


 , Manish Mishra 2
, Manish Mishra 21 Department of Physiology (APP), College of Medicine, University of Saskatchewan, Saskatoon, SK S7N 5A2, Canada
2 Department of Pharmacology, Dalhousie University, Halifax, NS B3H 4R2, Canada
Academic Editors: Karol E. Watson and Morris Karmazyn
Abstract
Hypercholesterolemia is involved in the development of atherosclerosis and is a
risk factor for coronary artery disease, stroke, and peripheral vascular disease.
This paper deals with the mechanism of development of hypercholesterolemic
atherosclerosis. Hypercholesterolemia increases the formation of numerous
atherogenic biomolecules including reactive oxygen species (ROS), proinflammatory
cytokines [interleukin (IL)-1, IL-2, IL-6, IL-8, tumor necrosis factor-alpha
(TNF-
Keywords
- hypercholesterolemia
- reactive oxygen species
- atherosclerosis
- cell adhesion molecules
- cytokines
- advanced glycation end products
- C-reactive protein
- nuclear factor-kappa B
- atherogenic biomolecules
Atherosclerosis affects medium and large-sized arteries and is characterized by
focal thickening of the intima of the arteries and deposition of lipid, resulting
in narrowing of the arteries. Atherosclerosis leads to cardiovascular diseases
[1]. There are numerous factors including hyperlipidemia [2, 3], diabetes [4],
hypertension, cigarette smoking [5], obesity [6], hyperhomocysteinemia [7], and elevated serum C-reactive
protein [8, 9] which are involved in the
development of atherosclerosis. The term hyperlipidemia refers to increased
levels of serum total cholesterol (TC), low-density lipoprotein-cholesterol
(LDL-C) and triglycerides (TG), or a combination of all the three. A major risk
factor for coronary artery disease is hyperlipidemia [3, 10]. CAD (coronary
artery disease) risk increases by 2% to 3% for every 1% increase in serum
cholesterol [11]. A 10% reduction of serum cholesterol reduces the risk of
CAD by half for men of 40 yrs of age and by 25% for men 60 yrs of age over 5 yrs
[11]. An increase of 10 mg/dL of LDL-C was associated with a 12% increase
in the risk of cardiovascular disease (CVD) [12]. The serum TG levels are
strongly associated with CAD [13, 14]. There is a strong inverse correlation of
high-density lipoprotein cholesterol (HDL-C) with atherosclerotic CAD. High serum
HDL-C levels reduce the rate of atherogenesis [15], while low levels of HDL-C
accelerate atherosclerosis [16]. The risk of CAD is increased by 2% to 3% for
every 1 mg/dL reduction in the levels of HDL-C [17]. The ratio of TC/HDL-C
Atherogenic biomolecules are defined as the biomolecules which are involved in the induction of atherosclerosis. This section describes the hypercholesterolemia-induced production of atherogenic biomolecules (Fig. 1).
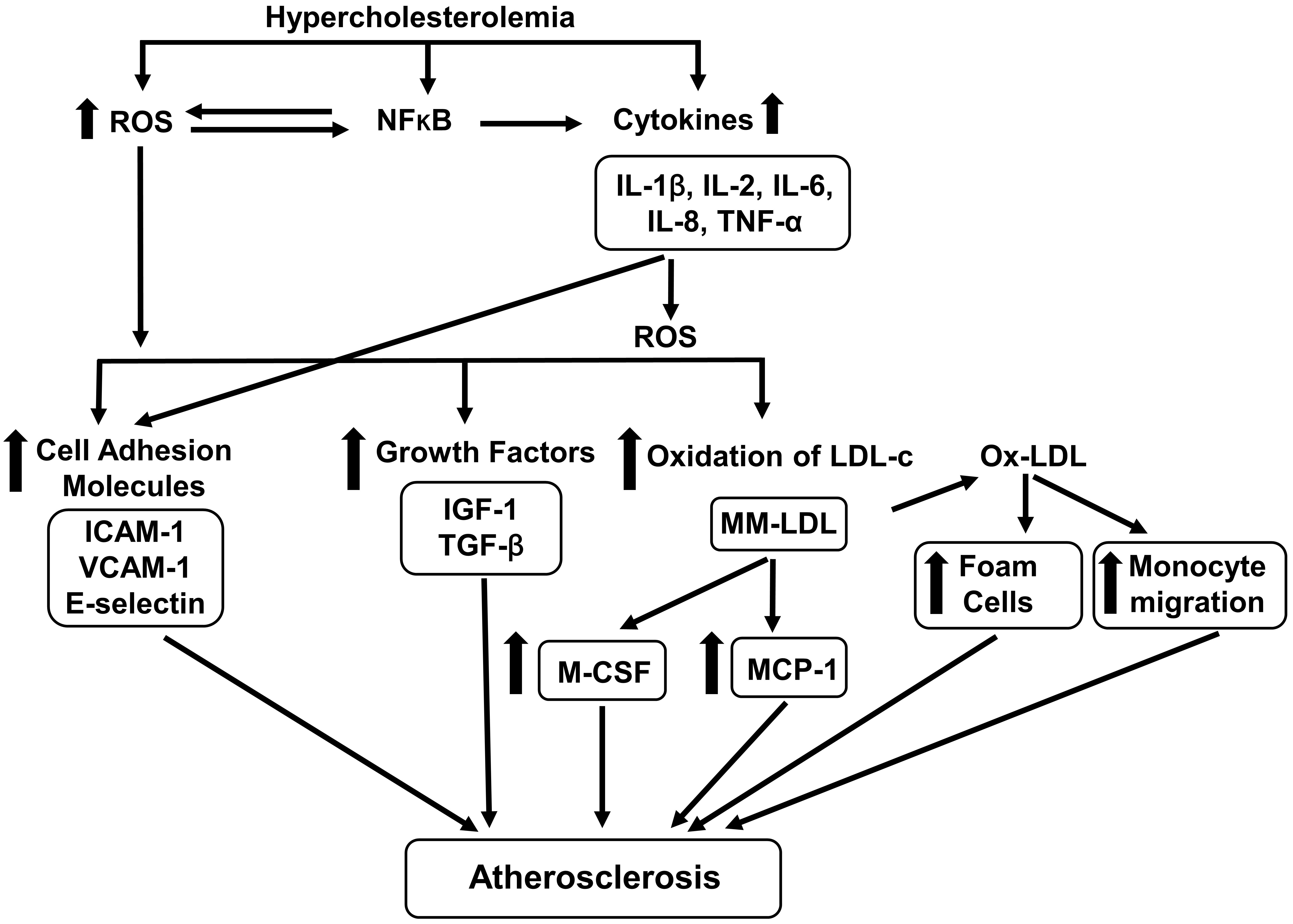 Fig. 1.
Fig. 1.Effects of hypercholesterolemia on atherogenic biomolecules. Hypercholesterolemia increases the generation of ROS (reactive oxygen species)
and cytokines [interleukin (IL)-1, IL-2, IOl-6, IL-8, tumor necrosis factor-alpha
(TNF-
There are various sources of hypercholesterolemia-induced increases in ROS. The
content of cholesterol in platelets, polymorphonuclear leucocytes (PMNLs),
endothelial cells, smooth muscle cells and monocytes are elevated by
hypercholesterolemia [28, 29, 30]. Thrombin, histamine, and adenosine diphosphate
(ADP) are released by cholesterol-rich platelets [31, 32]. Phospholipase A
Reduction in antioxidants would also elevate the serum levels of ROS. Superoxide
dismutase (SOD), catalase and glutathione peroxidase (GSH-Px) are enzymatic
antioxidants. Superoxide dismutase metabolizes superoxide anion to
hydrogen peroxide (H
ROS have numerous functions in the development of atherosclerosis. It activates
nuclear factor-kappa-B (NF-
Oxidation of LDL-C by ROS has numerous functions in the development of atherosclerosis [68, 69, 70, 71, 72]. LDL-C is mildly oxidized to form minimally modified LDL (MM-LDL) which is further oxidized to form maximally oxidized LDL (OX-LDL). MM-LDL activates smooth muscle cells and endothelial cells to produce monocyte chemoattractant protein-1 (MCP-1) which is involved in the migration of monocytes (leukocytes) from endothelial surface to subendothelial space. Monocytes possess LDL receptors which combine with native LDL, but the amount of native LDL is not enough to form foam cells. MM-LDL stimulates endothelial cells to generate monocyte colony-stimulating factor (MC-SF) which triggers monocyte differentiation into macrophages that develop receptor for OX-LDL. OX-LDL is taken up by differentiated macrophages to form foam cells. An overview on the formation of OX-LDL and its role in the development of atherosclerosis have been reported by Poznyak et al. [73]. Parthasarathy et al. [70] have reported that OX-LDL is present in the circulating blood. LDL oxidation takes place in the vascular wall [73]. Hashimoto et al. [74] have reported that transmigration of monocytes into subendothelial space is assisted by OX-LDL directly through a change in the endothelial junction. Other investigators [75] have reported that OX-LDL assists in the recruitment of monocytes through interaction of platelet with monocytes and endothelial cells. Macrophages are involved in the generation of numerous growth-regulating factors [76]. Plasma LDL has been shown to have a positive correlation with ROS release by mononuclear leucocytes (MNLs) and polymorphonuclear leukocytes (PMNLs) [77].
Triglycerides (TG) enhance the generation of ROS and secretion of TGF-
HDL-C has antiatherogenic properties. Plasma HDL-C has a negative correlation with ROS release by resting MNLs and PMNLs [77]. It has antioxidant activity [82] and has inhibitory effects on LDL oxidation [83]. HDL-C reduces the expression of MCP-1 [84] and prevents the CRP-induced upregulation of proinflammatory adhesion molecules [85].
Hypercholesterolemia-induced atherosclerosis is based on the oxidative hypothesis of atherosclerosis which has been accepted universally [71, 72, 76, 86]. The proposed mechanism of atherosclerosis produced by hypercholesterolemia is depicted in Fig. 2. Hypercholesterolemia augments the production of ROS [37, 38, 42, 43, 44, 45, 46] and cytokines [40, 41] which increase the expression of CAM [55, 56, 57, 58]. CAM [55, 56, 57, 58] in endothelial cells. The early step in the development of atherosclerosis is adherence of monocytes to endothelial cells [61] and which is achieved through CAM. CAM is involved in the rolling and adhesion of monocytes to the endothelial cells. Monocyte then transmigrates into subendothelial space [87]. MM-LDL produce monocyte chemoattractant protein-1 (MCP-1) in endothelial cells and vascular smooth muscle cells [88]. The migration of monocytes to the subendothelial space is assisted by MCP-1 [89]. OX-LDL increases the expression of cell adhesion molecules [90]. OX-LDL directly enhances the migration of monocytes to subendothelial space. Immigrating monocytes into the subendothelial space have LDL receptor but the rate of uptake of native LDL is not enough to produce foam cells [91]. MM-LDL stimulates endothelial cells to express MC-SF [92] that enhances the monocyte differentiation to form tissue macrophages which develop receptors for OX-LDL [92]. OX-LDL is a ligand for scavenger receptors which are expressed in tissue macrophages [93]. OX-LDL is taken up by tissue macrophage to form foam cells. Foam cells are involved in formation of numerous growth factors which enhance vascular smooth muscle cell proliferation and migration and fibrous tissue synthesis which helps in the development and progression of atherosclerosis. There is a development of fatty streaks in full-fledged atherosclerosis.
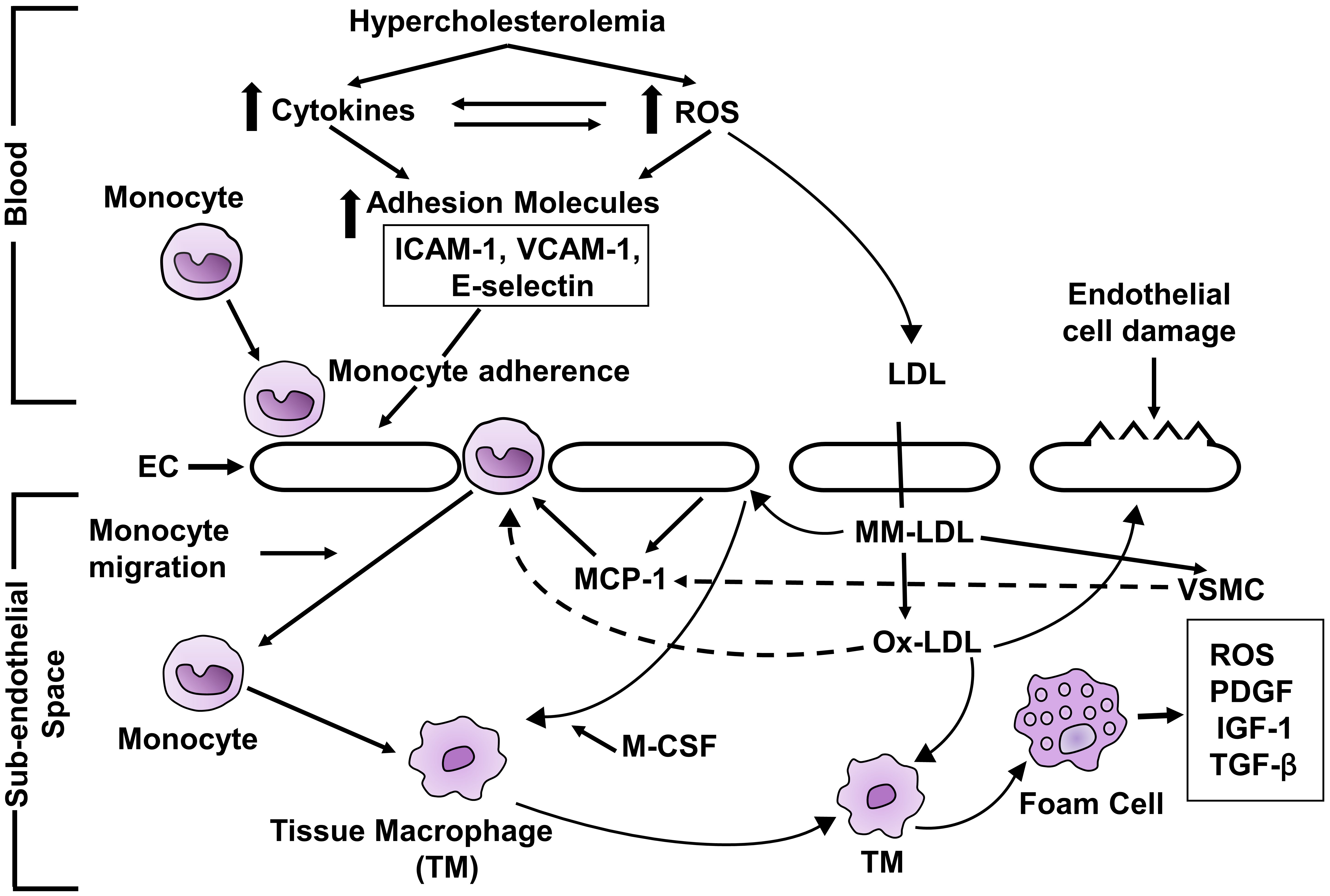 Fig. 2.
Fig. 2.Schematic diagram of mechanism of hypercholesterolemia-induced
atherosclerosis. ROS, reactive oxygen species; ICAM-1, intercellular adhesion
molecule-1; VCAM-1, vascular cell adhesion molecule-1; EC, endothelial cell; LDL,
low-density lipoprotein; MM-LDL, minimally modified LDL; OX-LDL, maximally
oxidized LDL; MCP-1, monocyte chemoattractant protein; VSMC, vascular smooth
muscle cell; MC-SF, monocyte colony stimulating factor; TYM, tissue macrophage;
PDGF, platelet-derived growth factor; IGF-1, insulin-like growth factor-1;
TGF-
As described above, hypercholesterolemia generates ROS. The question arises if hypercholesterolemia-induced ROS induces atherosclerosis. This section describes the increases in the levels of ROS and the indirect measures of ROS in hypercholesterolemic atherosclerosis. Indirect measures of ROS include lipid peroxidation products, malondialdehyde (MDA) [94, 95], aortic tissue chemiluminescence (AO-CL) [96], a polymorphonuclear leukocyte chemiluminescence (PMNL-CL) [97] and white blood cell chemiluminescence (WBC-CL) [97]. AO-CL is a measure of antioxidant reserve [96]. An increase in AO-CL suggests a decrease in the antioxidant reserve and vice-versa. Luminol-dependent chemiluminescence is a highly sensitive method for measurement of ROS generated by PMNLs and WBCs [97].
Hypercholesterolemic atherosclerosis was associated with increases in the serum [20, 97, 98, 99, 100, 101] and aortic MDA [19, 96, 97], PMNL-CL [96], WBC-CL [96, 98, 101] and aortic-CL [96, 97]. However, the aortic-CL has been observed to be reduced in certain studies [98, 99, 100, 101, 102]. Aortic-CL is a measure of both oxidative stress and antioxidant reserve in the tissue [103]. If hypercholesterolemia increases the indirect measure of ROS and produces atherosclerosis, then lowering serum levels of cholesterol would be associated with reduction in the extent of atherosclerosis and the levels of both direct and the indirect measure of ROS. We describe the agents which have both antioxidant and hypolipidemic effects on hypercholesterolemic atherosclerosis and ROS. Secoisolariciresinol diglucoside (SDG) a product of flaxseed reduced the serum levels of cholesterol and this reduction was associated with a reduction in the extent of hypercholesterolemic atherosclerosis, aortic MDA and aortic-CL [104]. Flax lignin complex, a byproduct of flaxseed reduced hypercholesterolemic atherosclerosis by 30%, and this effect was associated with a lowering of serum levels of cholesterol by 20%, serum MDA by 35% and aortic MDA by 58% in rabbits [99]. It is to note that both SDG and flax lignan complex have antioxidant activity [99, 105, 106]. Probucol, an antioxidant and cholesterol-lowering agent [107] decreased the extent of hypercholesterolemic atherosclerosis, and aortic tissue MDA, but had no effects on aortic-CL [96].
We now discuss the effects of antioxidants on hypercholesterolemic atherosclerosis and ROS. Since ROS is implicated in the formation of atherosclerosis, the antioxidants would reduce the evolution of hypercholesterolemic atherosclerosis and associated indirect measures of ROS. Vitamin E, an antioxidant [108], reduced hypercholesterolemic atherosclerosis and this was associated with a decrease in serum and aortic MDA but had no effect on serum cholesterol [20].
Sources of hypercholesterolemia-induced ROS include the synthesis of prostaglandins and leukotrienes [37, 38], activated complements [39, 44], PAF [42], and cytokines [45, 46]. Hence inhibitors of the enzyme of synthesis of prostaglandin and leukotrienes, PAF, cytokines and activated compliments would decrease the formation of hypercholesterolemic atherosclerosis and ROS levels. Inhibitors of cyclooxygenase which is involved in the synthesis of prostaglandin and leukotrienes such as aspirin [109], and indomethacin [110] were used in the prevention of hypercholesterolemic atherosclerosis and reduction of ROS. Aspirin did not affect the serum levels of cholesterol in rabbits with hypercholesterolemia but reduced atherosclerosis by 47% and this effect was associated with lowering of serum and aortic tissue MDA, release of ROS from WBC-CL, and aortic-CL [101]. Indomethacin decreased the extent of hypercholesterolemic atherosclerosis by 46% and this effect was associated with a decrease in aortic MDA and antioxidant reserve, but no change in the serum cholesterol, and WBC-CL [111]. Pentoxifylline an inhibitor of cytokines [112], and PAF [113, 114] had no effect on serum cholesterol but the extent of hypercholesterolemia-induced atherosclerosis was lowered by 38% and this effect was associated with a reduction in serum and aortic tissue MDA, and normalization of aortic-CL [115].
Are the atherogenic biomolecules such as serum/plasma/tissue levels of ROS,
NADPH-oxidase, NF-
The increases in the serum/tissiue levels of ROS in hypercholesterolemic rabbits
have been described in detail in section 4 of this review. Hypercholesterolemia
increases the activity of the oxidant producing enzyme system, NADPH-oxidase
[116], and xanthine oxidase [117]. Hypercholesterolemia activates NF-
AGEs are heterogenous groups of irreversible adducts produced from the nonenzymatic interaction of amino groups of protein, lipids, and nucleic acids with reducing sugars such as glucose, fructose, and glyceraldehyde [130, 131]. Receptors for AGE include RAGE, sRAGE, esRAGE, and cRAGE (cleaved RAGE). RAGE is bound to the cell membrane, while sRAGE, esRAGE, and cRAGE circulate in the blood. RAGE has two isoforms, esRAGE and cRAGE. cRAGE is cleaved from RAGE by proteolytic enzymes [132] and esRAGE is produced from alternate mRNA splicing of full-length RAGE [133]. sRAGE contains both cRAGE and esRAGE. sRAGE, esRAGE, and cRAGE lack the cytosolic and transmembrane domain and circulate in the blood. Interaction between AGE with RAGE produces atherogenic biomolecules [23, 134]. The binding of sRAGE, cRAGE and esRAGE with AGE does not activate intracellular signaling and does not produce atherogenic biomolecules. There is a competition between RAGE and sRAGE for binding with AGE [135]. Thus, sRAGE and esRAGE have protective effects against adverse effects of interaction of AGE with RAGE. AGE-RAGE stress, defined as the ratio of AGE/sRAGE has been coined by Prasad and Mishra [136], A high ratio of AGE/sRAGE indicates the presence and progression of atherosclerosis.
The serum levels of AGE and AGE/sRAGE were higher, while the sRAGE levels were
lower in hypercholesterolemic subjects than normocholesterolemic subjects [137].
The above investigators also reported that there was a positive correlation
between serum cholesterol levels and the levels of AGE and AGE/sRAGE, and a
negative correlation between serum cholesterol and sRAGE. Santilli et
al. [138] have also reported that hypercholesterolemic subjects had lower serum
levels of sRAGE than normocholesterolemic subjects. Hypercholesterolemia-induced
AGE would interact with RAGE to generate ROS [139], which would activate
NF-
The levels of AGE and RAGE were elevated in the wall of the carotid artery of Zucker diabetic rats, and these levels were further elevated in the balloon-injured carotid artery of these rats [140]. These authors also reported that sRAGE administration before and for 21 days post-balloon injury reduced the neointimal hyperplasia in the carotid artery. De-endothelialization of the carotid artery in wild type mice has been shown to elevate the expression of RAGE in injured arteries [141]. They also observed that use of sRAGE reduced neointimal hyperplasia in these mice. Wendt et al. [27] have shown that diabetes-accelerated atherosclerosis in apo-E deficient mice had increased expression of VCAM-1 in the aorta, and that sRAGE administration significantly reduced the atherosclerotic lesion in the aorta. Administration of sRAGE completely suppressed the accelerated and advanced atherosclerosis in apo-E deficient mice [142]. Serum levels of sRAGE were reduced in Non-ST-segment elevated myocardial infarction [25]. Serum levels of sRAGE were reduced in patients with restenosis following percutaneous coronary intervention (PCI) [26]. Low pre-PCI sRAGE levels in serum have been reported to be a predictor of post-PCI restenosis in NSTEMI patients [26]. AGE-RAGE stress has been reported to play a role in the development of coronary artery disease [134, 143] and carotid artery stenosis [144].
A hypercholesterolemic diet increases the serum levels of CRP [49]. CRP can
induce atherosclerosis through the generation of ROS [8, 145, 146] activation of
NF-
Hypercholesterolemia increases the production of ROS which sets the stage for the production of other atherogenic biomolecules [27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48] leading to the formation of atherosclerosis. Reduction in antioxidant enzymes by high blood cholesterol would also elevate the ROS levels [50]. Hypercholesterolemia-induced atherosclerosis is associated with increases in the serum/plasma/tissue levels of direct and indirect measures of ROS [19, 20, 96, 97, 98, 99, 100, 101, 104]. Blockade of the ROS with antioxidant (vitamin E) [20], hypolipidemic and antioxidant agents (SDG [104], flax lignan complex [99], and probucol [96]), cyclooxygenase inhibitors (aspirin) [101] and indomethacin [111], and inhibitors of cytokines and PAF (pentoxifylline [115]) decreased the development of hypercholesterolemic atherosclerosis and amount of ROS. The above data indicate that there is an association between hypercholesterolemic atherosclerosis and ROS, while lowering the serum cholesterol and blockade of sources ROS reduces the extent of atherosclerosis and ROS. It is to note that hypercholesterolemia elevates the serum levels of AGE [137] and AGE/sRAGE [137], and lowers the serum levels of sRAGE [135, 136]. An increase in AGE and AGE/sRAGE, and a decrease in sRAGE in the serum have been implicated in the development of atherosclerosis [23, 134, 137]. Hypercholesterolemia has been reported to elevate the serum levels of CRP in human subjects [49]. A rise in C-reactive protein increases the serum levels of atherogenic biomolecules [146, 147, 148, 149, 150] and induces development of atherosclerosis [151]. It is surprising that there are limited publications on the effects of hypercholesterolemia on C-reactive protein and AGE-RAGE axis. Hypercholesterolemia increases the production of AGE, CRP, and ROS, and decreases the production of sRAGE all of which are implicated in the formation of atherosclerosis. Lowering of AGE and C-reactive protein, raising of sRAGE, and use of antioxidants may be considered as an adjunct therapy besides lipid lowering agents for the treatment of hypercholesterolemia.
Hypercholesterolemia induces atherosclerosis through increases in the
atherogenic biomolecules (ROS, NADPH-oxidase, NF-
KP designed the study and wrote the manuscript. MM made the figures and reviewed the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest. KP is serving as one of the Editorial Board members of this journal. We declare that KP had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Karol E. Watson and Morris Karmazyn.