






 , Sandra Klein 1,*
, Sandra Klein 1,*
1 Institute of Biopharmaceutics and Pharmaceutical Technology, Department of Pharmacy, Center of Drug Absorption and Transport (C_DAT), University of Greifswald, 17489 Greifswald, Germany
Abstract
Switching from branded to generic extended-release (ER) metoprolol formulations is common practice due to cost considerations. Although generics are approved as bioequivalent, subtle differences in formulation, such as coating composition, may influence drug release under real-life conditions across patients.
This study aimed to evaluate potential differences in drug release between the originator product Beloc-Zok 95 mg and randomly selected generic ER metoprolol succinate formulations available on the German market.
Formulations were assessed under variable, interindividual, physiologically relevant in vitro conditions simulating gastrointestinal transit.
All formulations demonstrated controlled drug release; however, the release profiles were not fully overlapping. One generic formulation consistently exhibited slightly slower release across all test scenarios. These findings indicate that minor differences in release behavior can occur even among bioequivalent ER formulations. While such variations are generally clinically negligible, they may contribute to pharmacokinetic and pharmacodynamic differences in sensitive individuals, potentially explaining reported adverse effects such as bradycardia, hypotension, chest pain, or increased blood pressure when switching to generic products.
The results underscore the importance of considering patient-specific variability when switching from branded to generic formulations. Incorporating physiologically relevant in vitro studies combined with physiologically based pharmacokinetic and pharmacodynamic modeling in future research may improve predictions of in vivo performance and support safer therapeutic decisions.
Graphical Abstract
Keywords
- dissolution
- drug release
- gastrointestinal transit
- generic substitution
- in vitro testing
Metoprolol is one of the most widely used
After oral administration, metoprolol is rapidly and almost completely absorbed from the gastrointestinal tract. Due to extensive first-pass metabolism, its systemic bioavailability is limited, generally remaining below 50%. It is a lipophilic drug with a plasma half-life of about 3–4 hours; however, its clinical effects on heart rate and blood pressure persist longer than predicted by plasma concentrations. Clinically relevant drug interactions must be considered, particularly in multimorbid patients. Metoprolol may enhance or prolong the effects of insulin and oral antidiabetic drugs and may mask symptoms of hypoglycemia; therefore, regular blood glucose monitoring is recommended. Concomitant use with other antihypertensive agents (e.g., nitrates, diuretics, vasodilators) may result in additive hypotensive effects. Combined administration with calcium channel blockers (especially non-dihydropyridines such as verapamil or diltiazem) or other antiarrhythmic agents can increase the risk of bradycardia, atrioventricular conduction disturbances, and cardiac depression. Caution is also advised when metoprolol is used together with sympathomimetic agents, as blood pressure responses may be altered [1].
Metoprolol was first introduced in the 1980s in the form of its tartrate salt (metoprolol tartrate) and Beloc (Astra) and Lopresor (Ciba) were the first commercial preparations to contain this active ingredient. Since the early 1990s, various generic metoprolol tartrate drug products for oral use have been available on the market in addition to the two originator preparations. These include immediate-release (IR) and extended-release (ER) preparations. The latter represent classical ER matrices with 12-hour first-order release kinetics, which generally require twice-daily administration.
In 1990, a novel ER formulation based on metoprolol succinate (metoprolol
hemisuccinate) was introduced under the brand name Beloc-Zok (Astra Zeneca),
offering once-daily dosing with a controlled-release profile consistent with
zero-order kinetics [2]. It was postulated by the developer that the transition
from the IR formulation Beloc to Beloc-Zok would result in a more uniform 24-hour
plasma concentration profile with reduced peak–trough fluctuations, thereby
potentially supporting therapeutic efficacy while minimizing adverse effects. The
Beloc-Zok formulation utilizes a multi-unit pellet system (MUPS) composed of
numerous coated pellets, each functioning as an individual diffusion unit. These
pellets are designed to release metoprolol succinate at a nearly constant rate
for approximately 20 hours, largely independent of physiological variables such
as gastrointestinal pH, motility, or food intake. Considering the plasma
half-life of metoprolol, this system achieves stable drug concentrations
throughout the day, minimizing peak–trough fluctuations and thereby enabling
sustained
From 2001 onward, additional ER formulations of metoprolol tartrate were approved, for which a 24-hour release profile consistent with zero-order kinetics was claimed, despite differences in salt form and formulation technology compared with Beloc-Zok. In these products, metoprolol tartrate is embedded within a hydrophilic polymer matrix that rapidly swells upon oral administration, forming a gel layer at the tablet surface that acts as a diffusion barrier; drug release occurs via diffusion of dissolved metoprolol tartrate through this gel layer and by matrix erosion during gastrointestinal transit, allowing the release kinetics to be approximated by zero-order behavior. With the exception of use in stable, chronic heart failure of mild to moderate severity, these formulations were positioned as generic substitutes [5].
With approvals in 2005 and 2009, generic ER drug products of metoprolol succinate became available that were designed to be therapeutically equivalent to Beloc-Zok. These generics contain the same salt form and employ a significantly different release mechanism compared to the tartrate-based formulations. While both the metoprolol tartrate “twice-daily” and “once-daily” ER formulations use matrix-based technologies, the succinate-based generics are rapidly disintegrating tablets that release multiple coated pellets upon disintegration. These pellets then release the active ingredient in a controlled manner over approximately 24 hours, mimicking the release characteristics of the original Beloc-Zok formulation.
All currently approved generic metoprolol succinate formulations in Europe (EU) have demonstrated bioequivalence to the reference product Beloc‑Zok (EU), based on single-dose pharmacokinetic studies conducted under both fasting and fed conditions, and, in some cases, on multiple-dose studies under fasting conditions, all performed in healthy volunteers. These studies form the basis for regulatory approval but do not directly evaluate clinical interchangeability across diverse patient populations. While regulatory data support the assumption of therapeutic equivalence, real-world evidence indicates that some patients may experience differences in therapeutic response.
Reports from clinical practice worldwide, including patient experiences and anecdotal observations, suggest that some patients, and occasionally healthcare professionals, perceive generic ER metoprolol formulations as not fully equivalent to the branded product. Experience-based reports include variability in blood pressure and heart-rate control, recurrence of arrhythmias, palpitations, dizziness, fatigue, and other general symptoms, with some individuals reporting improvement after returning to the branded formulation or switching between generic manufacturers [6]. While these observations are anecdotal and cannot establish causality, they highlight perceived differences in therapeutic response following substitution. From a biopharmaceutical and clinical perspective, this raises important questions: Can these generics with differing release mechanisms truly serve as interchangeable substitutes in a diverse patient population under real-world conditions? Is the substitution of these formulations, which is frequently driven by cost considerations, rebate contracts, or temporary unavailability due to supply shortages, pharmacologically neutral and clinically safe?
This project aims to address these questions through a series of compendial and physiologically relevant in vitro dissolution studies on selected commercial metoprolol formulations. The objective is to critically evaluate the interchangeability of these products under simulated routine dosing conditions in diverse patient populations, with a particular focus on their predicted in vivo release characteristics and the clinical implications of formulation differences.
Metoprolol tartrate reference substance (# LRAC1767) was obtained from Sigma-Aldrich (St. Louis, MO, USA). The following metoprolol formulations, all of which are marketed in Germany, were randomly chosen and acquired from a local pharmacy: Beloc-Zok 95 mg (# W200480, Recordati, Ulm, Germany), Beloc-Zok forte 190 mg (# W200471, Recordati), Metoprololsuccinat AL 95 (# 00561, Aliud Pharma, Laichingen, Germany), Metoprololsuccinat AL 190 mg (# 20066691, Aliud Pharma), MetoHexal Succ 95 mg (# KL8228, Hexal, Holzkirchen, Germany), Metoprolol-Z AL 200 retard (# 01522, Aliud Pharma). Since a preliminary set of release studies performed with all listed drug products (data not shown) indicated that the release characteristics of the metoprolol succinate ER formulations were not altered by different dose strengths, i.e., 95 mg vs 190 mg, respectively, the subsequent release experiments were only performed with the dosing strength of 95 mg. For comparison with a matrix-based “once-daily” ER formulation, Metoprolol-Z AL (200 mg) was chosen as a representative of this class of products. The compositions of the formulations are given in Table 1. All other chemicals used in the study were of analytical grade and purchased commercially.
| Brand name | Drug | Excipients |
| Beloc-Zok 95 mg | Metoprolol succinate | silicon dioxide, microcrystalline cellulose, ethyl cellulose, hyprolose, hypromellose, sodium stearylfumarate (Ph.Eur.), macrogol 6000, hard kerosene, titanium dioxide |
| Metoprololsuccinat AL 95 mg | Metoprolol succinate | microcrystalline cellulose, macrogol 6000, magnesium stearate (Ph.Eur.) [vegetable], polyacrylate dispersion 30%, povidone K 90, highly dispersed silica, talc, sugar-starch pellets (consisting of corn starch, sucrose and D-glucose), hypromellose, macrogol 6000, titanium dioxide (E 171) |
| MetoHexal Succ 95 mg | Metoprolol succinate | crospovidone, glucose, hypromellose, lactose monohydrate, macrogol 4000, magnesium stearate (Ph.Eur.), corn starch, polyacrylate, silicon dioxide, sucrose, talcum powder, titanium dioxide (E 171), iron(III) hydroxide oxide × H2O (E 172) |
| Metoprolol-Z AL 200 | Metoprolol tartrate | microcrystalline cellulose, ethyl cellulose, hyprolose, hypromellose, macrogol 6000, magnesium stearate (Ph. Eur.), highly dispersed silica, talc, triethyl citrate, sugar-starch pellets (sucrose, corn starch, starch hydrolysate) titanium dioxide (E 171) |
In an initial set of experiments, drug release from the ER metoprolol
formulations was determined according to the official United States Pharmacopoeia
(USP) dissolution method for Metoprolol Succinate ER Tablets [7]. Accordingly,
the drug products were tested in USP apparatus II (DT 728, Erweka, Langen,
Germany) in 500 mL of USP phosphate buffer pH 6.8 [8] maintained at
37.0
In the next set of experiments, the impact of physiological concentrations of bile components on metoprolol release from the ER formulation was studied. For this purpose, an amount of 2.24 g of FaSSIF powder (biorelevant.com Ltd, London, UK) was added to 1 L of the USP phosphate buffer pH 6.8 used in the first part of the study to obtain a pH-modified FaSSIF medium (FaSSIFmod) with a final taurocholate and phospholipid concentration of 3 mM and 0.75 mM, respectively. The experiments were performed in an off-line Paddle apparatus (DT 820, Erweka) and samples of 5 mL were withdrawn by an automated syringe pump (SP 840 S and FRL 800, Erweka) at predetermined time points via a 10 µm poroplast cannula filter (Erweka) using 10 mL glass syringes (Fortuna Optima, Poulten & Graf, Wertheim, Germany). Following an additional manual filtration step through a 0.45 µm cellulose acetate syringe filter (Puradisc 30 CA-S, Puradisc 30 FP 30/0.45 CA-S, Whatman/GE Healthcare Life Sciences, Buckinghamshire, UK) the samples were subjected to HPLC-UV analysis.
The HPLC system consisted of a 1525 binary pump, a 2707 autosampler and a 2489
dual wavelength detector (Waters, Milford, MA, USA). The analysis was performed
on a LiChrospher RP-18e, 5 µm, 250
2.2.3.1 Simulation of Individual pH-Profiles & Residence Times in Different Gastrointestinal Sections
In the second part of the study, drug release of the metoprolol ER formulations
was studied in experiments simulating gastrointestinal pH-profiles of individual
patients. For this purpose, a test setup previously employed to estimate the
variability of in vivo drug release of enteric-coated formulations with
both local and systemic action was applied [9]. This setup is based on
individual gastrointestinal pH-profiles that were derived from an
in vivo study published by Koziolek et al. [10], who used a
telemetric capsule to record pH and temperature profiles of healthy adult fasted
subjects after intake of 200 mL of water. From the gastrointestinal pH profiles
of all subjects included in the study, four individual profiles characterized by
average or “extreme” pH conditions were selected for the in vitro
release experiments (Fig. 1). Experiments were performed as follows: Gastric
conditions were simulated in a Mini-Paddle apparatus [11]. To mimic gastric
conditions, 155 mL of Simulated Gastric Fluid without enzymes (SGFsp) at
a static pH corresponding to the mean gastric pH of the respective subject was
used as dissolution medium. The media temperature was 37.0
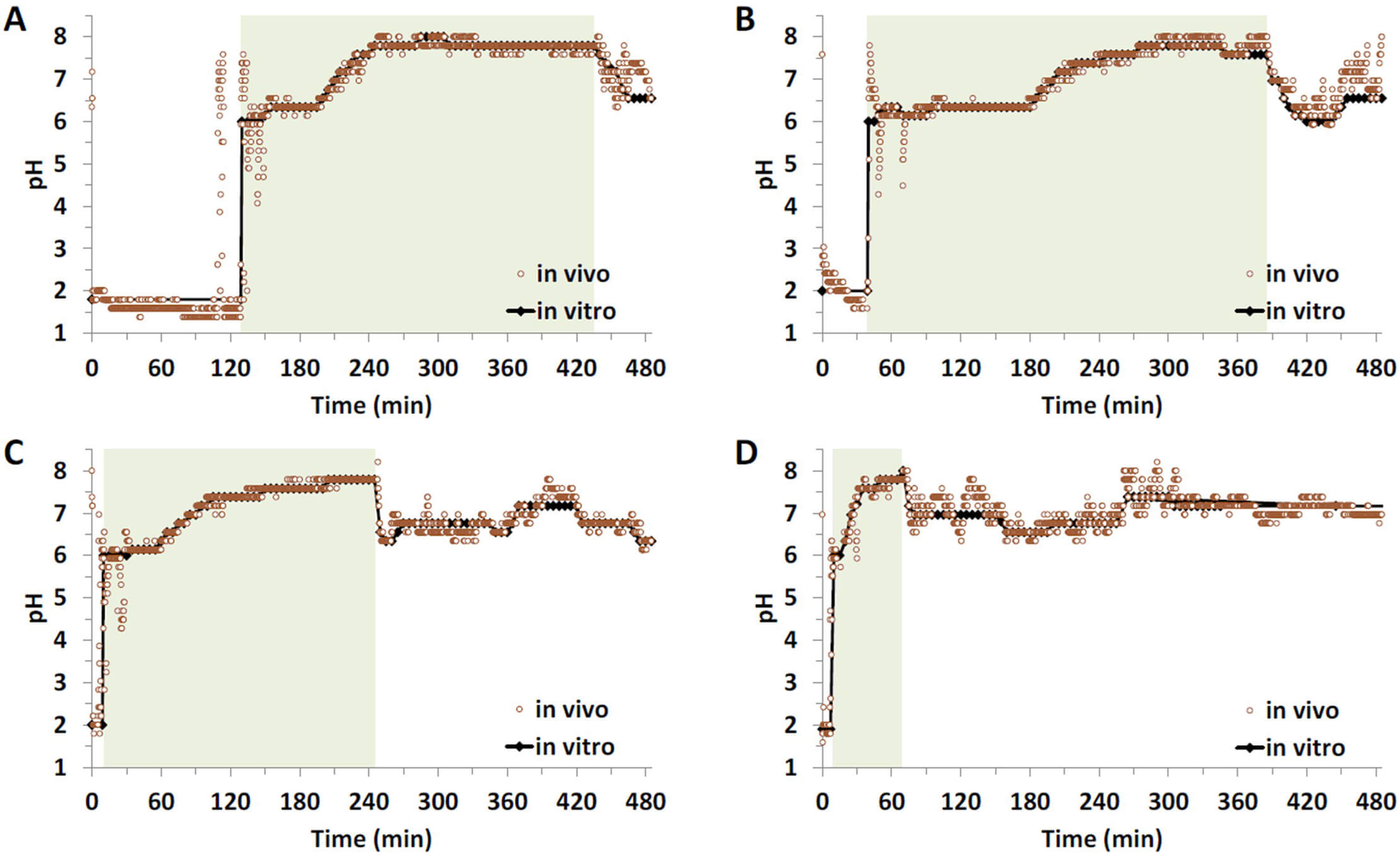 Fig. 1.
Fig. 1.
Gastrointestinal pH-profiles of four selected subjects and their translation into the corresponding in vitro profiles: long duration of high (distal) small intestinal pH (A), long duration of low (proximal) small intestinal pH-conditions (B), average pH-profile (C) and short gastric emptying time (GET) and colon arrival time (CAT) (D), shaded areas indicate the small intestinal transit.
2.2.3.2 Simulation of Gastrointestinal Motility and Continuous and Intermediate Contact with Fluid
In the final part of the study, drug release from the metoprolol ER formulations was investigated under various in vitro scenarios designed to simulate gastrointestinal pH conditions, motility, as well as residence of the dosage form in fluid-filled pockets and “dry” sections of the gastrointestinal lumen. For this purpose, a novel modified Reciprocating Cylinder apparatus (RRT10i, Erweka) was employed. Unlike the compendial instrument, this apparatus offers several new features that allow simulation of varying pH profiles and residence times in different gastrointestinal sections, as well as diverse motility patterns and residence in both fluid-filled and “dry” areas of the gastrointestinal tract [13]. In the present setup, pH conditions of an average healthy adult were simulated. The pH gradient and residence times in the different gastrointestinal sections were kept constant, while motility and periods of no movement were mimicked by varying the agitation speeds and patterns. Additionally, periods of rest were simulated both in fluid-filled pockets (i.e., within the medium) and in “dry” sections (i.e., above the medium surface within the vessel) (Fig. 2). The media used in these experiments as well as the simulated residence times in the different gastrointestinal sections are given in Table 2. To allow for this simulation, the media volume was reduced to 50 mL per vessel.
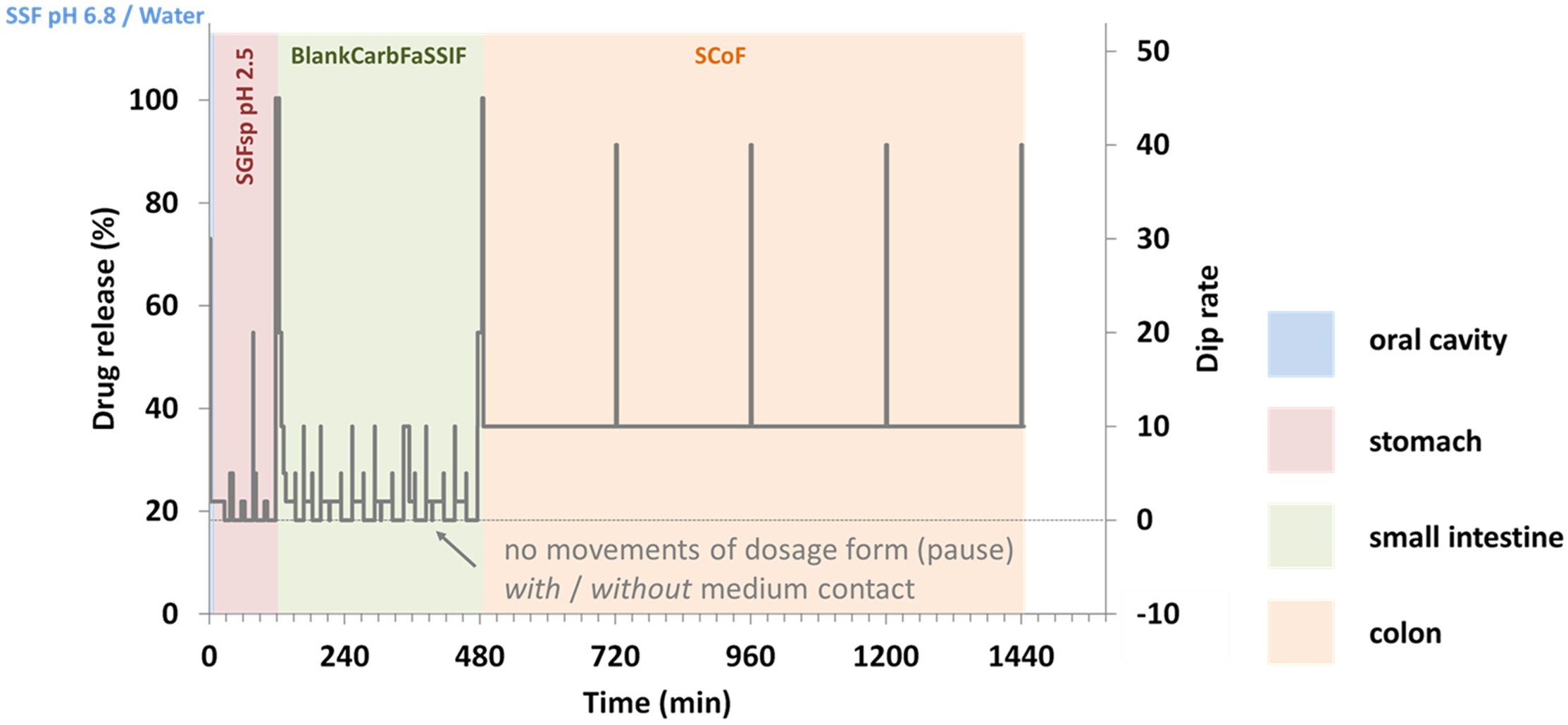 Fig. 2.
Fig. 2.
Detailed test design illustrating simulated gastrointestinal residence times, motility profiles, and fluid contact durations across different gastrointestinal sections.
| Row | Gastrointestinal segment | Medium | Residence time (min) |
| 1 | Esophagus | SSF pH 6.8 + Water (1 + 4) | 2 |
| 2 | Stomach | SGFsp pH 2.5 | 59 |
| 3 | Stomach | SGFsp pH 2.5 | 61 |
| 4 | Duodenum | BlankCarbFaSSIF | 15 |
| 5 | Proximal Jejunum | BlankCarbFaSSIF | 75 |
| 6 | Distal Jejunum | BlankCarbFaSSIF | 90 |
| 7 | Proximal Ileum | BlankCarbFaSSIF | 90 |
| 8 | Distal Ileum | BlankCarbFaSSIF | 90 |
| 9 | Colon | SCoF pH 5.8 | 240 |
| 10 | Colon | SCoF pH 5.8 | 240 |
| 11 | Colon | SCoF pH 5.8 | 240 |
| 12 | Colon | SCoF pH 5.8 | 240 |
In order to assess the impact of the test conditions on drug release from the
metoprolol ER formulations, the mean dissolution times (MDT) for each formulation
and each test condition were calculated as described in previous studies [14, 15].
Briefly, the area under the dissolution curve (AUC) was determined and subtracted
from the total area of the diagram (Atotal), which was obtained by
calculating the area of a rectangle defined by the y-axis, ranging from 0 to
100%, and the x-axis, ranging from 0 min to the time point of last sampling, as
side lengths to obtain the area between the curve (ABC). In the next step, the
MDT was calculated by dividing the ABC by 100%. The calculated MDTs were
analyzed for significant differences by means of a repeated measure analysis of
variance (ANOVA), followed by a Tukey post-hoc test to allow direct comparison
between two test conditions. A p-value
In the first set of experiments, drug release from the ER metoprolol formulations was assessed using the official USP dissolution method for Metoprolol Succinate ER Tablets [7]. Although the official method specifies only four sampling time points (1, 4, 8, and 20 h), it was adapted in this study to allow online concentration measurements every 30 minutes in order to obtain a complete dissolution profile. It should be noted that the formulations tested were not marketed in the United States (US) and therefore may not necessarily comply with USP specifications. Nevertheless, this approach provided a first comparison of the formulations using an official, standardized method. Fig. 3A shows the drug release profiles obtained under these conditions, i.e., in USP phosphate buffer at pH 6.8, expressed as the percentage release of the investigated active pharmaceutical ingredient (API) dose. All formulations exhibited controlled release. Interestingly, not all metoprolol succinate tablets based on ER multiparticulates displayed identical release profiles. The release profiles of Beloc-Zok 95 mg and Metoprololsuccinat AL 95 mg were nearly superimposable, whereas the profiles of MetoHexal Succ 95 mg and Metoprolol-Z AL 200, the latter being the only metoprolol tartrate ER matrix tablet included in this study, differed from those of the (other) succinate formulations but were very similar to each other.
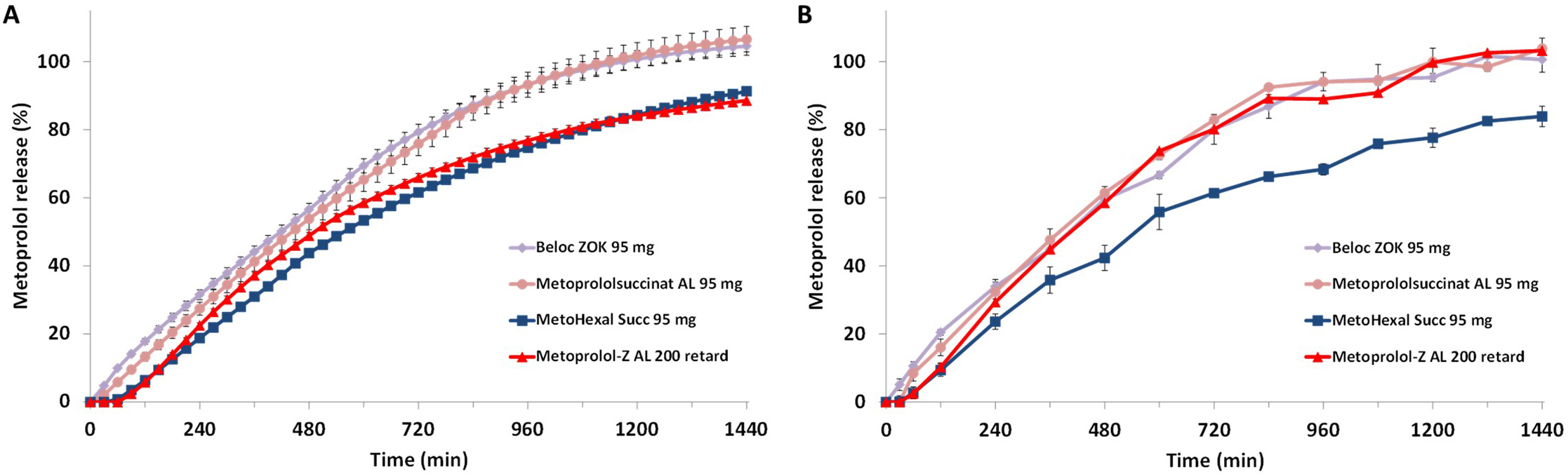 Fig. 3.
Fig. 3.
Metoprolol release from the ER formulations in USP Phosphate
Buffer pH 6.8 without (A) and with (B) FaSSIF powder added (FaSSIF𝐦𝐨𝐝), mean
of n = 3
While the first set of dissolution experiments was designed to provide an
initial overview of drug release from the tested formulations under typical
quality control conditions, the second set was modified to simulate more
biorelevant conditions corresponding to the average fasted small intestine. The
phosphate buffer at pH 6.8 was maintained, and physiologically relevant
concentrations of sodium taurocholate and phospholipid were added, corresponding
to those in Fasted State Small Intestinal Fluid (FaSSIF) [16]. Because the pH of
6.8 differs from that of the original medium (pH 6.5), this variant is referred
to as FaSSIFmod. Drug release profiles obtained in this medium were based on
fewer data points, as online UV quantification was not possible and HPLC analysis
was used instead, resulting in slightly different profile appearances (Fig. 3B).
In FaSSIFmod, Beloc-Zok 95 mg and Metoprololsuccinat AL 95 mg again
displayed nearly superimposable release profiles, with no statistically
significant differences compared to those in phosphate buffer pH 6.8. Similarly,
no statistically significant differences were observed for MetoHexal Succ 95 mg
when comparing the two experimental conditions. In contrast, Metoprolol-Z AL 200
showed a slightly faster release of the API starting at approximately 2 hours and
a higher cumulative release at the end of the test period compared to phosphate
buffer pH 6.8. Under these test conditions, the release profile of this
formulation rather resembles those of Beloc-Zok 95 mg and Metoprololsuccinat AL
95 mg. The difference in API release between the two media for Metoprolol-Z AL
200 was statistically significant (p
The second and more extensive part of the study aimed to investigate the drug release of the ER formulations under gastrointestinal conditions simulating fasted administration in individual patients, in order to better represent routine dosing across diverse patient populations in vitro. For this purpose, individual gastrointestinal pH profiles and residence times in the corresponding gastrointestinal sections were incorporated into the in vitro test. As the preceding test series had shown no significant influence of bile salts and phospholipids on the rate or extent of API release from the formulations under investigation, namely the metoprolol succinate formulations, the addition of bile salts to the gastric and small intestinal media was omitted in all individualized release experiments.
Although the individualized dissolution studies were designed to mimic true gastrointestinal transit profiles of an ER formulation in different patients, considering variations in pH conditions and residence times across distinct gastrointestinal segments to reflect highly diverse patient scenarios, an overall trend comparable to that observed in the preceding experiments was identified (Fig. 4A–D). Under all test conditions, Beloc-Zok 95 mg and Metoprololsuccinat AL 95 mg exhibited nearly identical release profiles. In contrast, the release profile of MetoHexal Succ 95 mg differed markedly but also showed no variability in response to the applied test conditions. Metoprolol-Z AL 200, the metoprolol tartrate formulation, also demonstrated controlled drug release under all test conditions but marginal differences in the shape of the release profiles across the different test scenarios.
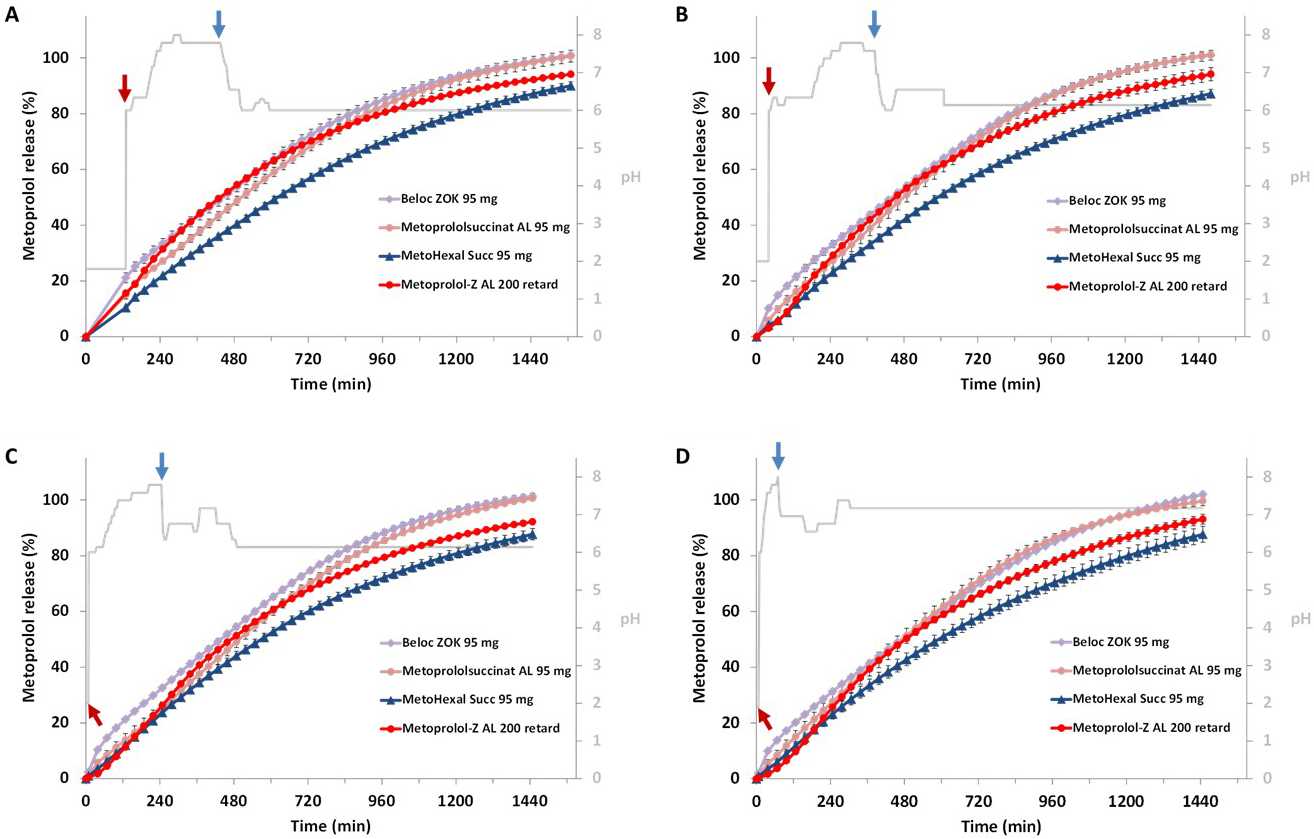 Fig. 4.
Fig. 4.
Metoprolol release from the tested ER formulations when
mimicking gastrointestinal pH profiles of individual subjects with a long
duration of high (distal) small intestinal pH (A), long duration of low
(proximal) small intestinal pH (B), an average gastrointestinal pH profile (C)
and a short gastric residence time and small intestinal passage (D); red arrows
indicate gastric emptying and blue arrows colon arrival; mean of n = 3
Also, in the second set of individualized release experiments, which assumed pH values and transit times representative of an average adult but simulated specific motility profiles as well as distinct resting phases of the dosage form in fluid-filled gastrointestinal segments or in comparatively “dry” regions, no relevant alterations in the release profiles were observed (Fig. 5A,B). All investigated formulations exhibited release behavior that was largely independent of patient-specific motility conditions and resting phases and closely resembled the profiles obtained under simulated average adult gastrointestinal passage conditions as shown in Fig. 4C.
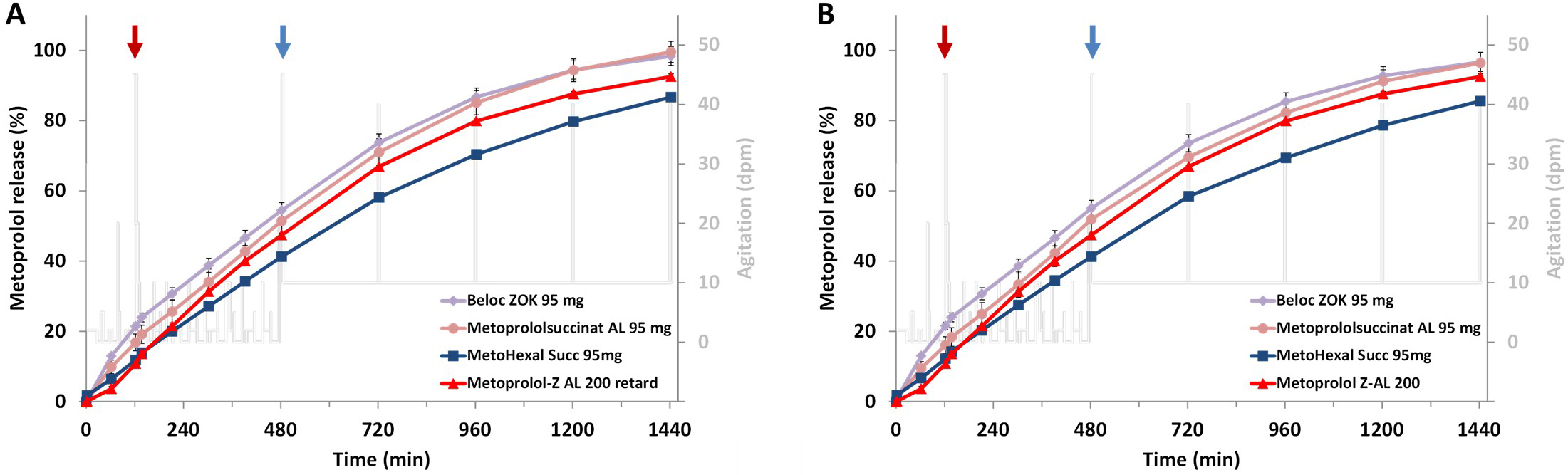 Fig. 5.
Fig. 5.
Metoprolol release from the ER formulations simulating a
discontinuous gastrointestinal passage in a healthy average adult with sequences
of no movement of the dosage form (agitation = 0 dpm) with medium contact while
pausing (A) and without medium contact while pausing (B), red arrows indicate
gastric emptying and blue arrows colon arrival; mean of n = 3
Considering all results obtained from the release experiments, all investigated formulations exhibited controlled drug release under all test conditions, with minimal dependence on simulated patient-specific factors. If variability was observed, it tended to be most apparent for the Metoprolol-Z AL 200 ER tablet, which was included in the test series solely for comparative purposes. Statistical analysis of the influence of various test parameters on drug release from this dosage form confirmed these observations. As shown in Fig. 6, statistically significant differences were observed, in particular when comparing drug release in FaSSIFmod, a bile salt- and phospholipid-containing medium, with all other test conditions. In contrast, no such differences were observed for MetoHexal Succ 95 mg, indicating that this formulation exhibited the most robust drug release among the metoprolol succinate ER formulations investigated. However, it should be noted that, unlike the other two metoprolol succinate formulations, this formulation did not achieve complete (i.e., 100%) release of the labeled drug dose in any of the experiments. These results were supported by a series of content uniformity and assay determinations conducted for each of the investigated formulations in parallel with each release experiment, using the respective analytical methods. Although not discussed in detail herein, the available data suggested that the drug content of the MetoHexal Succ 95 mg tablet was uniform and greater than the amount released after 24 h.
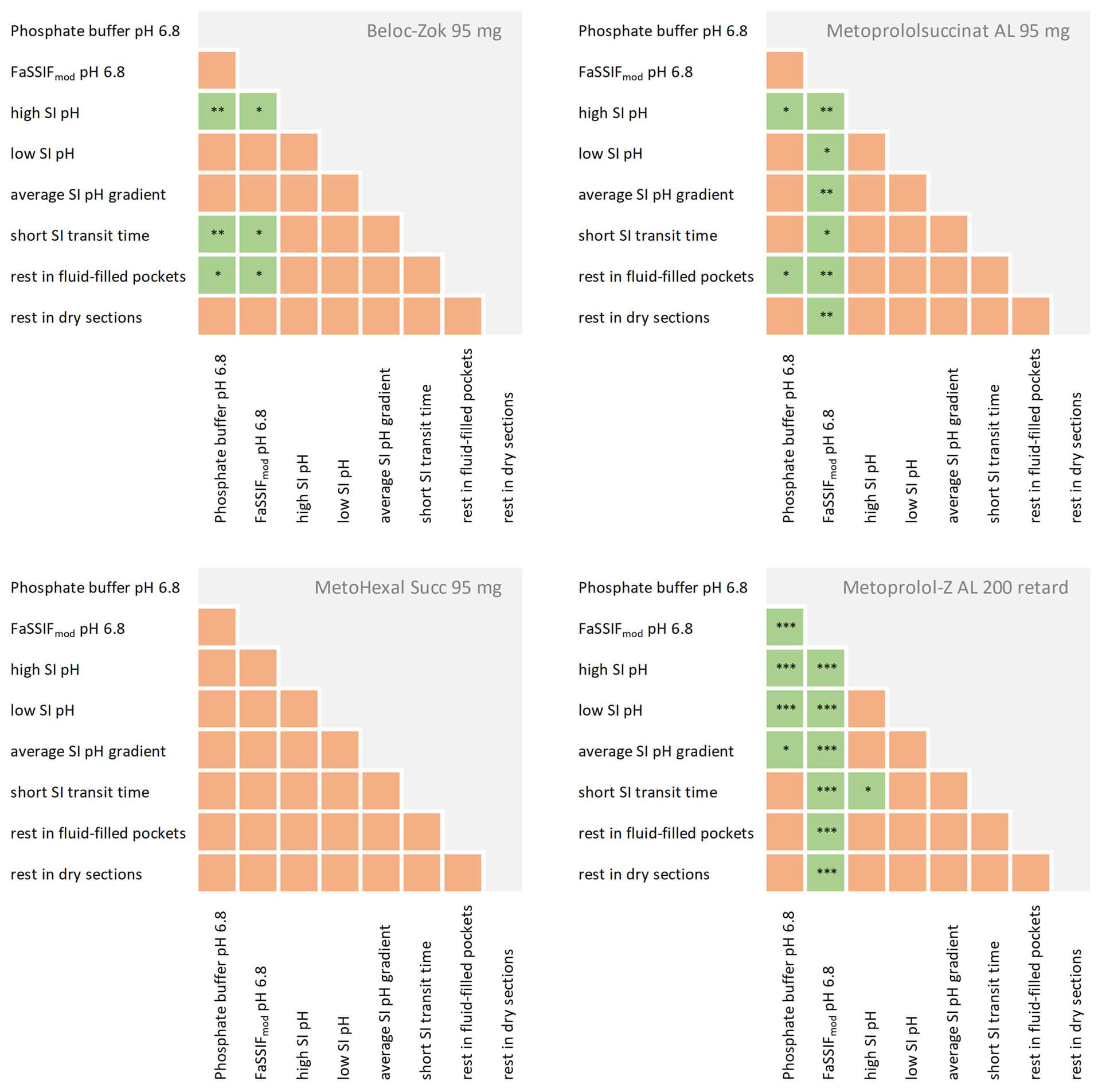 Fig. 6.
Fig. 6.
Statistical evaluation of the impact of different simulated
in vivo conditions on drug release from the metoprolol ER formulations,
statistically significant effects are highlighted in green (* p = 0.01 to 0.05
(significant), ** p = 0.001 to 0.01 (very significant), *** p
The discussion regarding the interchangeability of metoprolol ER formulations has been somewhat of a never-ending story. In particular, since the market introduction of Beloc-Zok, now over 35 years ago, questions and debates on this topic have repeatedly arisen. These issues tend to resurface whenever a new generation of generics enters the market. In the late 1990s and early 2000s, a major problem was that, due to cost constraints, Beloc-Zok was replaced by less expensive generics, which exhibited completely different release kinetics because, unlike Beloc-Zok, they were designed for twice-daily administration [5]. The patient-reported complaints in this context were entirely understandable, as administration of these metoprolol tartrate-based ER formulations according to the Beloc-Zok dosing schedule resulted in a markedly altered plasma concentration profile due to the omission of the second daily dose.
Once this issue was recognized, novel metoprolol tartrate ER formulations with purported zero-order release kinetics were introduced, reigniting the debate on interchangeability. Overall, under simulated gastrointestinal passage conditions in average adults, these formulations demonstrated release profiles similar to the corresponding Beloc-Zok formulation, despite differences in salt form [17]. However, it remained unclear whether this similarity would also be observed under conditions expected in individual patients. At that time, the issue was not further pursued, as German legislation mandated the substitution of brand-name drugs, which could only be avoided in exceptional cases.
With the market introduction of a new generation of generic products formulated according to the same concept as Beloc-Zok, namely tablets composed of compressed ER multiparticulates containing metoprolol succinate and designed as diffusion cells that deliver the drug at a relatively constant rate largely independent of physiological variations in the gastrointestinal tract, this question became relevant again and formed the basis of the present study. Based on the fact that the current “latest” generation of ER metoprolol succinate generics contains the same metoprolol salt, follows the same overarching formulation principle, and that the generic products available on the German market have been shown to be pharmacokinetically equivalent to the originator product Beloc-Zok, such a question initially appears to be entirely irrelevant. However, closer examination of the composition of these generics, two of which were randomly selected as components of the present experimental series, reveals that the coatings of the discrete units of these dosage forms consist of different polymers (Table 1). Under conditions present in the human gastrointestinal tract, these differences may result in at least slightly different drug release rates. For a highly soluble and highly permeable drug like metoprolol, for which in vivo release is the primary determinant of absorption, this can lead to differing plasma concentration profiles. In a mechanistic modeling study by Basu et al. [18], physiologically based pharmacokinetic simulations showed that changes in drug release rates due to formulation variables (such as the release-controlling polymer) can result in significant differences in maximum plasma concentration (Cmax) and other pharmacokinetic parameters, even when the active ingredient is the same, indicating that polymer and release characteristics are critical determinants of systemic exposure for ER formulations containing metoprolol succinate. Furthermore, experience from the US market has shown that different generic ER metoprolol succinate formulations were associated with an increased incidence of adverse effects in routine clinical use, leading to the eventual withdrawal of some of these products from the market [6, 19, 20].
Notably, despite differences in the coatings of the drug-releasing multiparticulates, Beloc-Zok 95 mg (ethylcellulose) and Metoprolol Succinate AL 95 mg (polyacrylate) both exhibited very similar release profiles under all test conditions. In contrast, the extent and rate of drug release from MetoHexal 95 mg was consistently lower under all experimental conditions, although, as for Metoprolol Succinate AL 95 mg, the release in this formulation is also controlled by a polyacrylate coating according to the declared excipients. In the present test series, only these two randomly selected generic formulations were investigated, and the objective was not to perform a statistical comparison with the originator product, as they are approved as bioequivalent. Rather, the study aimed to replicate realistic everyday in vivo conditions with a high degree of variability, in order to better interpret patient-specific complaints and reported adverse effects, such as bradycardia, hypotension, chest pain, increased blood pressure, etc., associated with generic formulations.
Considering the differences observed in some drug release profiles and the large number of generic formulations currently available for Belok-Zok, it can be assumed that slightly different in vivo release patterns in individual patients are likely and difficult to predict. This may lead to minor differences in absorption and the resulting pharmacokinetic parameters, which may be clinically irrelevant for many patients, but in selected individuals could result in significant effects if pharmacokinetic differences impact critical pharmacodynamic parameters. This could explain the issues frequently reported in the lay press and raises the question, particularly for well-controlled patients, of whether it is advisable to follow market-driven cost considerations and switch such patients to generic metoprolol formulations. Beyond potential adverse effects, such switches may also cause considerable uncertainty among patients and negatively affect adherence, which can further compromise therapeutic outcomes.
One approach to improving the therapeutic safety of generic drugs is the use of physiologically relevant in vitro release methods that do not merely aim to simulate the conditions of a standardized clinical study but can also reflect variable physiological conditions in individual patients, as employed in the present study. In combination with physiologically based pharmacokinetic and pharmacodynamic models, these methods are expected to provide better predictions of the spectrum of in vivo performance of such formulations in the future.
Some limitations of this study should be considered when interpreting the results. The primary objective was to investigate and compare the release behavior of selected metoprolol ER formulations under standardized, physiologically relevant in vitro conditions, in order to better understand potential reasons why individual patients may respond differently to the substitution of metoprolol ER products. The aim was not to establish in vitro–in vivo correlations or to demonstrate bio(in)equivalence. Accordingly, no IVIVC validation against clinical pharmacokinetic data was performed, and for the same reason no formal similarity testing of the dissolution profiles was conducted. The set of formulations investigated represents only a limited subset of the metoprolol ER products available on the market. The products were selected as a pragmatic cross-section rather than as a comprehensive survey, also reflecting variability in market availability and substitution practices that may limit the accessibility of specific formulations at a given time. In addition, only a single batch of each formulation was investigated, which may not fully capture potential batch-to-batch variability. An increased number of tested products would likely lead to a broader range of observed release profiles. The data presented already indicate variability and trends in release behavior across the included formulations, but they do not claim to comprehensively represent the full extent of variability across all available products. Finally, while the applied in vitro setup reflects key physiological aspects of the gastrointestinal environment as well as its variability, it cannot fully capture the complexity and interindividual differences of in vivo conditions and, in the present study, did not account for additional influences that may arise from the administration of the dosage forms with food. In light of these limitations, an increase rather than a reduction in variability could theoretically be expected. However, as this study is limited to an in vitro evaluation under controlled conditions designed to simulate selected in vivo scenarios, such effects remain speculative and are therefore not further elaborated here; the interpretation of the results should remain within the scope of the experimental setup.
Physiologically relevant in vitro release studies reveal that small differences in drug release among ER metoprolol formulations are possible despite bioequivalence. While these variations are generally minor, they may affect sensitive patients, highlighting the need for careful consideration when switching from branded to generic medicines. Future studies should incorporate such in vitro testing in combination with physiologically based pharmacokinetic and pharmacodynamic modeling to better predict in vivo performance and support safer therapeutic decisions.
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
FK: Formal analysis, Investigation, Methodology, Visualization. SK: Conceptualization, Project administration, Writing – original draft, review and editing. Both authors contributed to editorial changes in the manuscript. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflicts of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.