



 , Liliang Li 1,*
, Liliang Li 1,*1 Department of Forensic Medicine, School of Basic Medical Sciences, Fudan University, 200032 Shanghai, China
2 Department of Cardiology, Shanghai Institute of Cardiovascular Diseases, Zhongshan Hospital, Fudan University, 200032 Shanghai, China
†These authors contributed equally.
Academic Editors: Brian Tomlinson and Takatoshi Kasai
Abstract
Coronary artery spasm (CAS) is a transient reversible subtotal or complete occlusion induced by coronary hypercontraction and the critical cause of myocardial ischaemia with non-obstructive coronary arteries. During the past decades, our knowledge of the risk factors and pathophysiological mechanisms of CAS have been increasingly progressed, and various diagnostic approaches, including imaging technologies and novel biomarkers, have been proposed to serve well to diagnose CAS clinically. This review aims to summarize these research progresses on the risk factors of CAS and introduce current knowledge about the mechanisms accounting for CAS, including endothelial dysfunction, vascular smooth muscle cell hyperreactivity, and adventitial and perivascular adipose tissue inflammation. We also gathered the recently evolved diagnostic approaches and analyzed their advantages/disadvantages, in purpose of enhancing the diagnostic yield on the basis of ensuring accuracy.
Keywords
- coronary artery spasm
- risk factors
- endothelial dysfunction
- vascular smooth muscle cell hyperreactivity
- adventitial inflammation
- diagnostic approaches
In 1959, Prinzmetal et al. [1] first proposed the term “variant
angina” which is later evolved and re-named as coronary artery spasm (CAS). CAS
is generally considered as abnormal contraction of epicardial coronary arteries
causing myocardial ischemia and includes microvascular CAS in a broad sense.
Clinically, CAS is defined as a transient reversible subtotal or complete
occlusion of coronary arteries with
CAS is not a benign disease. Approximately 1–14% of AMIs are considered to occur in CAS patients, which could further lead to fatal arrhythmia, and even sudden cardiac death [5]. Thrombosis secondary to CAS may be another important cause of myocardial infarction [6]. Despite the area of CAS-induced myocardial infarction is small in general, spontaneous reperfusion after CAS subsiding also increases the risk of fatal arrhythmia [7]. VSA is the major clinical manifestation of CAS-induced myocardial ischemia. It is usually independent of effort occurring at rest with obvious circadian rhythm, namely more occurrences in the period from midnight to dawn [2]. ST-segment elevation or depression on ECG is one of the clinical features [2]. Compared with coronary atherosclerotic diseases (CAD), CAS is more prevalent in women, younger people and Asian populations, such as Japanese and South Koreans. With the utilization of invasive SPT, it is also not uncommon for VSA in some Western countries such as Germany and Australia [8, 9]. However, true prevalence needs further investigation due to the rare utilization of SPT in most countries, such as China, where SPT is cautiously performed only for clinical diagnosis in specialized medical centers.
Recent years have witnessed increasing advances towards our understanding of CAS. This review aims to introduce the recent knowledge on the risk factor, pathophysiological mechanisms of CAS and also highlights the latest advancements in clinical diagnosis of CAS, aiming at providing effective alternatives for invasive methods, especially for the countries where SPT is not performed routinely in the clinic.
There are a vast number of precipitating factors for CAS (Fig. 1), which can be divided as physiological and pharmacological categories. The former includes emotional stress, cold stimulation, hyperventilation, valsalva maneuver, and exercise etc., while the latter contains psychoactive drugs (such as cocaine, marijuana, and amphetamine), sympathomimetic agents (such as epinephrine, norepinephrine), parasympathomimetic agents (such as acetylcholine (Ach), pilocarpine), vasoconstrictors (such as thromboxane, ergonovine), alcohol consumption, and magnesium deficiency etc. [10, 11]. In addition, there have been reports about CAS induced by traditional Chinese medicine, including Di-Long (dried earthworm), Ma-Huang (plant of ephedra), and cucumis polypeptide (the combined extracts from deer horn and sweet melon seeds) [12].
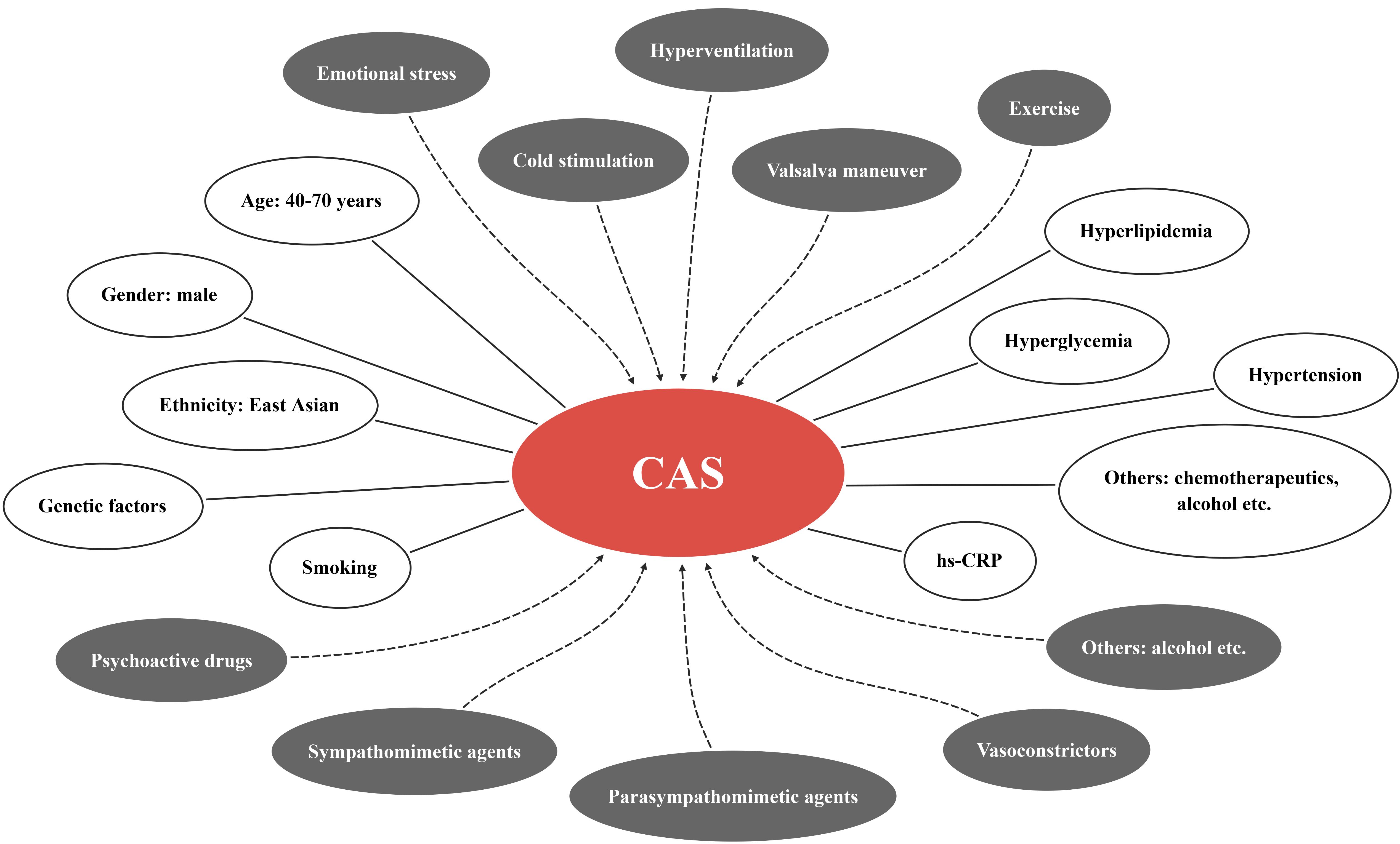 Fig. 1.
Fig. 1.Precipitating factors (grey ellipses) and clinical risk factors (white ellipses) of CAS. CAS, coronary artery spasm; hs-CRP, high-sensitivity C-reactive protein.
Unlike CAD, CAS patients seem to be more common among young people and women [7, 13]. However, male patients still account for the majority of CAS patients, and high prevalence is in the age range of 40–70 years [4]. As mentioned above, CAS is a highly prevalent disease in East Asia with ethnic and genetic diversity. It is worth noting that East Asian patients tend to present diffuse and multi-vascular CAS, while Caucasians tend to present focal CAS [14]. Smoking is an unequivocal risk factor for CAS and about 75% of CAS patients are smokers [15]. It was also reported that the proportion of smokers in CAS patients was 42.6%, but it still surpassed that in CAD patients [16]. The substances in cigarettes, such as carbon monoxide and nicotine, are able to damage blood vessels by increasing inflammation and oxidative stress, which explains why smoking is a high risk factor for CAS [17]. Although hyperlipidemia, hyperglycemia, and hypertension in CAS patients are less common than those in CAD patients [16], these metabolic disorders also contribute to the development of CAS. Serum high-sensitivity C-reactive protein (hs-CRP) is higher in CAS patients than that among healthy individuals, implicating the potential of hs-CRP to be a predictor of CAS [18]. Moreover, alcohol consumption [19] and chemotherapeutics [20] that destruct blood vessels through independent mechanisms have also been found to relate to CAS.
The pathogenesis of CAS is complicated and could be categorized as endothelial dysfunction (ED) in the intima, vascular smooth muscle cell (VSMC) hyperreactivity in the media, and adventitial and perivascular adipose tissue (PVAT) inflammation (Fig. 2).
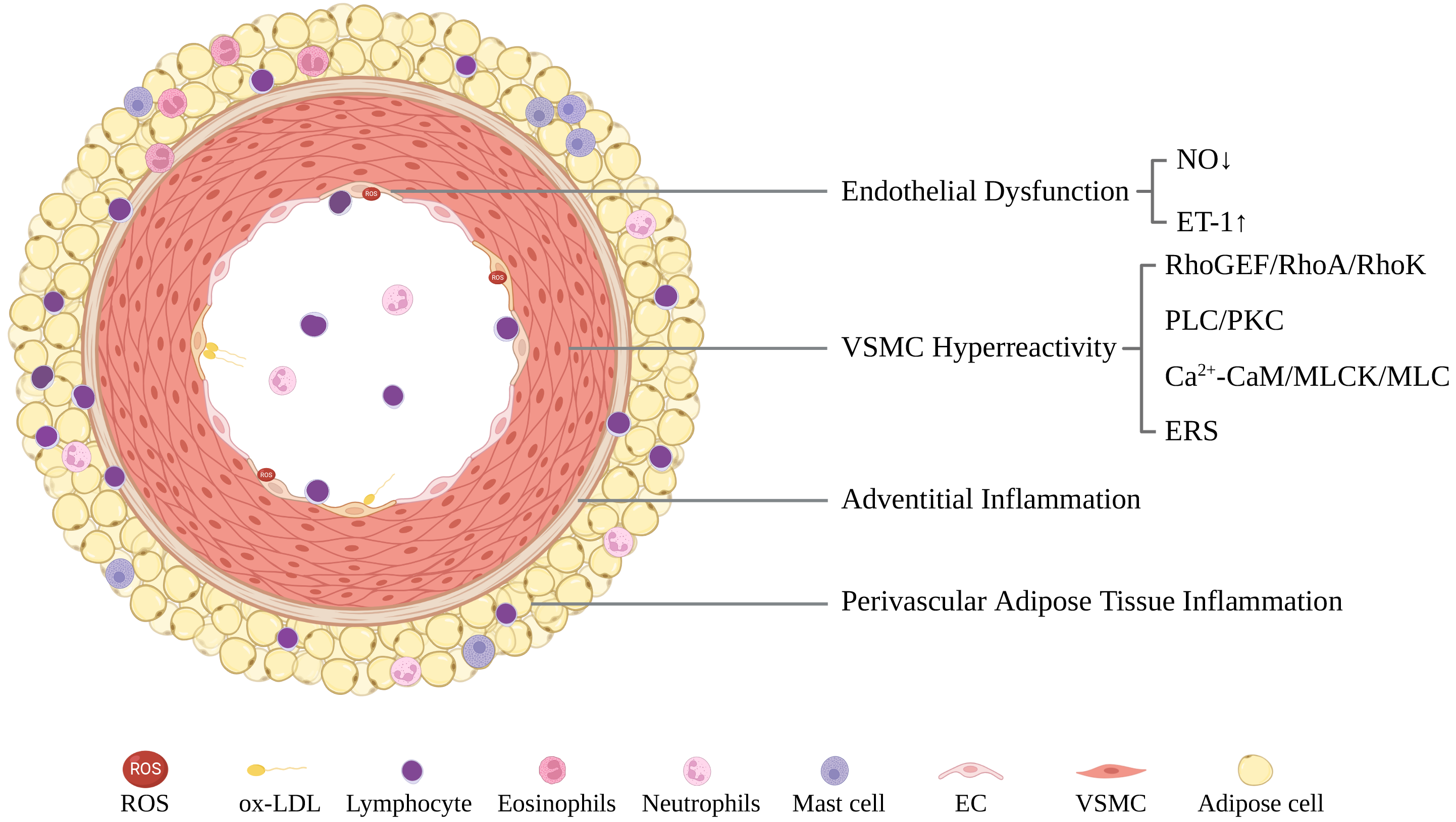 Fig. 2.
Fig. 2.A schematic illustration of CAS pathogenesis including endothelial dysfunction, VSMC hyperreactivity, and adventitial/perivascular adipose tissue inflammation. CaM, calmodulin; EC, endothelial cell; ERS, endoplasmic reticulum stress; ET-1, endothelin-1; MLC, myosin light chain; MLCK, MLC kinase; NO, nitric oxide; ox-LDL, oxidized low-density lipoprotein; PKC, protein kinase C; PLC, phospholipase C; RhoA, Ras homolog gene member A; RhoGEF, Rho guanine nucleotide exchange factors; RhoK, Rho kinase; ROS, reactive oxygen species; VSMC, vascular smooth muscle cell.
ED is defined as a series of phenotypes related to pathophysiological heterogeneous changes in vascular tone, permeability, inflammation, and de-differentiation by the ESC [21]. Clinical observations have shown that ED is associated with the pathogenesis of CAS. Nitroglycerin and isosorbide dinitrate, two endothelial-independent vasodilators, are highly efficient to relieve vasospasm angina during CAS [22, 23]. Nitrates are even prescribed as vasodilator agents after SPT [24]. In clinical angiography, it has been found that most of the spastic sites were in parallel to atherosclerotic plaque [25], and the coronary intima of CAS patients was remarkably thickened [26]. Immunohistological analysis of endomyocardial biopsy samples further showed that most CAS patients had endothelial cells (ECs) activation [27]. After removal of the endothelium, porcine coronary arteries successfully developed CAS with high cholesterol feeding [28].
At the molecular level, ED refers to disruption of homeostasis for endothelial
regulation of vascular tension, and defines the abnormal function of synthesis
and release of vasoactive substances such as nitric oxide (NO) and endothelin-1
(ET-1). Endothelial NO synthase (eNOS) dimer is the pivotal molecule for ECs to
physiologically produce NO. When high-risk factors are present, reactive oxygen
species (ROS) is increased in ECs due to stimulation by reduced nicotinamide
adenine dinucleotide phosphate (NADPH) oxidase [21, 29]. The increased ROS then
clears NO in ECs and converts it into peroxynitrite (ONOO
The ED in association with CAS is further supported by genetic evidence. The polymorphisms of NOS gene [37], aldehyde dehydrogenase 2 (ALDH2) gene [38], paraoxonase I gene [39], p22 phox gene in male [40], manganese superoxide dismutase (MnSOD) gene [41], and inflammatory factor interleukin-6 (IL-6) gene [40] all influence the NO synthesis, oxidative stress and inflammation. The polymorphisms of ET-1 gene are also related to CAS. Lee et al. [42] showed that CAS is related to the + 138delA, G8002A and Lys198Asn polymorphisms of the ET-1 gene. Ford et al. [43] observed that patients with coronary microvascular dysfunction have a higher frequency of the rs9349379-G allele and are associated with higher serum ET-1 levels.
Of note, Shimokawa [44] and Lanza et al. [45] presented evidence such as successful establishment of CAS animal models with normal endothelial function to show that ED might not be the key mechanism of CAS pathogenesis. Moreover, some CAS patients were resistant to nitrate treatment, which means supplementing NO cannot always mitigate CAS [46]. In addition, not all CAS patients have ED, and ED or inhibition of NO synthesis alone may be insufficient to cause CAS [45, 47], implicating that ED is an important yet unnecessary pathophysiological change of CAS.
While VSMC hyperreactivity is dependent on the cytoplasm Ca
The Ras homolog family (Rho) pathway activity has been observed to have
circadian rhythm, showing higher activity particularly at midnight and early
morning [48, 49], a time window that conforms to the circadian rhythm of CAS.
Also in CAS patients, intervention by Rho kinase (RhoK) inhibitors remarkably
reduced Ach-induced coronary contraction [50, 51], as well as the degree of
myocardial ischemia [52, 53], and further improve coronary artery relaxation
combined with nitroglycerin [54]. These data suggested that the Rho pathway plays
a pivotal role in the pathogenesis of CAS in human. Indeed, Rho guanine
nucleotide exchange factors (RhoGEFs) are a class of molecules with abundant
subtypes, which can activate Rho protein by converting GDP into GTP [55]. In
VSMCs, RhoGEFs are mainly regulated by G protein-coupled receptors (GPCRs) and
the activated RhoGEFs then transduce signals to the downstream Rho family member
A (RhoA), thereby modulating the Ca
Many etiologies can induce VSMC hyperreactivity by activating the RhoA/RhoK
pathway, such as oxidized low-density lipoprotein (oxLDL) [58, 59], chronic
hypoxia and ROS [60, 61, 62], inflammation [63, 64], hemorrhagic shock [65], and
chronic stress [66]. Galle et al. [58] observed that oxLDL augmented the
activity of RhoA in rabbit aorta, and thereby potentiating the contractile
responsiveness of aorta to Angiotensin (Ang) II. Bolz et al. [59] proved
that oxLDL increased the [Ca
Polymorphisms of RhoK gene also link with CAS. Kamiunten et al. [71] found that the missense mutation G930T resulted in the enhancement of RhoK activity in CAS patients and Yoo et al. [72] found that the GTCTG haplotype in 5 interesting single nucleotide polymorphisms (SNPs) might play a protective role in non-CAS patients.
Myosin light chain (MLC) phosphatase (MLCP) is one of the most important
downstream molecules of RhoK and its inactivation by RhoK enhances the
phosphorylation of MLC. Phosphorylated MLC (pMLC) was found at the spastic sites
and positively correlated with the degree of contraction in interleukin
1
Okumura et al. [74] cultivated the skin fibroblasts from CAS patients
and found that the phospholipase C (PLC) activity was enhanced and positively
correlated with the contractile hyperresponsiveness of coronary arteries,
proposing that the increased PLC activity may be involved in the pathogenesis of
CAS. The p122 protein, an agonist of PLC, was up-regulated in skin fibroblasts of
CAS patients [75]. Increased p122 protein promoted the basal and peak
[Ca
In addition, downstream PKC is also critically involved in the development of
CAS [79, 80]. Giardina et al. [81] proved that oxLDL enhanced the
Ca
Interestingly, in the porcine CAS model induced by IL-1
Calcium channel blockers (CCBs) have been well established as therapeutic agents
for CAS in clinic, suggesting that Ca
Calcium functions via binding with Calmodulin (CaM). The Ca
Endoplasmic reticulum stress (ERS) is defined as the accumulation of unfolded
and/or misfolded proteins in the endoplasmic reticulum (ER) that breaks the ER
homeostasis, and thereby activating the unfolded protein response (UPR) to
restore and maintain the ER homeostasis [97]. The causes of ERS encompass various
physiological or pathological stimuli such as hypoxia, starvation, oxidative
stress, imbalance of Ca
Choi et al. [98] found that hyperglycemia led to enhanced coronary
myogenic response and ED via triggering ERS in mice. Liang et
al. [99] showed that ERS inducers, such as tunicamycin (Tm), increased the
phosphorylation of MLC in VSMCs and enhanced the contractile responsiveness to
phenylephrine in aorta independent of endothelium. Zhang et al. [100]
observed that ceramide resulted in the VSMC hyperreactivity to phenylephrine
through ERS/COX-2/PGE2 pathway. We observed that an ERS inhibitor significantly
prevented VSMC contraction, whereas Tm aggravated the CAS-induced myocardial
ischemia in mice, and ERS regulated CAS possibly through the MLCK/MLC pathway
[101]. Ziomek et al. [102] also pointed out that Tm did not activate
Ca
Shimokawa and colleagues utilized IL-1
Of note, the vasoconstriction effect of PVAT inflammation seems to be
VSMCs-dependent [112]. For instance, Lynch et al. [113] revealed that
PVAT activated the BK
In the clinic, CAS may present in a variety of ways and is often asymptomatic, which causes CAS remaining a quite underdiagnosed and underreported disease with an average estimated delay of 3 months from presentation to diagnosis [7]. Currently, it is an urgency to develop accessible and practical diagnosis approaches for the disease. This section will introduce state-of-the-art diagnostic approaches (Tables 1,2) that might aid in clinical diagnosis of CAS.
| Imaging approaches | Advantages | Disadvantages | References |
| Coronary angiography (CAG) | Gold standard when performed under provocation testing | Confusion between CAD and CAS | [2, 116, 117, 118] |
| Omission in conditions of severe stenosis | |||
| Electrocardiogram (ECG) | Convenience, safety, availability, acceptability | Low specificity | [119, 120, 121, 122, 123] |
| Omission in resting intervals | |||
| Intracoronary imaging approaches | Exhibition of morphological and functional changes despite complex conditions | In theoretical stage | [117, 119, 124, 125, 126, 127, 128] |
| High requirements for equipment and operators | |||
| OCT | Better image quality and resolution to estimate intima | Interruption of the blood flow | [126, 128] |
| Tissue penetration: 2 mm | |||
| Safety worries | |||
| IVUS | Deeper penetration (4–8 mm) for accessing perivascular injury without interrupting the blood flow | Less resolution | [126, 129] |
| Positron emission tomography (PET) | Revelation of coronary vasomotor function and tissue image | Expensive | [109, 130] |
| High requirements for equipment | |||
| 18 |
Evaluation of inflammation of coronary perivascular adipose tissue | Expensive | [109] |
| High requirements for equipment | |||
| Myocardial contrast echocardiography (MCE) | Microvascular evaluation | Indirect functional information | [131, 132, 133] |
| Ignorance of minor systolic wall move | |||
| Low resolution | |||
| OCT, optical coherence tomography; IVUS, intravascular ultrasound. | |||
| Markers | Category | References |
| cystatin C | Endothelial dysfunction | [141, 142, 143, 144, 145] |
| xanthine oxidoreductase (XOR) | Endothelial dysfunction | [29, 146, 147, 148] |
| hs-CRP | Inflammation | [18, 148, 149, 150] |
| sCD40L | Inflammation | [18] |
| peripheral monocyte counts | Inflammation | [151] |
| Endothelin-1 (ET-1) | Vasomotor | [30, 152] |
| Serotonin (5-HT) | Vasomotor | [153, 154] |
| Neuropeptide Y | Vasomotor | [141, 155] |
| Lipoprotein(a) | perivascular adipose tissue metabolism | [148, 156, 157, 158, 159] |
| RhoK activity in circulating neutrophils | RhoK pathway | [49, 50, 66, 160, 161, 162, 163, 164, 165] |
| pMLC2 | Vascular smooth muscle cell hypersensitivity | [96, 101] |
| ox-LDL | Oxidative stress | [166, 167] |
| MDA-LDL | Oxidative stress | [166, 168, 169] |
| miR-17-5p, miR-92a-3p, miR-126-3 | MicroRNAs | [170, 171, 172, 173] |
Since the spontaneous coronary vasospasm at the time of angiography is only
occasionally observed [134], the current gold-standard diagnosis of CAS is
documentation by angiography with pharmacological provocative testing
via high-dose intracoronary administration of Ach, ergonovine, or
methylergonovine [2]. The typical positive response should include a transient
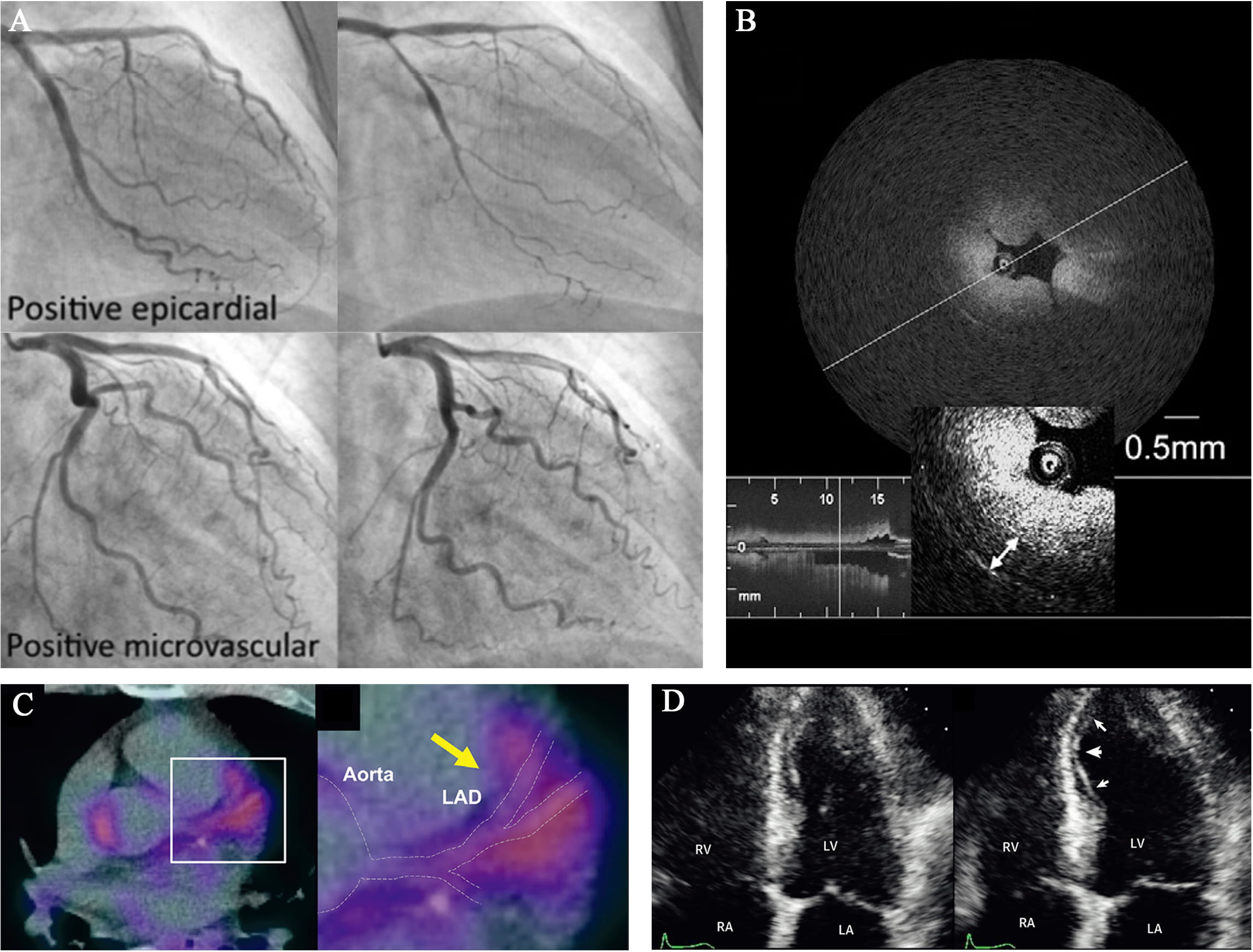 Fig. 3.
Fig. 3.Representative images of novel diagnostic approaches for CAS.
(A) Coronary angiograms of epicardial and microvascular CAS after spasm
provocation test (SPT) using intracoronary perfusion of Ach. Images from
Arrebola-Moreno et al. [131]. (B) Optical coherence tomography (OCT)
image of a spasm lesion after provocation. Medial thickening led to luminal
narrowing with intimal gathering. Image from Tanaka et al. [128]. (C)
CAG remains the gold standard for CAD [117]. However, except from the occasional attacks, the coronary artery shows normal appearance on resting CAG [116]. Therefore, if a patient is suspected with CAS, the angiography always accompanies with provocation testing to document the coronary spasm [134]. However, it is challenging to evaluate the interplay of the functional aspects and structural ones in patients with coronary artery atherosclerosis and the provocation testing is usually not performed in the presence of a significant epicardial stenosis. But studies approve that spontaneous attacks of coronary spasm can be superimposed on a relevant stenosis, illustrating the missing part in present clinical practice [118].
An ECG of CAS diversifies from completely normal to ST deviation, T, U, R wave abnormality and arrythmia, depending on the severity, duration of episodes and distribution of the spasm artery [119, 120]. Mild seizures could appear just normal in ECG, while total or subtotal spasm of a major coronary artery tend to cause a ST-segment elevation in the leads [120]. However, ST-segment depression also occurs when a less severe, subendocardial myocardial ischemia occurs, when a major artery receiving collaterals or a small artery is completely occluded [122]. These situations include most part of unstable angina/non–ST-elevation myocardial infarction (NSTEMI) cases, thus making ST-segment depression more frequent in CAS [14]. A previous study has shown that 45% of patients with angina at rest and ST-segment depression alone had CAS [123].
In addition to ST-segment changes, a peaked and symmetrical T wave appears in around 50% of cases during a focal proximal coronary spasm [119]. And other wave changes can occur including a delay in the peak and an increase in the height and width of R wave, a decrease in magnitude of S wave and negative U wave may also appear [22]. Various forms of arrhythmia including ventricular premature complex, ventricular tachycardia and/or fibrillation (mostly in case of anterior ischaemia), atrioventricular block (mostly in case of inferior ischaemia), asystole and supraventricular tachyarrhythmias may also be present [121]. In conclusion, ECG takes its advantage in convenience, safety, availability and high-acceptability.
However, even with ambulatory ECG monitoring, the attack may not appear during the monitoring periods, especially when the attack is not frequent [139]. Moreover, ECG does not provide direct or specific evidence of CAS [22]. Thus ECG monitoring is an auxiliary detection in clinic.
Intracoronary imaging, such as optical coherence tomography (OCT) and intravascular ultrasound (IVUS) [117], is capable of addressing not only the morphological changes of intima and media during vasospasm, but also providing information regarding the association of vasospasm with underlying atherosclerotic plaque, fibrous cap disruption, enhanced adventitial vasa vasorum [125, 127, 140], increased PVAT volume [109], inflammation, erosion or thrombus formation [119]. OCT analysis during CAS reveals a typical image of intimal bumps deforming the lumen, combining with intimal gathering (Fig. 3B), without alteration of the intimal area. Medial contraction is presented by an increment in medial thickness [124]. However, intracoronary imaging does not wildly spread in clinical practice due to the complex procedure and low specificity, and each approach has its advantages and disadvantages. OCT has better image quality and resolution, which enables estimations of intima [125, 126]. IVUS has a deeper penetration (4–8 mm versus 2 mm of OCT), which assists accessing perivascular injury. In addition, it is safer and easier to perform IVUS since there is no need to cut off the blood flow, rather than OCT which still needs an interruption [126].
PET is a well-validated technique that can not only help assess coronary
vasomotor function by providing non-invasive, accurate, and reproducible
quantification of myocardial blood flow and coronary flow reserve (CFR) in
humans, but also assist in revelation of coronary spasm tissue image [130].
Intriguingly, inflammatory changes of coronary PVAT assessed by
This non-invasive technique is able to provide indirect functional information about micro vessels and thus assists in diagnosing CAS (Fig. 3D). Ong et al. [132] documented a transient myocardial ischemia by myocardial contrast echocardiography during Ach-induced CAS. Similarly, Arrebola-Moreno et al. [131] has shown the MCE as a systematic evidence for 60% Ach-induced CAS, consistent with single photon emission computed tomography (SPECT) and ECG. However, there are still many limitations in MCE. Due to the restriction of supine position that all the transthoracic echocardiographic images are performed at, it is possible for operators to ignore the minor systolic wall motion [131]. Furthermore, MCE can only detect tissue perfusion in the addition of extra contrast because of the poor back scattering from red blood cells [130], which impairs specificity of the technique. In fact, few available studies of MCE are focused on CAS since the vast majority pay attention to the vasodilatation dysfunction [133].
Recently, non-invasive biochemical markers have been found to associate with the occurrence of CAS [141], including inflammatory factors, Lipoprotein a, Cystatin C, 5-HT, and ET-1 etc. (Table 2, Ref. [18, 29, 30, 49, 50, 66, 96, 101, 141, 142, 143, 144, 145, 146, 147, 148, 149, 150, 151, 152, 153, 154, 155, 156, 157, 158, 159, 160, 161, 162, 163, 164, 165, 166, 167, 168, 169, 170, 171, 172, 173]).
As mentioned above, ED has been demonstrated an underlying mechanism of CAS [45]. Several potential biomarkers are under investigation through this pathogenesis. It has been proved that cystatin C is a reliable marker of kidney dysfunction [142], and renal failure could lead to inactivation of eNOS [145], which is supposed to be a basic pathogenesis in CAS. In fact, 2 clinical studies conducted in Japan and Korea respectively found a promising relationship between a high level of cystatin C and the prevalence of CAS [143, 144]. Nevertheless, there are still questions since renal dysfunction is also related to atherosclerosis and CAD [141], thus further investigations are still required to identify cystatin C as the unique biomarker for CAS. Additionally, xanthine oxidoreductase (XOR) is a rate-limiting enzyme of purine metabolism, catalyzing the oxidation of hypoxanthine to xanthine and of xanthine to uric acid (UA) [146]. It has been elucidated that increased serum UA produces extra ROS [148], resulting in ED [144]. Previous studies also have revealed that XOR-induced ROS can lead to arterial smooth cell proliferation and migration, up-regulate the renin-angio-tensin system to cause vasoconstriction [147]. A recent prediction model including XOR activity showed significantly improved C index (0.771 versus 0.685 of baseline model), net reclassification index (0.612; 95% confidence interval, 0.237–0.986; p = 0.001) and integrated discrimination index (0.098; 95% confidence interval, 0.040–0.156; p = 0.001), and concluded that serum XOR level might be an effective biomarker of CAS [29].
Within the belief of an association between inflammation [64], vasomotor
dysfunction [45] and CAS, researchers keep finding evidence to prove inflammation
markers as potential predictors for CAS, such as hs-CRP and soluble CD40 ligand
(sCD40L). Hung et al. [149] showed that serum hs-CRP concentrations were
correlated independently to CAS in 116 Taiwanese patients with VSA (41% with
focal spasm) versus 66 control patients. Teragawa et al. [150] reported
that increased serum hs-CRP levels were an independent predictor of coronary
microvascular dysfunction by assessing coronary blood flow responses to Ach.
Masami et al. [148] found hs-CRP were significantly increased in the VSA
group (N = 441) than in the atypical chest pain group (N = 197). Ong et
al. [18] found elevated hs-CRP and sCD40L concentrations were significantly
(p
Except from hs-CRP and sCD40L as mentioned above, more biomarkers are found to be associated with CAS via inducing vasomotor dysfunction since decades ago. In 1990s, several laboratory teams viewed successively that the levels of ET-1 increased in blood during the episodes of CAS [30]. And bosentan, an antagonist of endothelin receptor, significantly relieved the severity and frequency of chest pain induced by CAS [152]. Until now, the relationship and pathogenesis of ET-1 in CAS almost disclose, but the clinical utility of ET-1 as a biomarker of the diseases is still on the way. In addition, 5-HT is proved to play an important role in vasocontraction and vasodilation [174]. Researchers found a high level of 5-HT in blood of patient with CAS during episodes as well as nonischemic intervals [153]. A recent study conducted showed an elevation of 5-HT in CAS patients without obstructed arteries [154]. Fortunately, no obvious contradictions occur in various studies so far. But there are still more work needing to be done about 5-HT before it gets to be applied in clinical practice because of lack of fresh evidence and clinical utility tests. Moreover, recent clinical studies found endogenous neuropeptide Y, another effective vasoactive factor, as a potential pathogenesis of CAS especially microvascular constrictions, for both patients without coronary stenosis and patients of ST-elevated myocardial infarction [155]. Intriguingly, as a co-transmitter of norepinephrine, neuropeptide Y is the only biomarker conformed to be correlated to microvascular spasm instead of epicardial ones [141], which indicates the potential differentiation between spasm in two sizes of coronary arteries and underlying different corresponding medication. Obviously, it will take a further more time from confirming the significant correlation between neuropeptide Y and CAS, to identify it as a well-qualified biomarker for clinical use.
Tsuchida et al. [158] have already reported that higher lipoprotein(a) level was associated with coronary vasomotion in VSA. Masami et al. [148] verified the relationship between serum lipoprotein(a) level and VSA again within 441 Japanese patients. Intriguingly, it has been suggested that the lipoprotein(a) level is related to racial and genetic backgrounds [159], which suggest it is difficult to control the lipoprotein(a) level with medications for the management of VSA in some way. However, a large-scale clinical study did not identify obvious relationship between lipoprotein(a) and the vasospastic response to the intracoronary Ach provocation test [157].
Accumulated evidence proves that enhanced RhoK activity plays a central role in the coronary VSMC hypersensitivity, which we have demonstrated in CAS pathogenesis above [50, 162]. Further investigations suggest that RhoK activity in circulating neutrophils maybe a potential biomarker for coronary spasm both in diagnosis and assessment of disease activity and efficacy of treatment [164]. In fact, a previous study showed an immediate, temporary increase of RhoK activity in circulating neutrophils in VSA patients after the Great East Japan Earthquake due to disaster-related mental stress [160]. And the cross-link between stress and CAS is indicated by another experimental study which found excessive sensitivity of VSMC to 5-HT under exposure to sustained elevation of serum cortisol level, resulting in coronary vasoconstrictive responses in pigs in vivo [66]. Moreover, there are some interesting biological coincidence between RhoK and CAS. For example, researchers found a circadian variation of RhoK activity in circulating neutrophils with a peak in the early morning, which showed strong association with alterations in coronary basal tone and vasomotor reactivity and might explain the onset preference of CAS [49]. Furthermore, the suppression effect on RhoK by estrogen may partly account for the higher incidence of vasospastic disorders in postmenopausal women [161]. Finally, RhoK activity in circulating neutrophils combining with the Japanese Coronary Spasm Association (JCSA) risk score substantially appears to be a better prognostic choice in risk stratification of VSA patient as compared with either alone [165]. Taking these issues into consideration, it seems that RhoK activity in circulating neutrophils has a strong potential to be developed into a useful biomarker for CAS with a broad versatility. Further investigations about mechanism, stability, detection time window and simplified measurement are required before it being applied to patients.
Oxidation of low density lipoprotein (LDL) produces ox-LDL, which has been proven as a well-established marker of oxidative disorder [141]. Meanwhile, oxidation of LDL is also a key factor in the process and plays a role throughout atherosclerosis as well as CAS pathogenesis [167]. Recently, malondialdehyde-modified low-density lipoprotein (MDA-LDL) is suggested as another marker of endothelial damage [168]. Observational studies reported a strong correlation between serum MDA-LDL levels and endothelial damage, assessed with flow-mediated dilatation [168]. High MDA-LDL levels harbor a predisposing atherosclerotic segment for coronary spasm to arise, which explains the higher chances of ergonovine-induced CAS [166]. MDA-LDL lowering therapy such as intensive statin treatment [169] may have the potential to treat CAS.
Human microRNAs (miRs) are small, single-stranded, endogenous noncoding RNAs that regulate gene expression at the post-transcriptional level by promoting the messenger RNA (mRNA) degradation or repressing certain coding mRNA translation [127]. It is recently reported that the significant higher expression levels of circulating miR-17-5p, miR-92a-3p, and miR-126-3p show discriminatory power in distinguishing patients with VSA from other CADs [170]. MiRs above are indicated to inhibit eNOS expression directly or via KLF2 gene [170, 171], resulting in impaired NO production and thus leaving the coronary arteries in risk of vasoconstriction, platelet aggregation, low-density lipoprotein metabolic abnormalities and VSMC proliferation disorder [172, 173].
During the last decades, our knowledge of CAS has been increasingly progressed due to advances in the research strategy and diagnostic approaches. This review summarized the clinical risk factors and molecular mechanisms of CAS pathogenesis, and introduce state-of-the-art diagnostic strategies including both clinical imaging approaches and currently under laboratory-testing biomarkers. More mechanistic studies are mandated to further uncover the development of CAS. The seemingly promising biomarkers exist contradictory results, which suggests a long way off from reaching the clinical practice. More rigorous studies are required for further improvement.
ZL and XL searched literatures and completed the original draft. XZ provided clinical comments on this review and provided meaningful discussion on the novel diagnostic approaches. CX and BY drew the figures and provided writing assistance. YS and LL conceived and designed the study, and revised the manuscript. All authors read and approved the final manuscript.
Not applicable.
We would like to express our gratitude to all those who helped us during the writing of this manuscript. Thanks to all the peer reviewers for their opinions and constructive suggestions.
This work was financially supported by the National Natural Science Foundation of China (No. 81871527 and 82070285), the Shanghai Health Committee Research Foundation (20194Y0066), the Zhengyi Scholar Foundation of School of Basic Medical Sciences, Fudan University (No. S25-15), and the Fudan Junzheng Scholar Foundation (No. 2193101011003).
The authors declare no conflict of interest.