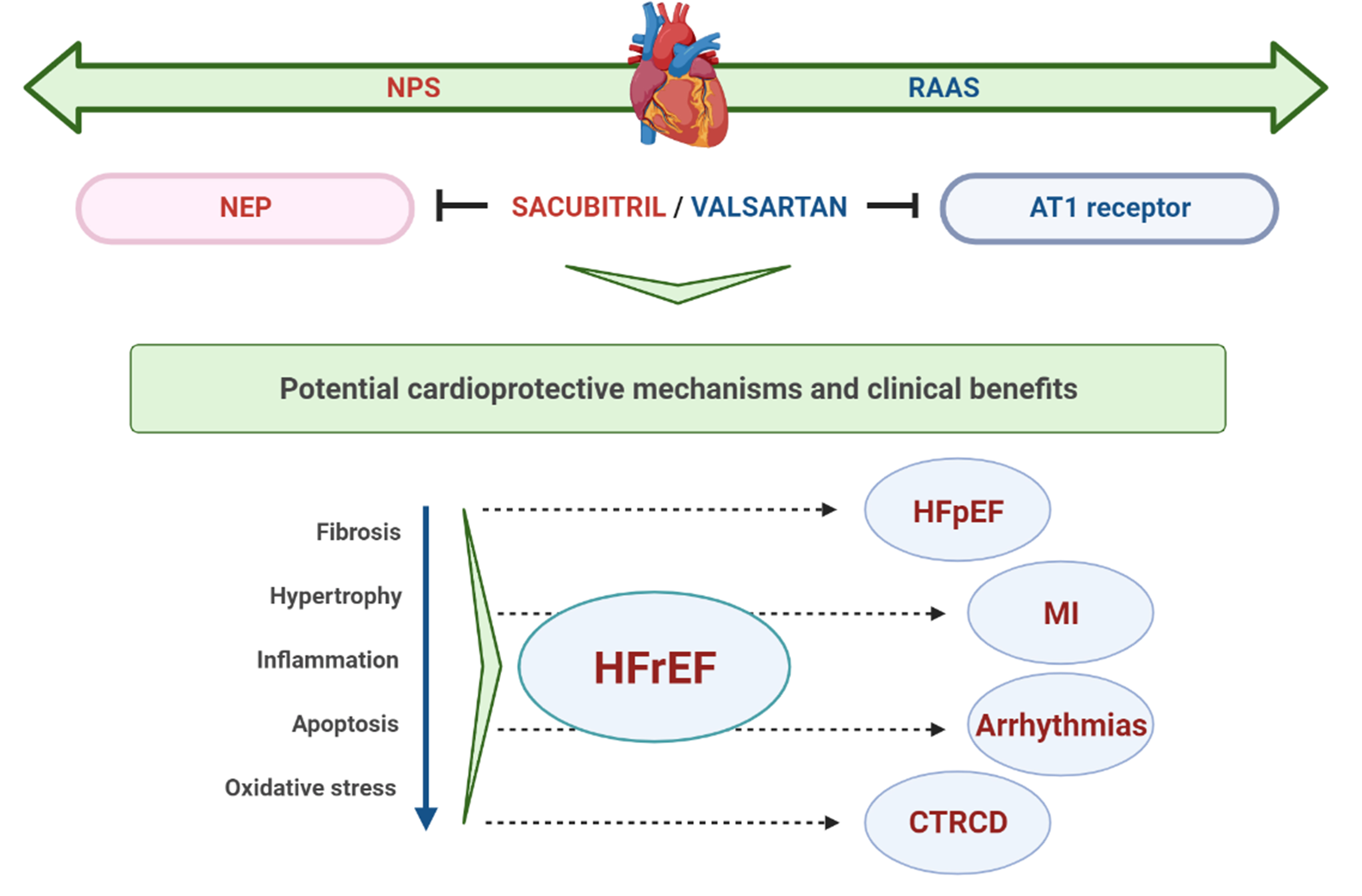 Fig. 1.
Fig. 1.Schematic representation of potential cardioprotective mechanisms and clinical benefits of S/V. NPS, natriuretic peptide system; RAAS, renin-angiotensin-aldosterone system; NEP, neprylisin; AT1, angiotensin II type I; HFrEF, heart failure with reduced ejection fraction; HFpEF, heart failure with preserved ejection fraction; MI, myocardial infarction; CTRCD, cancer therapy-related cardiac dysfunction.
| Trial, design | Study population (n), inclusion criteria | Intervention, median follow-up | Primary outcomes |
| PARADIGM-HF [8] multicenter, randomized, double-blind study | HFrEF patients (n = 8442) | S/V 200 mg BID or enalapril 10 mg BID | 20% reduction in composite of CV mortality and HF hospitalization with S/V |
| EF |
27 months | ||
| TRANSITION [52] multicenter, randomized, open-label, parallel-group study | HFrEF patients with ADHF (n = 1002) | S/V 200 mg BID | Around 50 % of HFrEF patients stabilized after ADHF achieved target dose of S/V |
| EF |
10 weeks | ||
| PIONEER-HF [53] multicenter, randomized, double-blind study | HFrEF patients with ADHF (n = 736) | S/V 200 mg BID or enalapril 10 mg BID | Significant reduction of NT-proBNP levels with S/V |
| EF |
2 months | ||
| PROVE-HF [54] multicenter, open-label, single-arm study | HFrEF patients (n = 794) | S/V 200 mg BID | Decrease in NT-proBNP levels correlated with improved echocardiographic markers of cardiac volume and function |
| EF |
12 months | ||
| PARAGON-HF [55] multicenter, randomized, double-blind, parallel group study | HFpEF (n = 4822) | S/V 200 mg BID or valsartan 160 mg BID | No significant difference in composite endpoint of total HF hospitalization and CV mortality between S/V versus valsartan |
| EF |
35 months | ||
| PARALLAX [56] multicenter, randomized, double-blind study | HFpEF (n = 2572) | S/V 200 mg BID or background medication (enalapril, valsartan, or placebo) | Significant reduction of NT-proBNP levels at 12 weeks, but no significant difference in 6MWT at 24 weeks |
| EF |
6 months | ||
| PARADISE-MI [57] multicenter, randomized, double-blind study, parallel-group study | post-MI (n = 4650) | S/V 200 mg BID or ramipril 5 mg BID | No significant difference in composite of CV mortality or incident HF between S/V and ramipril |
| EF |
23 months | ||
| Abbreviations: BID, twice a day; ADHF, acute decompensated heart failure. | |||
The landmark PARADIGM-HF trial was the largest HF trial ever, enrolling more
than 8 thousand ambulatory patients with HFrEF (New York Heart Association (NYHA)
class II–IV, EF
Since the PARADIGM-HF trial has been released, numerous sub-analyses have been performed concerning various aspects of S/V treatment regarding ACEi. While HF may occur due to various etiologies (ischemic or non-ischemic), S/V seems to be equally effective in diverse HFrEF population enrolled in PARADIGM-HF, irrespectively of specific pathophysiological state responsible for its development [59]. Furthermore, S/V is proved to be superior over enalapril in preventing the HFrEF clinical progression in surviving patients [60]. Moreover, in comparison to enalapril, an early benefit of S/V was also observed in regard to reduction of 30-day readmissions due to any cause and HF (by 26% and 38%, respectively) after discharge from HF hospitalization [61]. One observational study reported that S/V improved exercise capacity assessed by a 6-minute walk test (6MWT) in stable, symptomatic HFrEF patients [62]. Furthermore, in the open-label, PARASAIL study, HF patients (majority with NYHA II class) treated with S/V had improved mean 6-minute walking distance at 6-month follow-up, as well the quality of life [63]. However, there is limited data on S/V effects in patients with advanced HFrEF (NYHA class IV), while the PARADIGM-HF trial involved around 1% of patients in this NYHA category at baseline [8]. However, not so promising came the recent results from double-blind, randomized LIFE trial which demonstrated that in comparison to valsartan, S/V was not superior in reducing NT-proBNP levels nor in improving clinical outcomes (number of days alive, out of hospital, and free from HF events) in comorbid HFrEF patients (NYHA class IV) [64]. On the other hand, early initiation of S/V seems feasible in patients HFrEF confronted with recent acute decompensation either in a hospital setting or early after discharge, as reported in the TRANSITION and PIONEER-HF studies [52, 53]. The ESC Expert Consensus 2019 guidelines and the 2019 ACC Expert Consensus Decision Pathway for Hospitalized Patients reported that this agent might be considered in hospitalized patients with new-onset HFrEF or decompensation of chronic HF [65, 66].
As a common feature in HFrEF, cardiac dilatation is a predictor of poor outcome, while its reversal improves patients’ prognosis [67]. Thus, reverse cardiac remodeling is a significant marker of effective treatment and a predictor of better prognosis for HFrEF patients [68]. In the PARADIGM-HF trial, the risk of CV death or HF hospitalization increased by 10% for each 5% drop in LVEF, suggesting LVEF as an important outcome predictor [69]. One retrospective cohort by Almufleh and colleagues [70], reported that S/V improved LVEF (an increase from 25% to 33%) and reversed cardiac remodeling after 3-months of follow up compared to standard treatment. Similarly, Bayard and colleagues [71] observed that 3-month treatment with S/V significantly improved several echocardiographic parameters in patients with HFrEF, including LVEF (+3.6% in absolute value). Interestingly, more significant LV improvement has been observed in those with lower LV dilatation at baseline, suggesting that S/V could provide more prominent effects on LV when administered at the early HFrEF stage [71]. Most recently, a study by Paolini and colleagues [72] reported that during a 2-year follow-up, S/V treatment led to reverse cardiac remodeling in more than half of enrolled HFrEF patients, which usually occurred within 12 months after drug initiation. The authors also suggested that early ARNI implementation could prevent or postpone the ICD implantation in these patients.
The absence of a decrease in NT-proBNP levels in patients treated for HF is thought to be associated with poorer LV function and size [73]. In the PROVE-HF (Prospective Study of Biomarkers, Symptom Improvement and Ventricular Remodeling During Entresto Therapy for Heart Failure Study) trial, a decrease in circulating levels of NT-proBNP correlated with improved markers of cardiac function and volume in patients with HFrEF treated with S/V, suggesting reverse cardiac remodelling as its possible mechanism of benefit [54].
Among adverse events observed in PARADIGM-HF, only hypotension was significantly more common in patients treated with S/V than in those assigned to enalapril [8]. However, more patients who discontinued their study medication due to adverse events were receiving enalapril, while no difference in rates of hypotension-related therapy discontinuation was observed between these two groups. As for the other adverse events observed in PARADIGM-HF, mild angioedema was more common in patients treated with S/V, but without significant difference regarding those receiving enalapril. Interestingly, fewer patients treated with S/V experienced cough, elevated serum potassium, and elevated serum creatinine levels in comparison to those in the enalapril group [8].
In the PARADIGM-HF trial, the target dose of S/V was 97/103 mg twice a day, while the target dose of enalapril was 10 mg twice a day. However, almost half of the patients in both groups required a dose reduction, which was associated with a higher risk of major CV events in comparison to those who maintained on target doses [74]. Although, patients intolerant to maximal S/V doses, who were therefore prescribed with lower S/V doses still had more benefit than those receiving lower enalapril doses [74]. Nevertheless, there is still a large discrepancy between clinical trials and real-world studies in terms of initiation, titration, and adherence to S/V treatment. One study reported slow adoption of S/V in the real setting in the first 18 months after its FDA approval for HFrEF management, suggesting high cost as an important limiting factor for drug initiation and continuation [75]. The results from the CHAMP-HF Registry pointed out that 27% of HFrEF patients were not prescribed with an adequate guideline-directed treatment (ACEi/ARB or S/V) regardless of the absence of contraindications [76]. Furthermore, only 13% of HFrEF patients were using S/V, while only 14% were on maximal target doses [76]. Results from a large retrospective cohort conducted in Germany showed that two-thirds of patients treated with S/V were initially prescribed with the lowest dose, while up-titration was attempted in less than 50% of patients during the following 6 months [77]. The authors pointed out that it is necessary to identify the barriers responsible for missing S/V up-titration, as well as the importance of raising awareness among physicians on this matter [77]. The real-world data from Taiwan also showed that HFrEF patients were prescribed with lower doses than those included in the PARADIGM-HF trial [78]. There are various reasons for S/V underutilization, such as high cost, clinical factors (including older age, low BP, renal impairment, etc.), or even physician-related therapeutic inertia or fear from side effects [79]. Although clinical studies should not be a strict instruction for the use of certain drugs, but rather a guide to an individualized approach to each patient [80], raising awareness about timely implementation and adequate dose titration of guideline-directed medical treatment should be of particular interest, considering the observed benefits. In the future, randomized controlled trials are needed to determine the optimal dose of S/V [80].
Another important fact that needs to be addressed is that PARADIGM-HF trial had two phases of run-in period (enalapril active run-in period and LCZ696 active run-in period) with different duration of time, in whom the patients who tolerated both medications were randomized into the trial [81], which could suggest that individuals particularly susceptible to the hypotension were excluded from the study before randomization [82]. This part of the study design is of great importance, while run-in periods limit the benefits to a specific group of patients who are hard to be recognized in the clinical practice [83]. Furthermore, in PARADIGM-HF, patients assigned to S/V received valsartan in maximal dose in comparison to those on half-maximal enalapril dose, which can also be a reason for debate the origin of observed effects [83].
It seems that HFrEF patients would have particular benefit from both S/V and an SGLT2i [79], since these drug classes have a different mechanisms of action in patients with HF and their cardioprotective benefits are independent of each other [84]. Cardioprotection observed with SGLT2i is suggested to be regardless of neuro-hormonal antagonism [84] and it is most probably achieved with natriuretic/diuretic effect of these agents, but also other systemic, hemodynamic, and direct cardiac effects seem to have important involvement [85].
According to the latest ESC guidelines, in the absence of contraindications or intolerance, a cornerstone treatment for HFrEF patients includes ACEi/ARNI, BB, and MRA, up-titrated if possible to the target doses in clinical trials or maximally tolerated doses. In addition to optimal pharmacological treatment, SGLT2i (empagliflozin or dapagliflozin) are also recommended to HFrEF patients to reduce the risk of CV death and HF mortality, irrespectively from the presence of diabetes [4]. The present evidence suggests that HFrEF patients would have the most benefit from early initiation of the 4-drug treatment strategy (including ARNI, BB, MRA, and SGLT2i) in regard to death, HF hospitalization, and symptoms reduction [86]. A recent ACC consensus reported that the first-line therapy in patients with new-onset of symptomatic HFrEF should include ACEi/ARB/ARNI and BB, which need to be timely up-titrated to target or maximally tolerated doses [10]. In addition, adding an MRA and/or SGLT2i should be performed carefully in regard to estimated glomerular filtration rate and plasma potassium levels [10]. Furthermore, Packer and McMurray proposed a novel three-step strategy for the initiation of 4 drugs in euvolemic HFrEF patients during 4 weeks [87]. The first step would include concomitant initiation of BB and SGLT2i, the second step involves the initiation of ARNI, and the third step the initiation of MRA, with the possibility of an individualized approach if necessary [87]. The authors stated that this kind of therapeutic strategy would speed up the initiation of all 4 medications and improve their efficacy and tolerability in HFrEF patients [87].
In addition, another novelty in HFrEF treatment is FDA approval of vericiguat, an oral soluble guanylyl cyclase activator, for reducing the risk of CV mortality and HF hospitalization in patients with HFrEF worsening [88]. One network meta-analysis involving major trials with S/V, SGLT2i, and vericiguat, reported that in HFrEF patients these three drug classes had similar effects on outcomes, with only dapagliflozin being superior against vericiguat in HF hospitalization risk [89]. Interestingly, both S/V and vericiguat share a similar signaling pathway, cGMP-PKG pathway, and therefore may induce more prominent hypotensive effect which could protect from resistant hypertension (HTN) and HF development [90]. However, future investigations need to provide more detailed research regarding interaction between these two agents.
Many patients with HF suffer from HFpEF, which accounts for 22–73% of total HF cases, depending on the used definition [5]. It is associated with similarly poor survival rates as HFrEF [91]. HFpEF is more frequent in women and the elderly, with HTN, DM, and obesity being among common risk factors in this population of patients [92]. Despite the armamentarium of therapeutics proven effective for treating HFrEF, previous studies on HFpEF haven’t succeeded to develop an agent powerful enough to reduce morbidity and mortality in these patients [93].
The PARAMOUNT-HF trial, a phase II trial designed to assess the effects of ARNI against ARB in HFpEF patients, revealed promising results. In 301 HFpEF patients (NYHA II–IV) enrolled in this study, S/V was superior to valsartan in reducing the NT-proBNP levels at 12-weeks, and in lowering left atrial size and improving NYHA class at 36-weeks of treatment [51]. Moreover, BP reductions were also more significant at both 12 and 36-weeks of treatment in a group of patients receiving S/V compared to those assigned to valsartan [51]. A further sub-analysis of the PARAMOUNT-HF trial demonstrated a decrease in high sensitive Troponin I (hsTnI) levels in HFpEF patients treated with ARNI in regard to ARB, suggesting that S/V has the potential to ameliorate myocardial injury in HFpEF patients [94]. These long-awaited results have shed the light on HFpEF management and encouraged further research on S/V in this population of patients.
The PARAGON-HF trial, which enrolled more than 4 thousand patients with HFpEF
(EF
There could be several explanations for different outcomes of S/V treatment
efficacy in HFrEF and HFpEF patients. Large inconsistency in therapeutic response
between HFrEF and HFpEF may actually originate from different pathophysiological
drivers in these two entities or even in phenotypic diversity across the HFpEF
spectrum [96]. Furthermore, the active comparator to S/V was enalapril in
PARADIGM-HF, while in PARAGON-HF it was valsartan, which could have a potential
influence on observed outcomes [8, 55]. In their study, Solomon and colleagues
[97] performed a pooled analysis of combined data from both trials in order to
access the effectiveness of S/V across the EF spectrum. The authors pointed out
that the efficacy of S/V varies by EF values, with the most prominent benefits
seen in patients with EF below normal (mid-range or borderline EF). These
observations could be due to later therapy initiation in patients with higher EF
or prevalence of cardiac stiffness in those with EF
Cardiac fibrosis is undoubtedly recognized as one of the significant pathophysiological drivers in HFpEF, which occurs independently of its etiology [98]. Therefore its targeting could probably ensure significant cardioprotection. In a study by Zile and colleagues [99], S/V decreased profibrotic biomarkers in patients with HFrEF enrolled in PARADIGM-HF. Moreover, Cunningham and colleagues [100] provided further valuable evidence on antifibrotic features of S/V in the setting of HFpEF. As it was shown, HFpEF patients enrolled in the PARAGON-HF trial had increased biomarkers of extracellular matrix (ECM) dysregulation, which were associated with the risk of further HF events in this population. At the same time, S/V favorably affected these biomarkers revealing its antifibrotic potential [100].
In addition, there is an ongoing, randomized, double-blinded PARAGLIDE study
investigating the effects of S/V versus valsartan alone on NT-proBNP values, as
well as clinical outcomes, safety, and tolerability in HFpEF patients (EF
MI remains the most common cause of HF, which may develop due to diverse pathophysiological mechanisms depending on the time of its occurrence [102]. Although several studies confirmed lower rate of HF in post-MI patients with implementation of primary percutaneous coronary interventions, only a few had a longer follow-up period [102]. After demonstrating the beneficial effects of S/V in a diverse population of patients with HFrEF, further investigations were dedicated to determining its impact in patients following acute MI (AMI). Moreover, in the RECOVER-LV trial, which involved 93 patients with LV systolic dysfunction late after MI, S/V has shown no superiority over valsartan regarding reverse cardiac remodeling effects [103]. On the other hand, the data from the most recent meta-analysis involving four studies pointed to the beneficial effects of early S/V treatment after acute MI reflected through improved LVEF and reduced MACE incidence in comparison to ACEi [104]. However, S/V showed no superiority in reducing the incidence of cardiac death, HF hospitalization, MI or adverse side effects [104]. The SAVE-STEMI trial compared the efficacy of S/V against ramipril in patients with ST-segment elevation MI [105]. While there was no significant difference between these two treatment strategies after 1 month, treatment with S/V for 6 months prove to be more effective in decreasing MACE, as well as in improving EF and LV remodeling in these patients [105]. Most recently, in a study by Chen and colleagues [106], combined treatment with S/V and bisoprolol seem to be more effective in improving cardiac function and lowering the rate of adverse events during cardiac rehabilitation of patients with AMI and left-sided HF after PCI in comparison to bisoprolol monotherapy.
Finally, the most recent evidence emerged from the PARADISE-MI, the first large
trial which compared the efficacy of ARNI versus ACEi in post-MI patients with LV
dysfunction (EF
Sudden cardiac death (SCD) represents the major cause of death in HF patients [107]. As the most robust parameter related to SCD, reduced LVEF is considered as an indication for implantable cardioverter-defibrillator (ICD) implantation for primary prevention of SCD [108]. Importantly, HFrEF patients assigned to S/V lived up to 2 years longer with less possibility to die from SCD or HF worsening than those prescribed with enalapril [109, 110]. The exact mechanism responsible for reduced SCD in HFrEF patients treated with S/V is not entirely clarified, while there are conflicting results in the present literature regarding its antiarrhythmic potential [108, 111, 112, 113, 114, 115].
In a study by Vincent and colleagues [112], which involved 108 of patients prescribed with S/V six patients presented with ventricular arrhythmic storm early after S/V initiation, which required drug discontinuation. Paradoxically, one observational study involving 167 patients with dilated cardiomyopathy (ischemic/non-ischemic etiology) and dual-chamber ICD reported that treatment with S/V reduced the incidence of atrial and ventricular arrhythmias and improved ICD electrical atrial parameters during 12-month follow up [116]. Similarly, de Diego and colleagues [117] reported a lower rate of non-sustained ventricular tachycardia and premature ventricular contraction (PVC) in HFrEF patients treated with S/V than those prescribed with ACE/ARB. Furthermore, a correlation between plasma NT-proBNP levels and hourly PVC rate was observed and decreased with S/V treatment [117]. In a study by Martens and colleagues [118], HFrEF patients with implanted ICD or cardiac resynchronization therapy (CRT) receiving S/V had a lower rate of SCD, which could be at least partially driven by the reduced onset of ventricular tachyarrhythmia’s. Reverse cardiac remodelling observed during treatment with S/V treatment could be one of the contributing mechanisms responsible for reduced risk of arrhythmias.
One retrospective cohort reported increased peak atrial longitudinal strain (PALS) in patients with HF (NYHA II or II–III) with a history of atrial fibrillation (AF) prescribed with S/V [119], as well as reduced rate of AF episodes in comparison to standard therapies, during 12-months follow up. While PALS is considered a marker of reservoir function of atrial chambers, its augmentation under S/V treatment emphasize the beneficial effects of this agent on atrial filling and thus more pronounced LV ejection during systole [119]. Furthermore, same group of authors reported that at ventricular level S/V increases both LVEF and global longitudinal strain in HFrEF patients [120], with possible ability for reverse remodeling of atrias and regulation of heart rhythm [121]. Beneficial effects of S/V in the setting of AF may be explained by its favourable influence on electroanatomic atrial remodelling, and it should be one particular field of examination in future studies.
In previous years, significant progress has been made in the treatment of
patients with malignant diseases. However, along with improved survival in these
patients, prolonged exposure to cancer therapies led to an increased risk of
their adverse effects [122]. Cancer therapy-related cardiac dysfunction (CTRCD)
is considered in case of reduced EF of more than 10% from baseline to an EF
Although the PARADIGM-HF trial demonstrated beneficial effects of S/V in patients with HFrEF of diverse etiology, the evidence on its efficacy and safety in patients with CTRCD is lacking. Initially, potential cardioprotective effects of S/V were reported in a few case reports and case-series studies involving patients with malignant diseases [126, 127, 128, 129]. Most recently, in a study by Gregorietti and colleagues [130], authors reported potential benefit from S/V in the population of patients with breast cancer and cardiac dysfunction, reflected through improved LVEF, LV diameters, and diastolic dysfunction, as well as symptoms and 6MWT parameters. Furthermore, one multicentric retrospective study involving 67 CTRCD patients treated with S/V revealed promising results [131]. The use of S/V was associated with reverse cardiac remodelling (improved LVEF and LV volumes) along with improved exercise tolerance and reductions in NT-proBNP levels [131]. Moreover, tolerability of S/V was reported as good, with only a few patients (6%) experiencing an adverse event [131]. Similarly, significant improvement in LV volumes and LVEF assessed by cardiac magnetic resonance, along with reductions in NT-pro-BNP levels were observed in CTRCD patients treated with S/V [132]. Although these studies involved small number of patients, promising observations should encourage further investigations to assess more robust conclusions on the use of S/V in cancer survivors with CTRCD.
Multiple preclinical studies showed the protective effects of S/V on cardiac function in the setting of various CV pathologies. It is well established that NEP inhibition may influence the circulating levels of peptides other than NPs, which may additionally contribute to favourable effects of S/V in the setting of different CV pathologies. Therefore, various animal studies tried to unravel mechanistic aspects of S/V contributing to its cardioprotection (Table 2, Ref. [133, 134, 135, 136, 137, 138, 139, 140, 141, 142, 143, 144, 145, 146, 147, 148, 149, 150, 151, 152, 153, 154]). In the next section, we summarized the current preclinical knowledge on potential mechanisms of S/V in CVD modelling.
| Animal model | Dose/duration | Main molecular mechanism(s) and effects | Cardiac and hemodynamic effects | Ref. |
| post-MI (LAD ligation) in SD rats | 68 mg/kg daily, 4 weeks | Reduction of fibrosis rate in peri-infarct and remote myocardium | Reduction of HW, hypertrophy and fibrosis, LV remodeling, LVEDd; improvement of LV function and EF | [133] |
| MHD in C57BL/6J mice | 100 mg/kg daily, 16 weeks | Reduction of fibrosis, hypertrophy, and collagen production in heart; reduction of myocardial oxidative stress | Reduction of total wall thickness, LV mass, LVEDP, E/Em; improvement of E/A ratio, Em, RPP | [134] |
| ISO-exposed Wistar rats | 60 mg/kg daily, 1 week | Reduction of cardiac interstitial fibrosis and expression of TGF- |
Reduction of serum NT-proBNP, SBP; attenuation of the LVEDP and Dp/dt max increase | [135] |
| MI (LAD-ligation) in SD rats | 68 mg/kg daily, 4 weeks | Inhibition of myocardial fibroblast proliferation and collagen synthesis through downregulation of TGF- |
Reduction of LVEDd, LVEDs, IVSd, LVPWd; increase of EF, FS | [136] |
| HFrEF in diabetic C57BL/6J mice | 60 mg/kg daily, 4 weeks | Reduction of LV fibrosis; decreased expression of TGF- |
Reduction of serum NT-proBNP, HW/BW; improvement of EF, SV, CO, SW | [137] |
| HFpEF in Dahl/SS rats | 68 mg/kg daily, 4 weeks | Inhibition of cardiac fibrosis by suppressing the TGF- |
Reduction of serum NT-proBNP, SBP, LV/BW, (Wet lung-Dry lung)/BW, LA/BW, IVSd, LVPWd, LA; correction of LV mass, E/A and E/E′; improvement of EF, FS, LV DD | [138] |
| DOX-exposed Wistar rats | 68 mg/kg daily, 4–6 weeks | Altered extracellular matrix remodeling secondary to a reduction in myocardial MMP activity | Preservation of EF and FS | [139] |
| HFpEF in ZSF1 obese rats | 60 mg/kg daily, 12 weeks | Reduction of perivascular fibrosis, decrease of Collagen I and III, ANP and BNP expressions, decrease of MMP-2 activity, increase of cGMP levels and phosphor-titin levels | Reduction of serum NT-proBNP, HW, LVESP, LVEDP, and LV stiffness, MAP in aorta, RV volume capacity; improvement of DD, EF and endothelial-dependent vasodilation in carotid arteries | [140] |
| Debanding surgery in C57BL/6 J mice with aortic banding | 60 mg/kg daily, 4 weeks | Inhibition of NF-κB-mediated NLRP3 inflammasome activation | Reduction of HW/BW, LV mass, LVESd; improvement of EF, FS | [141] |
| MI (LAD ligation) in C57BL/6J mice | 20 mg/kg daily, 4 weeks | Suppression of pro-inflammatory cytokines and ECM degradation by macrophages | Reduction of rate of death due to LV rupture, LVEDd, LVESd, plasma aldosterone, aldosterone/cGMP ratio; increase of survival rate, FS, plasma cGMP levels | [142] |
| TAC in C57Bl6/J mice | 57 mg/kg twice daily, 4 weeks | Inhibition of Rho signaling via stabilization of ANF-induced PKG signaling | Reduction of SBP, LV mass, systolic and diastolic internal dimensions and volumes; improvement of EF, FS; preservation of E/E’ values | [143] |
| Post MI (LAD ligation) in SD rats | 60 mg/kg daily, 4 weeks | Reduction of cardiomyocyte hypertrophy, cardiac fibrosis and collagen I expression in the non-infarct and border zone, expressions of ANP, |
Reduction of HW, LV weight, LA weight, lung weight, LV filling pressures, LVESV, LVEDP, LVPW thickness, LV diastolic wall strain, LV compliance; improvement of EF, FS, ESPV relationship; preservation of dP/dt max and dP/dt min normalized to LVEDV | [144] |
| TAC in C57BL/6 mice | 60 mg/kg daily, 4 weeks | Inhibition of inflammatory response in blood and heart tissues, reduction of cardiac fibrosis and hypertrophy, improvement of ventricular remodeling | Reduction of LVEDs, LVEDd, LVPWs, LVPWd, IVSs, IVSd, LV mass; improvement of EF, FS | [145] |
| Post-TAC in C57BL/6 mice | 60 mg/kg daily, 4 weeks | Reduction of cardiac fibrosis and preservation of cardiomyocyte morphology | Reduction of LA, EF, IVSd, LVPWd, LVEDd, LVESd | [146] |
| AF rabbit model | 10 mg/kg twice daily, 3 weeks | Attenuation of atrial electrical and structural remodeling probably via calcineurin/NFAT pathway, preservation of cardiomyocyte morphology | Reduction of serum NT-proBNP, AF incidence; preservation of rapid pacing-induced atria and RV enlargement | [147] |
| diabetic CMP in C57BL/6 mice | 60 mg/kg daily 16 weeks | Inhibition the HG- or diabetes-induced JNK/p38MAPK phosphorylation and NF-kB nuclear translocation; decrease of apoptosis, oxidative stress, fibrosis, collagen I, and collagen III levels | Reduction of serum NT-proBNP; improvement of LV contractility and diastolic function | [148] |
| MI (LAD ligation) in SD rats | 68 mg/kg daily, 1 week | Inhibition of TAK1/JNK signalling cascade; reduction of interstitial fibrosis, collagen volume fraction, serum levels of inflammatory factors (IL‑1 |
Reduction of myocardial injury and improved ventricular remodeling | [149] |
| EAM in BALB/c mice | 20 mg/kg daily, 2 weeks | Inhibition of Th17 cell differentiation (independent from the NLRP3 inflammasome pathway); Reduction of inflammatory markers | Reduction of HW/BW, pathological scores of heart sections and cTnT | [150] |
| MCT-induced PH and Hypoxia-induced PH in SD rats | 68 mg/kg daily, 2 weeks | Increase of ANP and CNP; Restoration of the down-regulated NPRs protein expression, preservation of cGMP content of lung tissues; decrease of IL-1 |
Reduction of MAP, mPAP, PVR, RV weight to LV + S weight ratio, pulmonary artery wall thickness, fully muscularization of pulmonary arterioles and improved non-muscular arterioles | [151] |
| DOX-induced dilatative CMP in Balb/c mice | 60 mg/kg daily, 4 weeks | Preservation of mitochondrial function via reduced activity of fission protein Drp1; reduction of myocardial hypertrophy, fibrosis and cell size, apoptosis and cardiomyocyte contractile dysfunction | Reduction of serum NT-proBNP, HW/BW; improvement in HW/TL, EF, LVEDd, LVESd | [152] |
| TAC-induced pressure overload HF In C57BL/6 | 20 mg/kg daily, 4 weeks | Anti-hypertrophic effect by ameliorating oxidative stress via the Sirt3/MnSOD pathway; reduction of cardiac hypertrophy, fibrosis, ANP, BNP, |
Reduction of HW/BW, LW/BW, HW/TL; improvement of EF, FS and hypertrophy contractile dysfunction | [153] |
| HF by I/R injury (LAD ligation) in SHR | 68 mg/kg daily, 4 or 6 weeks | Reduction of extension of infarct border zone, collagen volume fraction, collagen I and collagen III expressions, TIMP2, TGF- |
Reduction of serum NT-proBNP, LVEDd, LVESd, LVEDP; improvement of EF | [154] |
| Abbreviations: AF, atrial fibrillation; ANP, atrial natriuretic peptide; BNP,
brain natriuretic peptide; BW, body weight; Ccl2, C motif chemokine ligand 2;
cGMP, cyclic guanosine monophosphate; CMP, cardiomyopathy; CNP, C-type
natriuretic peptide; CO, cardiac output; Col1a1, collagen type 1 alpha 1; cTnT,
cardiac Troponin T; DBP, diastolic blood pressure; DD, diastolic dysfunction;
DOX, doxorubicin; EAM, experimental autoimmune myocarditis; ECM, extracellular
matrix; EF, ejection fraction; ESPV, end-systolic pressure volume relationship;
FS, fractional shortening; HFpEF, heart failure with preserved ejection fraction;
HFrEF, heart failure with reduced ejection fraction; HW, heart weight; HW/TL,
heart weight/tibial length; IL, interleukin; ISO, isoproterenol; IVSd,
interventricular septum thickness at the end of diastole; IVST, intraventricular
septum thickness; LA, left atrial internal dimensions; LAD, left anterior
descending artery; LV, left ventricle; LVEDd, left ventricle end-diastolic
diameter; LVEDP, left ventricle end-diastolic pressure; LVEDV, left ventricle
end-diastolic volume; LVESd, left ventricle end-systolic diameter; LVESP, left
ventricle end-systolic pressure; LVESV, left ventricle end-systolic volume;
LVPWd, left ventricular posterior wall thickness at the end of diastole; LVPWs,
left ventricular posterior wall thickness at the end of systole; LVSP, left
ventricle systolic pressure; MAP, mean arterial pressure; MCT, monocrotaline;
| ||||
Cardiac fibrosis as a frequent companion of heart diseases, leads to dilatation,
cardiomyocyte hypertrophy, and apoptosis, with HF as an ultimate
pathophysiological event [155]. Accumulation of activated myofibroblast at the
injury site stands for a significant driver of the fibrotic process in cardiac
tissue [98]. In recent years, antifibrotic properties of S/V have been
investigated in various preclinical studies involving majorly MI and HF
modelling. Early investigations in this field were reported in a study by von
Lueder and colleagues [133], where chronic S/V treatment, initiated one week
after MI induction in rats, preserved cardiac function and remodelling by
reducing myocardial hypertrophy and fibrosis in peri-infarcted and non-infarcted
remote myocardium. Similar findings were reported in a study by Kusaka and
colleagues [156], where, comparing to valsartan, treatment with S/V inhibited
cardiac fibrosis and hypertrophy in rats with metabolic syndrome and HTN.
Furthermore, Croteau and colleagues [134] demonstrated that S/V was superior over
valsartan in improving diastolic function and reducing cardiac interstitial
fibrosis in obesity-related metabolic heart disease in mice. In a study by
Miyoshi and colleagues [135], authors reported decreased cardiac fibrosis along
with reduced mRNA expressions of transforming growth factor (TGF)-
Present literature evidence supports the role of TGF-
Matrix metalloproteinases (MMPs) are members of a zinc-dependent enzyme family
responsible for degrading specific molecules that constitute extracellular matrix
(ECM). Various studies implied the role of MMPs overexpression in the setting of
LV remodeling and myocardial dysfunction, while inhibiting MMPs activity may be a
promising therapeutic approach for preventing HF [158]. In a study by Boutagy and
colleagues [139], cardioprotective effects of chronic S/V treatment were at least
partially mediated by reduced activity of cardiac MMPs in doxorubicin-induced
cardiotoxicity. Increased activity of MMP-2 by 25% was observed in the HFpEF rat
model compared to controls, while 12-week treatment with S/V attenuated its
activity by around 30% [140]. Similarly, a decrease in
In their research, Burke and colleagues [143] reported that in the
pressure-overload HF model in mice, S/V exerted antifibrotic effects by directly
affecting cardiac myofibroblast via PKG-dependent inhibition of RhoA, which is
involved in myofibroblast transition and activation. In a study by Kompa and
colleagues [144], 4-week treatment with S/V decreased cardiac hypertrophy and
fibrosis to a similar extent as perindopril alone. At the same time, gene
expressions of ANP and
Suo and colleagues’ [146] study showed that S/V treatment was superior to valsartan in preserving left atrial and left atrial appendage remodelling and reduced atrial fibrosis in the pressure-overload mice model. Beneficial effects of S/V treatment were also observed in the AF rabbit model, where this agent preserved atrial structural remodelling by reducing cardiac fibrosis [147]. Furthermore, in rats with induced pulmonary hypertension (PH), 3-week treatment with S/V prevented RV remodelling by its favourable effects on RV pressure, hypertrophy, the orientation of collagen and myofiber as well as tissue stiffening [159].
Up to date, there is little evidence supporting the anti-inflammatory potential of S/V on its observed cardioprotection in clinical studies [160]. However, according to the previous basic researches, treatment with S/V decreased the circulating levels of proinflammatory cytokines, such as MMP-8, IL-6, and monocyte chemoattractant protein (MCP)-1 in ApoE2/2 mice in regard to valsartan [161]. Similarly, treatment with S/V reduced proinflammatory mediators in the blood and hearts of TAC mice [145]. In a study by Ge and colleagues [148], S/V alleviated diabetic cardiomyopathy in mice, which was an effect partially driven by its ability to inhibit inflammation. Furthermore, S/V was more effective than enalapril in improving survival by inhibiting inflammatory response in post-AMI setting in mice [142].
It is well known that Nucleotide-Binding Domain-Like Receptor Protein 3 (NLRP3)
inflammasome signalling, primarily involved in inducing secretion of
proinflammatory cytokines, contributes to the pathophysiological cascade of
various chronic diseases such as HF [162]. On the other hand, it was previously
proposed that the TGF‑
Cardiac apoptosis has an important role in various CVDs, including MI and HF [164]. Several preclinical studies highlighted the antiapoptotic effects of S/V as potential mediators of improved cardiac structure and function. In a study by Xia and colleagues [152], 4-week treatment with S/V improved DOX-induced cardiomyopathy in adult mice. In this report, cardioprotective effects of S/V are thought to be (at least partially) driven by its ability to reduce the activity of fission protein dynamin-related protein 1 (Drp1) involved in the apoptotic cascade and thus preserve mitochondrial function [152]. In a study by Peng and colleagues [153], pressure overload HF was induced by a transverse aortic constriction in mice, which were further treated with S/V. Authors reported that a 4-week treatment with S/V decreased myocardial apoptosis assessed by TUNEL staining, along with significantly reduced expressions of apoptotic markers (Bax and Bcl-2) [153]. Similarly, diabetic cardiomyopathy mice administered with S/V had attenuated protein expression of apoptotic markers (cleaved caspase-3 and Bax/Bcl-2 ratio), along with improvement in cardiac dysfunction and remodelling [148]. S/V treatment also tended to reduce myocardial apoptotic rate in pressure unloaded mice, although these effects did not reach a significant level [141].
Oxidative stress, as an imbalance between ROS production and antioxidative defense capacity [165], is suggested as one of the contributors in the pathophysiological cascade of various heart conditions, including HF [166]. In ISO-induced MI in rats, combined administration of sacubitril and valsartan in high dose preserved myocardial tissue damage and reduced infarcted area [167]. The authors further reported that the potential cardioprotective mechanism of S/V involves its ability to reduce oxidative stress [167]. Furthermore, Croteau and colleagues [134] reported that S/V decreased oxidative stress in the myocardium more prominently than valsartan, an effect mirrored through reductions of oxidized lipid 4-Hydroxy-2-nonenal (4-HNE) in mice with metabolic heart disease.
Oxidative stress can activate nuclear factor (NF)-
Given the adopted knowledge on S/V cardioprotection, it would be of great interest to further investigate its effectiveness and safety in different CV pathologies. Moreover, evidence on simultaneous administration of S/V and other existing and developing CV therapeutics could provide novel insights into possible synergistic benefits. Bearing in mind its antiinflamatory potential, it would be important to conduct more preclinical studies concerning the efficacy of S/V in the setting of inflammatory heart diseases, including myocarditis, while this agent may improve both cardiac dysfunction and inflammatory response in this clinical setting. Of note, discovering other signalling pathways affected by S/V should be of particular interest for basic researches, while it can provide additional understanding of its cardioprotective mechanisms.
MN—wrote the manuscript and table design; IS, JJJ, IC—revised the manuscript; JS, JJ, SS, NM, VS—performed research of the literature and text editing; DD, Stefani B, Sergey B, VJ—text supervising. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
For the creation of Fig. 1, we used BioRender.com.
This work was supported by Faculty of Medical Sciences, University of Kragujevac (Junior project No. 03/18 and No. 33/20).
The authors declare no conflict of interest. Vladimir Jakovljevic is serving as one of the Guest Editors of this journal. We declare that Vladimir Jakovljevic had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Filippos Triposkiadis and Davide Bolignano.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.