







 , Hanrui Sun 1,2,†, Jiabin Zhou 3, Jiayu Shi 2, Peng Xu 4, Chen Wang 1, Chuchu Chen 5, Pengyang Gu 2, Xinglin Wang 6, Qing Gong 1,2, Anzhen Xu 1,2, Yanbo Li 1,2, Qi Lu 1,2,*
, Hanrui Sun 1,2,†, Jiabin Zhou 3, Jiayu Shi 2, Peng Xu 4, Chen Wang 1, Chuchu Chen 5, Pengyang Gu 2, Xinglin Wang 6, Qing Gong 1,2, Anzhen Xu 1,2, Yanbo Li 1,2, Qi Lu 1,2,*
1 Medical School, Nantong University, 226000 Nantong, Jiangsu, China
2 Department of Cardiology, Affiliated Hospital of Nantong University, 226000 Nantong, Jiangsu, China
3 Department of Cardiovascular Medicine, Affiliated Wuxi People’s Hospital of Nanjing Medical University, Wuxi People’s Hospital, 214023 Wuxi, Jiangsu, China
4 Department of Cardiology, Nantong Sixth People’s Hospital, 226000 Nantong, Jiangsu, China
5 Department of Cardiology, Jiangyin Hospital of Traditional Chinese Medicine, 214400 Wuxi, Jiangsu, China
6 Department of Cardiology, Fujian Medical University Union Hospital, 350001 Fuzhou, Fujian, China
†These authors contributed equally.
Abstract
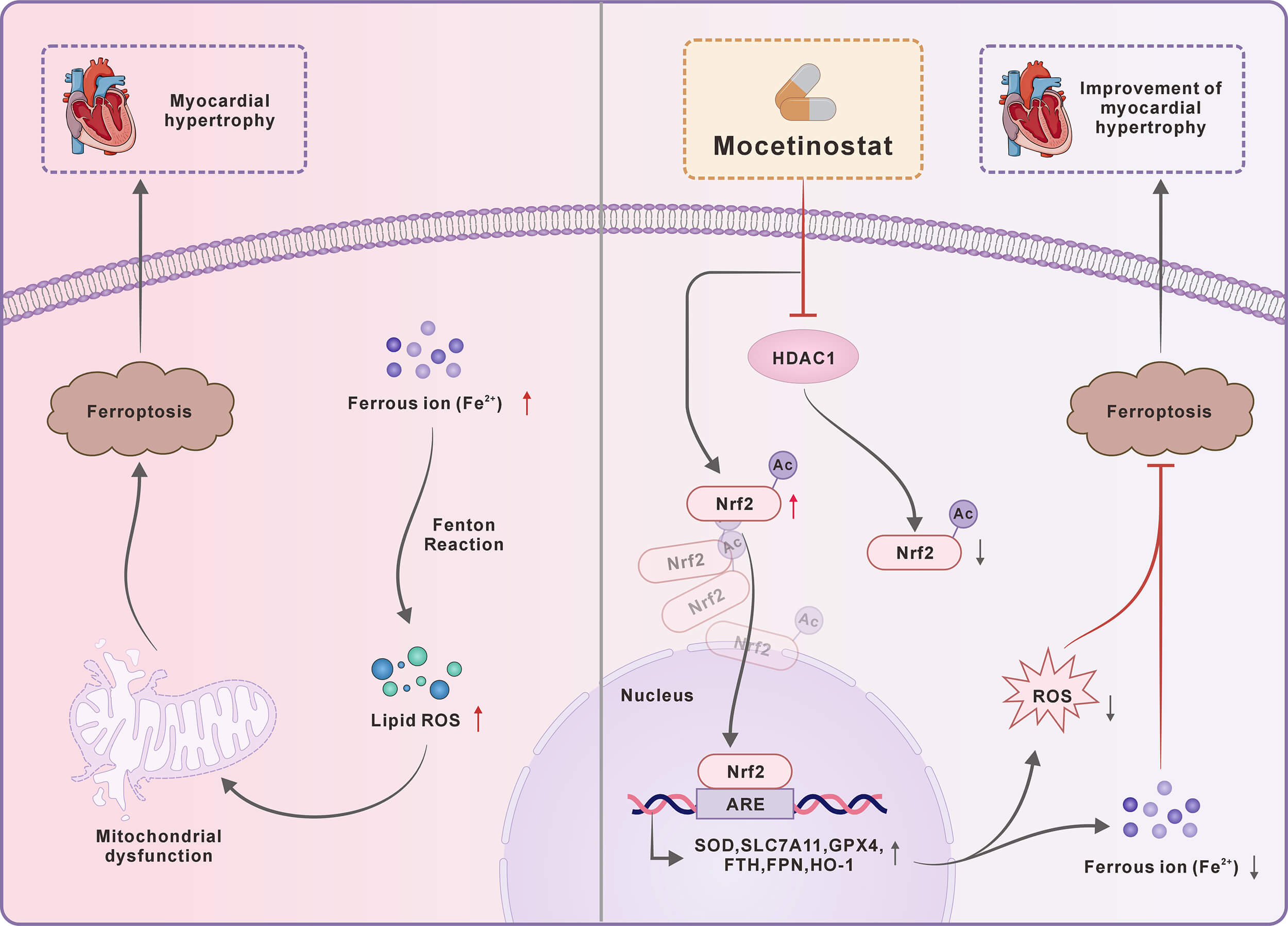
Histone deacetylase 1 (HDAC1) exacerbates ventricular remodeling and heart failure by promoting myocardial peroxidative damage. Thus, this study aimed to investigate whether the HDAC1 inhibitor mocetinostat alleviates pathological cardiac hypertrophy via suppressing ferroptosis and to elucidate the potential mechanisms involving the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway.
Primary cardiomyocytes were stimulated with phenylephrine (PE) to induce hypertrophy and ferroptosis in vitro, with or without mocetinostat treatment. The Nrf2 inhibitor ML385 was used to verify pathway specificity. In vivo, a mouse model of pressure-overload-induced cardiac hypertrophy was established using a transverse aortic constriction (TAC)-induced approach. Mocetinostat was administered to evaluate its therapeutic effects. Ferroptosis markers including lipid peroxidation, iron accumulation, and levels of ferroptosis-related proteins, were assessed. The acetylation and nuclear translocation of Nrf2, as well as the expression of the associated downstream targets (Solute carrier family 7 member 11 (SLC7A11), glutathione peroxidase 4 (GPX4), ferroportin, ferritin heavy chain 1 (FTH1), heme oxygenase-1 (HO-1)), were analyzed.
Mocetinostat treatment significantly ameliorated PE-induced cardiomyocyte hypertrophy and ferroptosis in vitro and attenuated TAC-induced cardiac hypertrophy and fibrosis in vivo. Mechanistically, mocetinostat facilitated Nrf2 acetylation and promoted Nrf2 nuclear translocation, leading to the transcriptional activation of downstream antioxidant targets and subsequent inhibition of lipid peroxidation. The protective effects of mocetinostat were abrogated by the Nrf2 inhibitor ML385.
This study demonstrates that mocetinostat attenuates pathological cardiac hypertrophy by inhibiting ferroptosis through activation of the Nrf2 pathway . These findings indicate the potential of mocetinostat as a therapeutic strategy for delaying heart failure progression.
Graphical Abstract
Keywords
- myocardial hypertrophy
- ferroptosis
- histone deacetylase inhibitors
- nuclear factor erythroid 2-related factor 2
Myocardial hypertrophy is an initial adaptive response to cardiac injury or stress. Early pathological hypertrophy is characterized by concentric remodeling and diastolic dysfunction, coupled with cardiomyocyte death, microvascular rarefaction, and widespread perivascular/interstitial fibrosis driven by activation of collagen type I (COL1) and myofibroblasts [1]. If left unmanaged, this chronic, dysfunctional compensation triggers progressive cardiac remodeling, leading to systolic dysfunction, chamber dilation, and congestive heart failure [2]. Heart failure severely impairs quality of life while imposing high readmission rates and substantial economic costs [3]. Given the limited efficacy of current therapies, novel strategies are essential to attenuate disease progression and improve patient prognoses.
Ferroptosis, a widely studied unique cell death mechanism in cell biology, has distinct characteristics and is tightly regulated by cellular iron levels [4]. Unlike conventional cell death pathways (e.g., apoptosis and necrosis), ferroptosis is uniquely characterized by extensive lipid peroxidation and distinct signaling cascades. Morphologically, it is characterized by shrunken mitochondria, elevated bilayer density, disrupted outer membrane, and diminished cristae [5]. Mechanistically, intracellular accumulation of ferrous iron (Fe2+) drives the Fenton reaction to produce highly reactive hydroxyl radicals [6] and excessive reactive oxygen species (ROS), which then trigger lethal lipid peroxidation. Impairment of core antioxidant defenses, specifically glutathione (GSH) and glutathione peroxidase 4 (GPX4), critically exacerbates this oxidative damage [7]. The process is tightly regulated by multiple factors [e.g., GPX4, ferritin heavy chain 1 (FTH), heme oxygenase-1 (HO-1)] and the acyl coenzyme A synthase long-chain family member 4 pathway [7,8,9]. Crucially, ferroptosis is implicated in the pathogenesis of myocardial hypertrophy. Mice with transverse aortic constriction (TAC) or isoproterenol-induced cardiac hypertrophy exhibit reduced myocardial GPX4 and active ferroptosis [10]. Solute carrier family 7 member 11 (SLC7A11) normally suppresses ferroptosis via cystine-dependent GSH synthesis, and its depletion exacerbates angiotensin II-induced hypertrophy and fibrosis, establishing ferroptosis as a core mechanism in pathological remodeling [11]. Furthermore, nuclear receptor coactivator 4 drives ferroptosis via ferritinophagy-mediated iron release, while its cardiomyocyte-specific knockout attenuates TAC-induced hypertrophy and heart failure [12]. Collectively, these findings establish ferroptosis as a promising therapeutic target for mitigating pathological myocardial hypertrophy.
Acetyltransferase promotes the enzymatic addition of acetyl groups to lysine residues. Reversible lysine acetylation is a key post-translational modification linked to numerous physiological and pathological cellular functions (e.g., transcriptional modulation, DNA repair, cell cycle control, signaling, protein folding, and cell death) [13]. This modification influences protein function by governing stability, enzymatic activity, intracellular distribution, and coordination with other post-translational events [14]. Deacetylation acts as the reverse of acetylation, dynamically regulating protein function by removing acetyl groups from lysine residues, a key post-translational modification. We previously established that histone deacetylase 1 (HDAC1)—a class I enzyme mainly found in the nucleus—is critical for regulating ferroptosis in heart failure. Concurrently, the authors identified significant HDAC1 upregulation in a TAC-induced heart failure model [15]. Recent evidence indicates that HDAC1/2 overexpression drives cardiac dysfunction, with HDAC1 specifically regulating myocardial development and contractility, whereas class I HDAC inhibitors attenuate pathological hypertrophy, delay pressure overload-induced remodeling, and improve hemodynamic and cardiac function [16]. These inhibitors also improve cardiac function in murine myocardial ischemia [17]. Therefore, to delineate how HDAC1 modulates ferroptosis during heart failure progression, the HDAC1 inhibitor mocetinostat (MGCD0103) was selected for subsequent investigation.
Mocetinostat is a selective class I HDAC inhibitor, originally developed as an anticancer agent. It has been investigated in clinical trials for various hematological malignancies, including Hodgkin’s lymphoma, acute myeloid leukemia, and myelodysplastic syndromes. Mocetinostat shows the strongest inhibition of HDAC1, with roughly twice the potency of HDAC2, while displaying weak to no activity against other HDAC isoforms [18]. A recent investigation demonstrated that mocetinostat modulates the interleukin-6 (IL-6)/signal transducer and activator of transcription 3 (STAT3) pathway in fibroblasts and attenuates cardiac fibrosis in mice with myocardial infarction [19]. Moreover, a study in rats showed that mocetinostat administration following TAC improved left ventricular function and attenuated remodeling with a reduction in cardiomyocyte apoptosis and fibrosis [20]. Mechanistically, HDAC1 suppresses 6-phosphofructokinase, muscle type (PFKM) gene expression in doxorubicin (DOX)-treated cells by inhibiting H3K27 acetylation, which plays a key role in glycolytic flux. By relieving HDAC1-mediated inhibition, mocetinostat increases PFKM expression, which enhances both glycolysis and oxidative phosphorylation, thereby improving mitochondrial respiration and protecting against DOX-induced cardiotoxicity [21]. Therefore, we hypothesize that inhibition of HDAC1 may exert beneficial effects on pathological cardiac hypertrophy.
Nuclear factor erythroid 2-related factor 2 (Nrf2) notably regulates the cellular response to ferroptosis [22]. Physiologically, Nrf2 undergoes continuous ubiquitination and degradation mediated by Kelch-like ECH-associated protein 1 (Keap1), thereby maintaining its low basal expression level [23]. Following cellular stress, conformational inactivation of Keap1 leads to Nrf2 stabilization, nuclear translocation, and subsequent activation of downstream defense genes to restore homeostasis [24]. Nrf2 fundamentally defends against ferroptosis by upregulating the SLC7A11 antiporter, which promotes glutathione synthesis and enables GPX4 to suppress lethal lipid peroxidation [25]. Furthermore, the Nrf2/HO-1 pathway activation confers resistance to oxidative damage, an effect attenuated by HO-1 suppression [26]. Nrf2 also prevents iron toxicity by inducing ferritin and ferroportin (FPN) to maintain intracellular iron homeostasis [27,28]. Notably, acetylation disrupts the Nrf2-Keap1 interaction, reducing Nrf2 degradation and bolstering these cellular defenses [29]. Nevertheless, the specific mechanisms of HDAC1 in regulating ferroptosis in pathological cardiac hypertrophy remain undefined. This study provides the first evidence that the HDAC1 inhibitor mocetinostat mitigates pathological cardiac hypertrophy by suppressing ferroptosis, with the involvement of the Nrf2 pathway.
Pathogen-free male C57BL/6 mice (8 weeks, 20-25 g) were supplied by the Experimental Animal Center of Nantong University and housed in a climate-controlled facility (21 ± 1 °C, 55–60% humidity) with free access to food and water. All animal protocols got approval from the Animal Ethics Committee of Nantong University and followed the Guide for the Care and Use of Laboratory Animals.
All mice were specific-pathogen-free (SPF) and healthy prior to surgery; no signs of illness were observed before the experiment. Successful TAC was confirmed by echocardiography 6 weeks after surgery, as evidenced by a significant increase in IVSd and in the left ventricular internal diameters (LVIDd and LVIDs), together with a marked reduction in EF and FS. To ensure a high success rate, all TAC surgeries were performed by a single experienced operator who had successfully completed more than 800 TAC procedures.
Based on scientific randomization principles, mice were evenly grouped (n = 6): TAC surgery + corn oil, TAC surgery + mocetinostat, sham operation + corn oil, and sham operation + mocetinostat. TAC was used to induce and confirm left ventricle (LV) overload. Following a 6-h surgical observation period, the mice were returned to their home cages. Anesthesia was induced via facemask with 4% isoflurane (cat. no. R510-22, RWD Life Science, Shenzhen, China) and maintained with 1.5% isoflurane. Subsequently, a median sternotomy was performed to expose the thoracic cavity, the thymus was removed, and the aortic arch and its branches were identified. At the site of stenosis, a 27-gauge needle was positioned parallel to a 5-0 suture located beneath the aortic arch. Uniform aortic stenosis was created by ligating the aorta over a needle, which was subsequently removed. The thoracic cavity was closed using a purse-string suture technique. Sham controls underwent identical surgical preparation and aortic suture placement, but without ligation. Postoperatively, the animals were observed on a daily basis, with free access to food and water.
Mocetinostat was administered at 10 mg/kg, as previously described [19]. Preliminary results indicated that a dose of 10 mg/kg was both safe and effective. The compound was injected intraperitoneally every other day beginning on postoperative day 2. Overall animal survival exceeded 80% in each group. Surviving mice at 6 weeks after surgery were subjected to echocardiography. After inducing deep anesthesia with 5% isoflurane for at least 1–2 min, we euthanized the mice by cervical dislocation. Specifically, upon confirmation of complete unconsciousness, cervical dislocation was rapidly performed, and death was verified prior to experimental procedures.
Postoperative care and analgesia: Mice received buprenorphine (0.1 mg/kg, subcutaneous) every 12 h for 48 h post-surgery to alleviate pain.
Exclusion criteria were established a priori: (1) accidental death during surgery, (2) failure to develop TAC-induced cardiac hypertrophy (no significant increase in HW/BW ratio or echocardiographic evidence of hypertrophy), (3) body weight loss >20%, and (4) reaching humane endpoints (severe distress, moribund state). Of the 7 mice initially assigned to the TAC group, one died of acute heart failure within 30 min after surgery and was excluded. No other exclusions occurred in the other groups. The final numbers analyzed were: Sham (6), Sham + Moc (6), TAC (6), TAC + Moc (6).
Upon anesthetization, mice were placed supine and maintained at a constant temperature throughout the procedure. Heart rate (HR) was continuously monitored. Ultrasound coupling gel was applied to the anterior chest wall, and cardiac images were obtained with the Vevo 2100 high-resolution ultrasound system (FUJIFILM VisualSonics, Toronto, Canada) fitted with an FMS-250 transducer operating at 13–24 MHz. The standard parasternal long-axis view of the LV was obtained to measure various parameters of LV structure and function. Parameters of LV systolic function were calculated using built-in software. Three to five consecutive cardiac cycles were recorded for each measurement, and results were averaged to increase accuracy.
To evaluate cardiomyocyte morphology, mouse cardiac tissues were subjected to H&E staining. After dehydration using a graded ethanol series (100%, 95%, 80%, and 70%), samples were cleared in 100% anhydrous xylene, and embedded in paraffin at 60 °C. The blocks were sectioned (5 μm) using a microtome (Thermo Fisher Scientific, Waltham, MA, USA). Following deparaffinization in 100% anhydrous xylene, the sections underwent rehydration through gradient ethanol series (100%, 95%, 80%, and 70%), and then rinsing in distilled water. For H&E staining, slides underwent hematoxylin treatment, 1% hydrochloric acid/70% ethanol differentiation, 0.1% ammonia water blueing (2 min), and eosin counterstaining. After that, following rapid dehydration using an ascending ethanol series (80%, 95%, 100%), the sections were subjected to xylene clearing and synthetic resin mounting. Images were obtained utilizing a light microscope (Axio Lab.A1, Zeiss, Oberkochen, Germany).
We performed Masson’s trichrome staining on mouse heart sections for cardiac tissue fibrosis assessment. Briefly, deparaffinized sections underwent Weigert’s hematoxylin staining (10 min) and rinsed thoroughly. Then sections were incubated in Masson’s trichrome solution for 10 min, washed with 0.5% acetic acid, and differentiated in 1% phosphomolybdic acid for 10 min until COL fibers appeared distinct. Following counterstaining with aniline blue and then dehydration through a graded ethanol series, the sections were cleared in xylene, mounted with a resinous medium, and imaged using a light microscope (Axio Lab.A1, Zeiss, Oberkochen, Germany).
WGA staining was performed on mouse cardiac sections to outline cardiomyocyte boundaries and determine cross-sectional area (CSA). Briefly, deparaffinized and rehydrated sections were incubated (1 h, room temperature) with a fluorescein-conjugated WGA probe in the dark. Following rinsing to remove unbound probe, the slides were counter-stained (5 min, room temperature) with 4′,6-diamidino-2-phenylindole (DAPI) to visualize nuclei. After two phosphate-buffered saline (PBS) washes, sections were coverslipped, followed by imaging using a fluorescence microscope (Axio Lab.A1, Zeiss, Oberkochen, Germany).
Cells were stained with BODIPY 581/591 C11 (Beyotime, Beijing, China) for cellular lipid peroxidation assessment. Briefly, after discarding the culture medium and PBS rinsing, cells underwent incubation (30 min, 37 °C) with 1 mL of staining solution per well. After two PBS washes, cells were covered with 2 mL of fresh PBS, and imaged using a confocal microscope (LSM 800, Zeiss, Oberkochen, Germany).
CCK-8 assay was performed for cell viability determination. Cells were plated onto 96-well plates and cultured in an incubator (24 h, 5% CO2), followed by treatment of different doses of mocetinostat (0.05, 0.1, 0.25, 0.5, 1, 2.5, 5, and 10 µM). At each concentration, cells were plated in six duplicate wells. After the designated treatment period, each well was added with 10 µL of CCK-8 solution, followed by incubation for an additional 2 hours. Then the absorbance at 450 nm was determined to quantify cell viability.
H9c2 cardiomyocytes (Shanghai Institute of Biological Sciences, Chinese Academy of Sciences, Shanghai, China) were cultured in high-glucose Dulbecco’s Modified Eagle Medium containing 10% fetal bovine serum and penicillin/streptomycin in a humidified environment (37 °C, 5% CO2). The medium was refreshed every 2–3 days. Upon reaching 80–90% confluence, cells were washed using PBS, dissociated using 0.25% trypsin-EDTA, and neutralized with complete medium. Following centrifugation (1000 ×g, 3 min), cells were subcultured at a 1:3 to 1:6 ratio. All experiments utilized cells between passages 5 and 10 to ensure phenotypic consistency. All cell lines were validated by STR profiling and tested negative for mycoplasma.
All instruments were sterilized. Neonatal mice underwent sternotomy for cardiac extraction, with the surgical site disinfected using 75% ethanol. The harvested hearts were rinsed in PBS, transferred to sterile glass containers, and finely minced into small tissue fragments. Following 5 min of enzymatic digestion in 10 mL of collagenase solution at 37 °C with constant stirring, the fragments were centrifuged (1000 ×g, 5 min). To achieve complete tissue dissociation, the digestion–centrifugation cycle was repeated seven times, followed by differential adhesion and filtration of the resulting cell suspension onto fresh culture plates using a cell strainer. Primary cardiomyocytes were validated by both morphological identification (spontaneous beating) and flow cytometry analysis of cTnT expression (purity >95%), and all cell preparations tested negative for mycoplasma (Supplementary Materials). Prior to treatment, cells were serum-starved (24 h). Following 48 h, the medium was replaced with serum-free culture medium. Cardiomyocytes were treated (48 h) with 50 µM PE to induce hypertrophy [30], with or without subsequent incubation with mocetinostat (0.1 μM) for 24 h.
Total RNA was extracted from heart tissue fragments and neonatal mouse cardiomyocytes utilizing TRIzol reagent. RNA was reverse transcribed into cDNA utilizing a commercial kit (Thermo) as instructed. The reaction mixture was prepared by combining the required components, including the ChamQ Universal SYBR qPCR Master Mix, followed by thorough mixing and brief centrifugation. qPCR was subsequently conducted utilizing a 0.1 mL PCR plate on a real-time PCR instrument (Thermo). All cDNA samples were analyzed in technical triplicate to ensure accuracy and reproducibility. Following amplification, the 2-ΔΔCt method was applied for relative gene expression calculation. The primer sequences used were as follows:
Atrial natriuretic peptide (ANP)
F: 5′-CTTCTTCCTCGTCTTGGCCTTT-3′
R: 5′-TCCAGGTGGTCTAGCAGGTTCT-3′
B-type natriuretic peptide (BNP)
F: 5′-GCTGCTGGAGCTGATAAGAGAA-3′
R: 5′-CGATCCGGTCTATCTTGTGCC-3′
β-myosin heavy chain (β-MHC)
F: 5′-CCCAGAAACAAGTGAAGAGCCT-3′
R: 5′-GTTCCACGATGGCGATGTTC-3′
Actin alpha 1 (ACTA1)
F: 5′-ATGTGCGACGAAGACGAGAC-3′
R: 5′-ACCCATACCTACCATGACACCCT-3′
Connective tissue growth factor (CTGF)
F: 5′-CATCTCCACCCGAGTTACCAA-3′
R: 5′-CCGCAGAACTTAGCCCTGTATG-3′
COL1 alpha 1 chain (COL1a1)
F: 5′-GAGAGGTGAACAAGGTCCCG-3′
R: 5′-AAACCTCTCTCGCCTCTTGC-3′
β-actin (internal control)
F: 5′-GTGACGTTGACATCCGTAAAGA-3′
R: 5′-GTAACAGTCCGCCTAGAAGCAC-3′
Total protein of cells and mouse cardiac tissues was isolated, quantified, mixed with loading buffer, and denatured by boiling (100 °C, 10 min). Equal amounts of protein were separated via sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis in Tris-Glycine-SDS buffer (80 V for 30 min, then 120 V for 60–90 min). Then proteins were wet-transferred to polyvinylidene difluoride membranes (100 V, 60 min) on ice, using a transfer buffer containing 20% methanol. After blocking (1 h, room temperature) in 5% skim milk in Tris-buffered saline with Tween-20 (TBST), the membranes underwent incubation (overnight, 4 °C) with primary antibodies. Following TBST washes, the membranes underwent incubation (2 h, room temperature) with horseradish peroxidase (HRP)-conjugated secondary antibodies. An enhanced chemiluminescent reagent was employed for target protein bands visualization. A chemiluminescence system (VILBER, Marne-la-Vallée, France) was used for imaging. Quantification was conducted via densitometry using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
The primary antibodies used are listed below:
HDAC1 (YA395) Rabbit mAb 1:1000 (HY-P80149; MedChemExpress, Monmouth Junction, NJ, USA);
Nrf2 (D1Z9C) Rabbit mAb 1:1000 (#12721; Cell Signaling Technology, Danvers, MA, USA);
GPX4 (YA399) Rabbit mAb 1:1000 (HY-P80450; MCE);
SLC7A11 (D2M7A) Rabbit mAb 1:1000 (#12691, CST);
HO-1 (HMOX1)(E3F4S) Rabbit mAb 1:1000 (#43966, CST);
Ferroportin (FPN/SLC40A1) (C2) Rabbit mAb 1:2000 (ab239583; Abcam, Cambridge, MA, USA);
Ferritin Heavy Chain 1 (FTH1) (D1D4) Rabbit mAb 1:1000 (4393, CST);
Nrf2 (Acetyl Lys599) Rabbit pAb 1:1000 (AP63260; Abcepta, San Diego, CA, USA);
β-Actin Rabbit mAb 1:1000 (ab8227, Abcam);
Lamin B1 (D9V6H) Rabbit mAb 1:1000 (#13435, CST);
GAPDH (D16H11) Rabbit mAb 1:1000 (#5174, CST);
Keap1 (D6B12) Rabbit mAb 1:1000 (#8047,CST);
Goat Anti-Rabbit IgG H&L (HRP) secondary antibody 1:5000 (ab205718, Abcam).
Mitochondrial morphology was evaluated. Briefly, mouse cardiac tissues were immersed (4 °C, 2–4 h) in 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4), followed by three 15-min washes in the same buffer. Samples were post-fixed in 1% osmium tetroxide (4 °C, dark, 1 h) and rewashed. Next, tissues were dehydrated through a graded ethanol series (30%, 50%, 70%, 80%, 90%, 95%, and 100%; 20 min per step), transitioned through propylene oxide, and infiltrated with graded propylene oxide and epoxy resin mixtures (2:1, 1:1, and 1:2; 2–4 h each). Following overnight infiltration in pure resin, samples were embedded and polymerized stepwise at 37 °C, 45 °C, and 60 °C. For ultrathin sections (70–90 nm), uranyl acetate and lead citrate were employed for staining, followed by rinsing. A transmission electron microscope (Talos 12, Massachusetts, USA, Thermo Fisher Scientific) was employed for imaging to assess myofibrillar and mitochondrial ultrastructure.
To observe primary cardiomyocyte morphology, cells were washed with PBS and fixed (20 min) in 4% paraformaldehyde. Following three washes, cells were permeabilized (5 min, room temperature ) with 0.1% Triton X-100 and subsequently blocked against non-specific binding for 30 min. Then the cells were incubated (30 min) with phalloidin (Thermo) away from light, and nuclei were counterstained with DAPI. Following a final PBS wash, coverslips were mounted with an anti-fade medium and imaged utilizing a fluorescence microscope (Axio Observer Z1, Zeiss, Oberkochen, Germany) at 493/517 nm excitation/emission wavelengths.
MMP was detected using the fluorescent probe JC-1 (Thermo). Briefly, cardiomyocytes underwent incubation (37 °C, 20 min) with the JC-1 working solution and then two washes with the JC-1 staining buffer. After that, cells were imaged using a fluorescence microscope (Axio Observer Z1, Zeiss, Oberkochen, Germany) at maximum excitation wavelengths of 514 nm (JC-1 monomers) and 585 nm (JC-1 aggregates) to detect JC-1 fluorescence signals.
Fluorescent probe 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) (Abcam) was employed for intracellular ROS evaluation. Briefly, cells underwent incubation (20 min, 37 °C) with the DCFH-DA working solution in serum-free culture medium away from light. After three washes, the cells were pre-warmed PBS and imaged using a fluorescence microscope (Axio Observer Z1, Zeiss, Oberkochen, Germany).
MDA levels were measured using a commercial lipid peroxidation assay kit (MDA Assay Kit, Beyotime) based on the TBA colorimetric method. The assay was performed according to the manufacturer’s instructions. Briefly, samples were mixed with the MDA detection working solution, heated at 100 °C for 15 min, and centrifuged. The supernatant was measured at 532 nm using a microplate reader. MDA concentrations were calculated from a standard curve.
The BCA assay was conducted for protein concentration determination. Then the intracellular Fe2+ content was assessed following the protocol provided with the Fe2+ detection kit (ELabscience Bionovation Inc., Houston, TX, USA). The optical density at 593 nm was recorded using a microplate reader, and the Fe2+ levels were computed utilizing a standard curve of known Fe2+ concentration.
Cellular GSH levels were measured in freshly prepared cell lysates according to the kit protocol (Beyotime). After two ice-cold PBS washes, the cells were pelleted at 4 °C. After discarding the supernatant, the cell pellet was lysed with a deproteinization buffer and vortexed, followed by freeze-thaw cycles of the sample. First, the sample was rapidly immersed in liquid nitrogen for 2 min. Subsequently, it was removed from the liquid nitrogen and immediately transferred to a constant-temperature environment at 37 °C to restore a near-physiological state. Then the thawed sample was quickly placed in an ultra-low-temperature freezer (–80 °C, 2 min) and incubated on ice (5 min). After that, samples underwent centrifugation (10,000 ×g, 10 min, 4 °C), with the supernatant collected. The absorbance at 405 nm was determined. The GSH content in the sample was calculated based on the standard curve.
A commercial assay kit (Beyotime) was employed for detecting SOD activity. Cell samples were washed with pre-chilled PBS and homogenized, followed by centrifugation (12,000 ×g, 15 min, 4 °C), with the supernatant collected for subsequent analysis. All reagents were prepared freshly as per the manufacturer’s protocol and mixed in the prescribed order and ratios. The mixed solution was added dropwise to the test sample with gentle agitation, followed by the addition of the reaction initiation reagent. The mixture was then vortexed thoroughly. Subsequently, the reaction mixture was transferred to a dark environment and incubated (37 °C, 30 min). Afterward, the absorbance was measured at 560 nm, and the enzymatic activity was determined based on the recorded absorbance values.
ATP levels were measured using a bioluminescence assay kit (Beyotime) according to the manufacturer‘s instructions. Briefly, cells were lysed with the provided lysis buffer, and the lysates were centrifuged at 12,000 × g for 5 min at 4 °C. Then, 100 µL of ATP detection reagent was mixed with 20 µL of the supernatant in a 96-well plate. After a 5-min incubation at room temperature, the luminescence intensity was measured.
Data were expressed as the mean ± standard error of the mean. GraphPad (version 9.3.0; La Jolla, CA, USA) was employed for data analysis. Group comparisons were conducted utilizing the Student’s t-test or one-way analysis of variance (ANOVA) with Bonferroni post hoc tests. A p-value < 0.05 indicated statistical significance.
To determine the suitable concentration of mocetinostat for treating cardiomyocytes in vitro, we conducted a CCK-8 assay using concentrations ranging from 0.05 µM to 10 µM. Our results identified 0.1 µM as the optimal dose for further studies, as it provided the best protective effect against PE-induced injury. However, concentrations of 0.25 µM or higher caused a modest but consistent decrease in cell viability compared with the control, indicating mild cytotoxicity at these doses (Fig. 1A). Phalloidin staining in primary mouse cardiomyocytes revealed that while PE stimulation induced significant hypertrophy, mocetinostat treatment effectively prevented this enlargement and maintained near-normal cellular morphology (Fig. 1B,C).
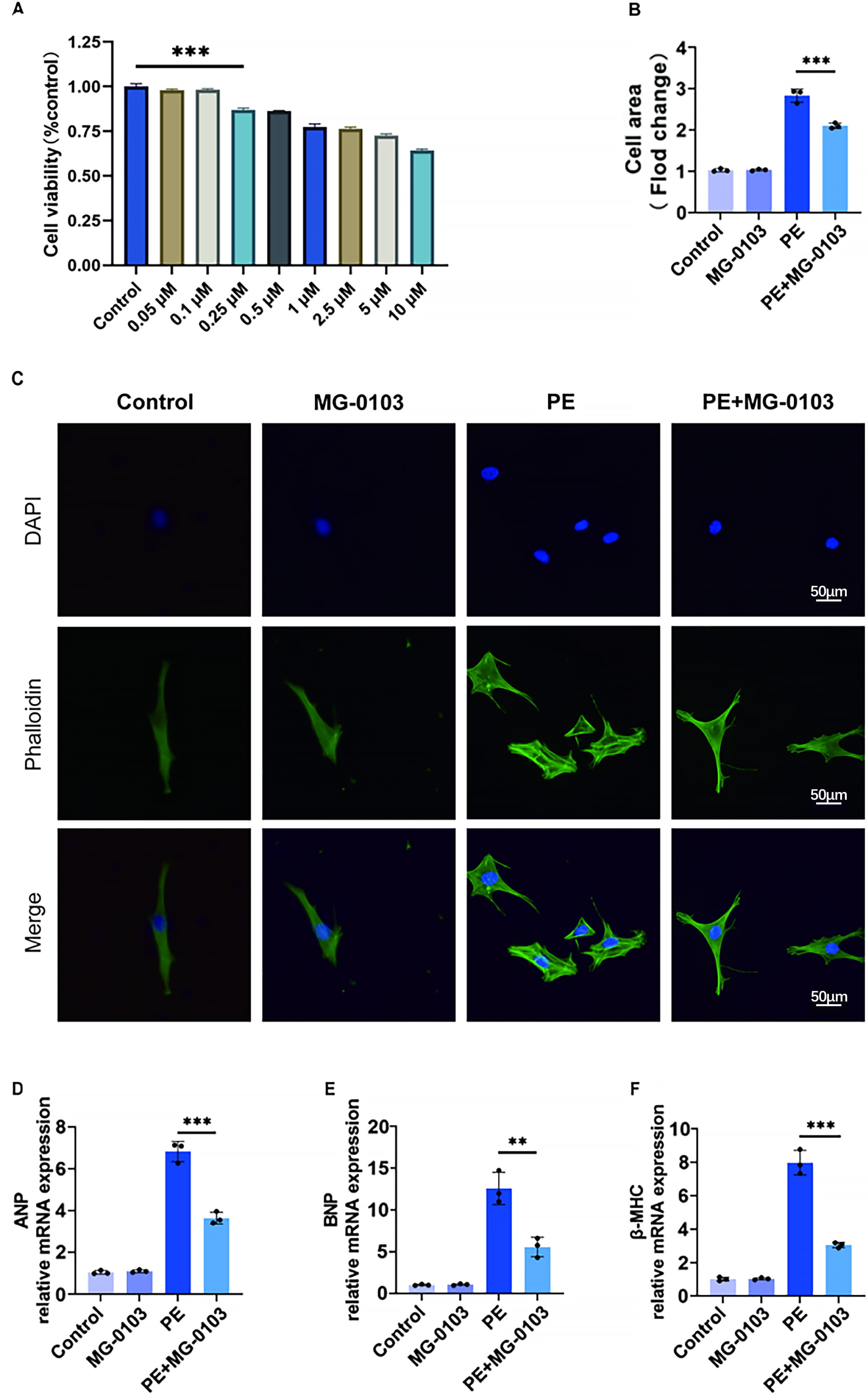 Fig. 1.
Fig. 1. Mocetinostat protects cardiomyocytes from PE-induced hypertrophy in vitro. (A) Effect of varying mocetinostat concentrations (0.05–10 µM) on H9c2 cardiomyocyte viability. (B,C) Phalloidin-stained images of primary cardiomyocytes and quantitative analysis of primary cardiomyocyte surface area. Scale bar: 50 µm. (D–F) Relative mRNA expression of pathological hypertrophy biomarkers (ANP, BNP, β-MHC) in primary cardiomyocytes assessed by qPCR. **p < 0.01, ***p < 0.001. N = 3 independent experiments. Data were analyzed via ANOVA with Bonferroni post hoc tests. H9C2, H9c2 rat cardiomyocyte; ANP, Atrial natriuretic peptide; BNP, B-type natriuretic peptide; β-MHC, β-myosin heavy chain; ANOVA, analysis of variance.
To further assess the hypertrophic response, qPCR was conducted to measure the expression of pathological markers (ANP, β-MHC, and BNP). PE stimulation upregulated all three genes, whereas mocetinostat pretreatment effectively reversed these transcriptional changes (Fig. 1D–F). These results indicate that mocetinostat confers substantial protection against PE-induced cardiomyocyte hypertrophy in vitro.
To elucidate how mocetinostat suppresses ferroptosis in hypertrophic cardiomyocytes in vitro, lipid peroxidation and ROS levels were assessed using BODIPY 581/591 C11 and DCFH-DA probes, respectively. PE stimulation markedly increased oxidized fluorescence, indicating enhanced lipid peroxidation. Notably, mocetinostat intervention partially attenuated this effect. Furthermore, PE exposure induced a pronounced elevation in intracellular ROS levels, which was similarly mitigated by mocetinostat co-treatment (Fig. 2A,B).
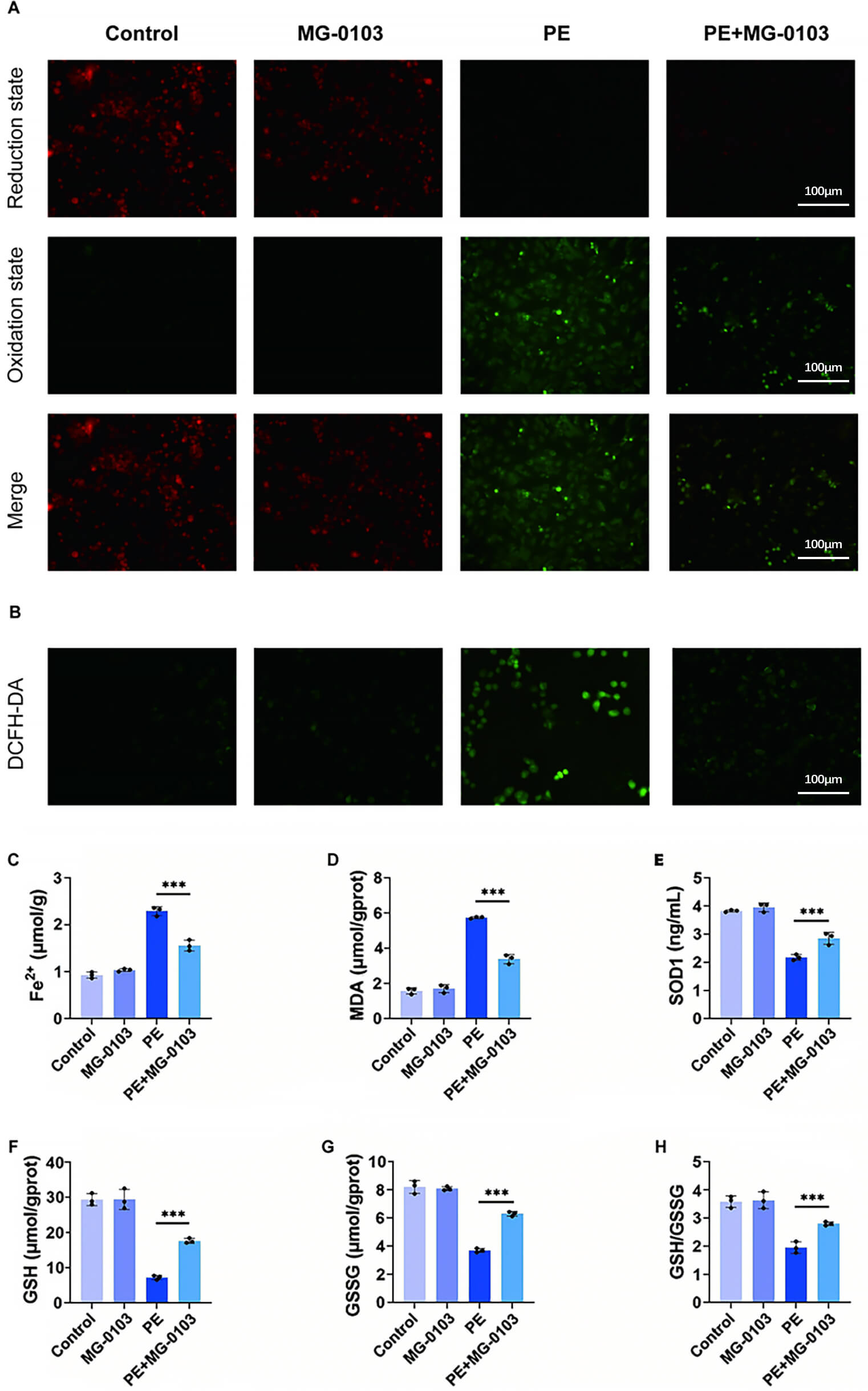 Fig. 2.
Fig. 2.Mocetinostat suppresses ferroptosis in PE-stimulated cardiomyocytes. (A) Analysis of lipid peroxidation in cardiomyocytes. Scale bar: 100 µm. (B) Measurement of ROS level. Scale bar: 100 µm. (C,D) Quantitative analysis of Fe2+ (C) and MDA (D) levels in primary murine cardiomyocytes. (E–H) Determination of SOD (E), GSH (F), GSSG (G), and GSH/GSSG ratio (H) in primary mouse cardiomyocytes. ***p < 0.001. N = 3 independent experiments. Data were compared via one-way ANOVA with Bonferroni post hoc tests. ROS, reactive oxygen species; MDA, Malondialdehyde; SOD, Superoxide Dismutase; GSH, glutathione; GSSG, Glutathione oxidized.
Quantitative measurements demonstrated that PE-stimulated cells exhibited significantly higher levels of intracellular Fe2+ and the lipid peroxidation marker MDA than control cells, indicating a disrupted redox balance. Mocetinostat intervention effectively alleviated these PE-induced increases (Fig. 2C,D). Further analysis revealed that PE treatment significantly impaired cellular antioxidant defense capacity, as evidenced by the decreased levels of SOD, GSH, oxidized glutathione (GSSG), and a reduced GSH/GSSG ratio (Fig. 2E–H). Following mocetinostat treatment, these key antioxidant indicators were markedly restored.
Mocetinostat’s effects on the MMP in PE-treated primary cardiomyocytes was assessed using JC-1 fluorescence staining. Under physiological conditions, JC-1 aggregates within healthy mitochondria and emits red fluorescence, whereas injury-induced depolarization prevents this aggregation, causing JC-1 to emit green fluorescence [31]. Both control and mocetinostat-treated cells exhibited strong red fluorescence, whereas PE stimulation markedly increased green fluorescence. Notably, mocetinostat co-treatment significantly attenuated this green fluorescent signal (Fig. 3A). Furthermore, the ATP bioluminescence assay demonstrated that while PE treatment markedly reduced intracellular ATP content, mocetinostat intervention effectively restored ATP levels (Fig. 3C). These observations indicate that mocetinostat mitigates mitochondrial dysfunction during pathological cardiac hypertrophy.
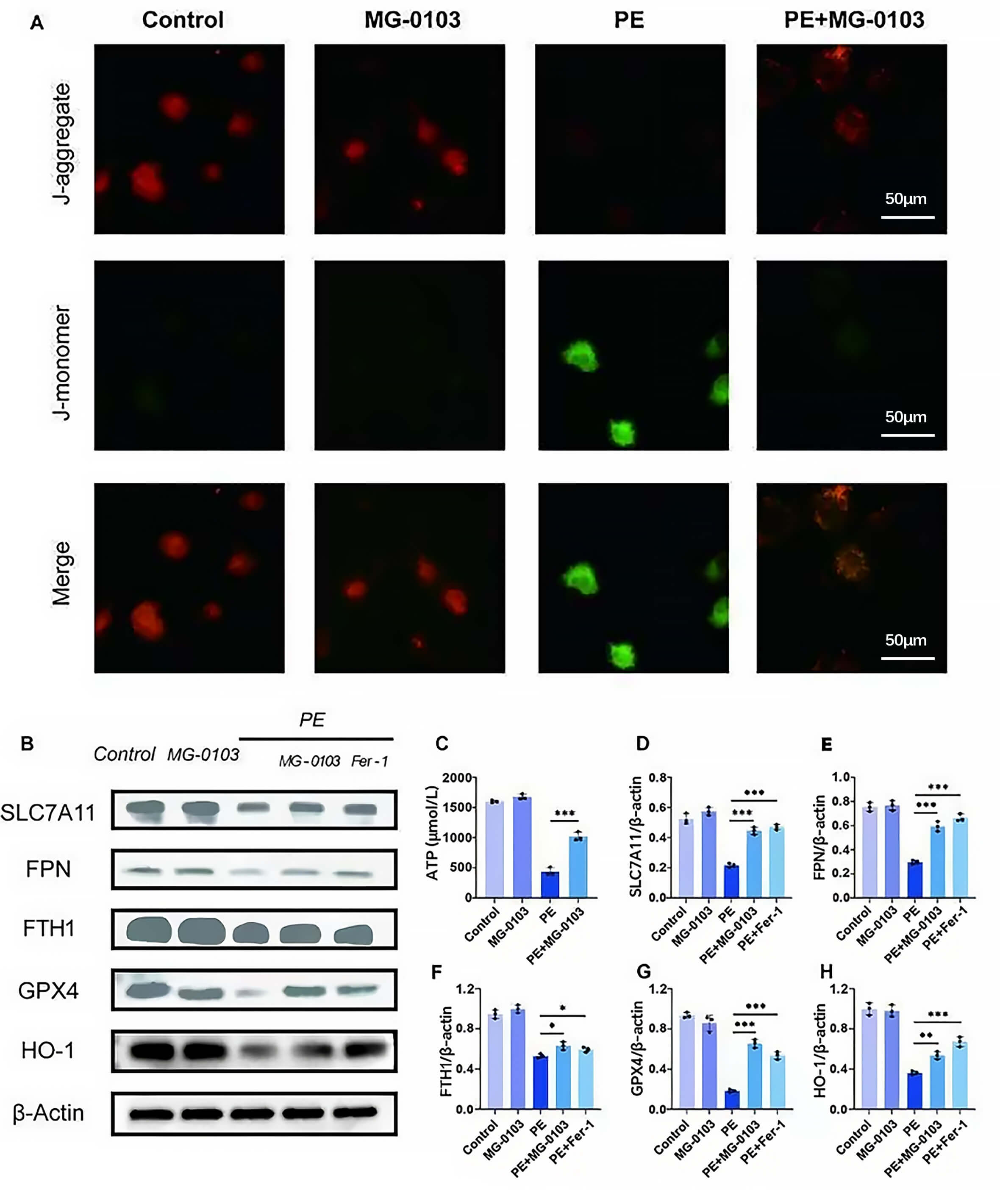 Fig. 3.
Fig. 3.Mocetinostat restores mitochondrial membrane potential and ferroptosis suppressor proteins. (A) JC-1 fluorescence imaging of primary cardiomyocytes. Scale bar: 50 µm. (C) Measurement of ATP concentration in H9c2 cells. (B,D–H) Western blotting and subsequent densitometric analysis were performed to determine the expression levels of SLC7A11, FPN, FTH1, GPX4, and HO-1 in primary cardiomyocytes. *p < 0.05, **p < 0.01, ***p < 0.001. N = 3 independent experiments. Data were compared via one-way ANOVA with Bonferroni post hoc tests. JC-1, 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide; ATP, Adenosine Triphosphate; SLC7A11, Solute carrier family 7 member 11; FPN, ferroportin; FTH, ferritin heavy chain 1; GPX4, glutathione peroxidase 4; HO-1, heme oxygenase-1.
Subsequently, the specific ferroptosis inhibitor ferrostatin-1 (Fer-1) was utilized as a positive control. The data showed that the level of key ferroptosis suppressor proteins (SLC7A11, FTH1, FPN, GPX4, and HO-1) was significantly downregulated in the PE-induced hypertrophy model. Crucially, mocetinostat intervention rescued these protein levels relative to the Fer-1 treatment group (Fig. 3B,D–H). Collectively, these findings provide compelling evidence that mocetinostat strengthens ferroptotic resistance and preserves mitochondrial function.
Mice were subjected to TAC surgery for investigation of HDAC1’s impact on cardiac hypertrophy in vivo. At 6 weeks post-TAC, Western blot analysis confirmed the marked upregulation of myocardial HDAC1 expression (Fig. 4A,B). Echocardiography revealed that TAC surgery markedly impaired cardiac function, as evidenced by reduced ejection fraction (EF) and fractional shortening (FS), alongside increased interventricular end-diastolic thickness (IVSd), and left ventricular diameters (LVIDd, LVIDs). Notably, mocetinostat treatment largely corrected these functional and structural abnormalities (Fig. 4C–H). Furthermore, the increased heart weight to body weight (HW/BW) and heart weight to tibia length (HW/TL) ratios observed in TAC mice were significantly reversed by mocetinostat, indicating a potent inhibitory effect on cardiac hypertrophy (Fig. 4I,M,N). Histological analysis via WGA and H&E staining showed that mocetinostat alleviated the significant increase in cardiomyocyte cross-sectional area (CSA) induced by TAC (Fig. 4J,K,O). Masson’s trichrome staining further demonstrated that mocetinostat markedly reduced myocardial fibrosis (Fig. 4L,P). As shown in Fig. 5, at the molecular level, TAC-induced hypertrophy was associated with increased expression of markers including ANP, BNP, β-MHC and Acta1, as well as fibrosis-related genes CTGF and COL1a1. Mocetinostat intervention significantly downregulated these markers compared to the untreated TAC group, suggesting its ameliorative effect on pathological myocardial hypertrophy (Fig. 5A–D). Furthermore, mocetinostat intervention also resulted in a significant downregulation of fibrosis markers (Fig. 5E,F). Taken together, mocetinostat exerts significant cardioprotective effects against TAC-induced myocardial hypertrophy and fibrosis in mice.
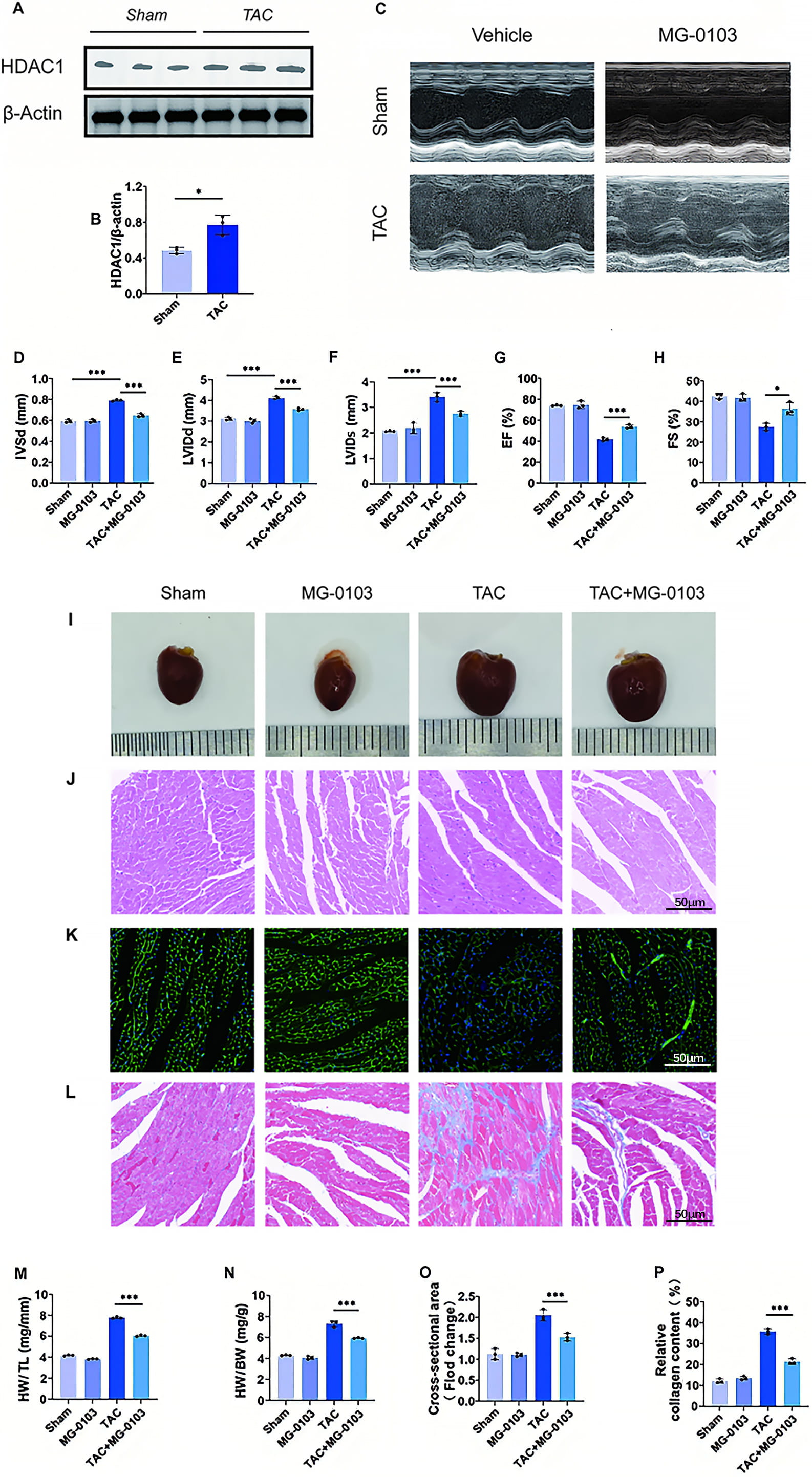 Fig. 4.
Fig. 4.Mocetinostat attenuates TAC-induced cardiac hypertrophy and fibrosis in vivo. (A,B) HDAC1 expression was shown in the myocardial tissue of both normal and pathologically hypertrophied animals using Western blot and quantitative analysis. (C–H) Long-axis M-mode echocardiography of mice, with quantitative evaluations of IVSd, LVIDd, LVIDs, EF and FS. (I) Macroscopic appearance and morphology of mouse hearts. (J) H&E staining of mouse hearts for cardiomyocyte morphological assessment. Scale bar: 50 µm. (K) WGA staining for cardiomyocyte CSA. Scale bar: 50 µm. (L) Masson’s trichrome staining for myocardial fibrosis detection. Scale bar: 50 µm. (M,N) Measurement of the ratios of heart weight (HW) to body weight (BW) and HW to tibia length (TL). (O) Quantitative CSA analysis. (P) Quantitative analysis of myocardial fibrosis. *p < 0.05, ***p < 0.001. N = 3 independent experiments. Data were compared via one-way ANOVA with Bonferroni post hoc tests. IVSd, interventricular end-diastolic thickness; LVIDd/LVIDs, left ventricular diameters; EF, ejection fraction; FS, fractional shortening; H&E, hematoxylin and eosin; WGA, Wheat Germ Agglutinin; CSA, cross-sectional area.
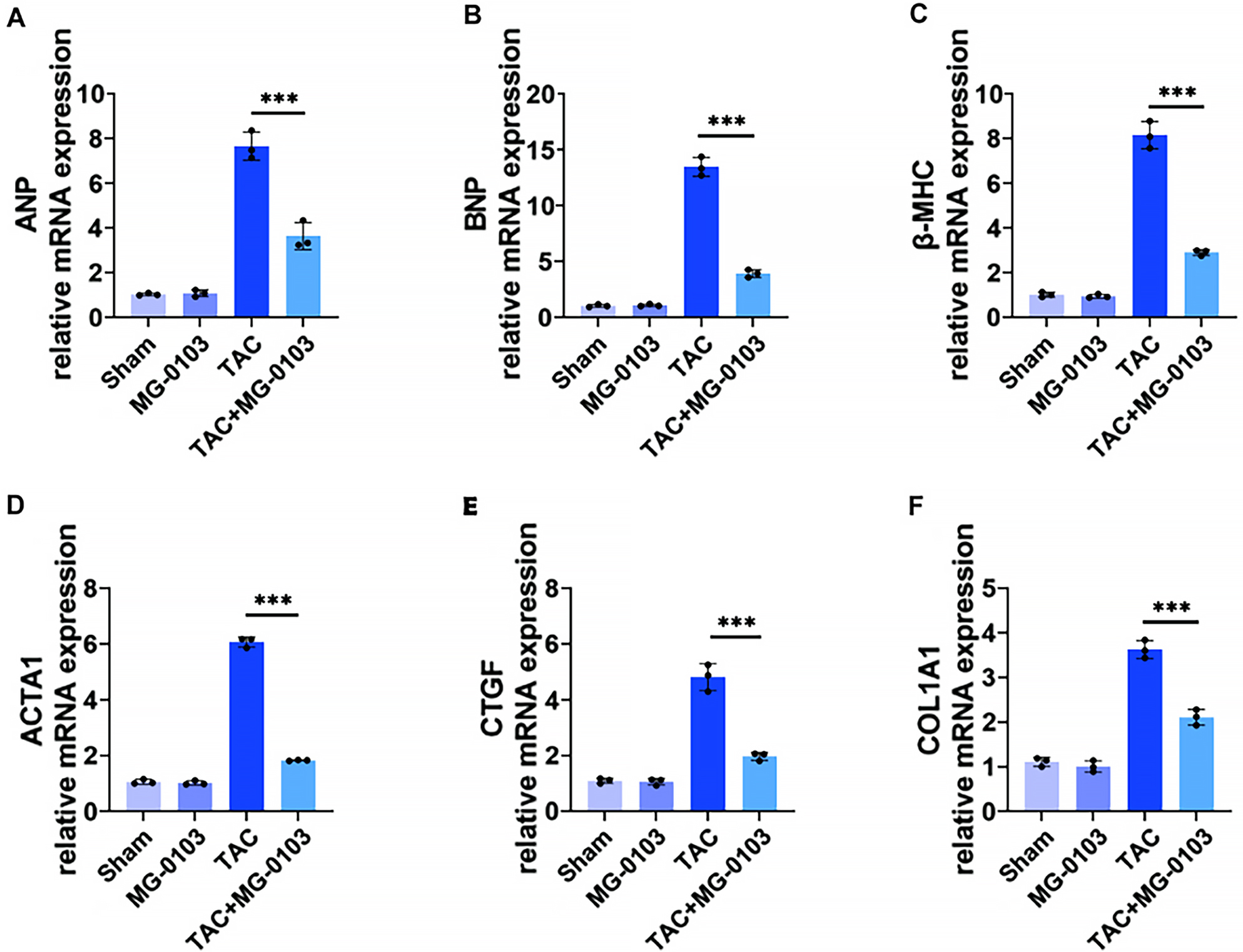 Fig. 5.
Fig. 5.Mocetinostat downregulates hypertrophy and fibrosis markers in TAC-mouse hearts. (A–D) qPCR analysis of mRNA expression for pathological hypertrophy biomarkers: ANP, BNP, β-MHC, ACTA1. (E,F) qPCR analysis of mRNA expression for myocardial fibrosis markers: CTGF and COL1a1. ***p < 0.001. N = 3 independent experiments. Data were compared via one-way ANOVA with Bonferroni post hoc tests.
To evaluate mitochondrial ultrastructure, cardiac tissues were examined using transmission electron microscopy. TAC surgery induced marked mitochondrial disarray, characterized by outer membrane disruption and partial or complete cristae loss. By contrast, mocetinostat treatment preserved cristae density and markedly reduced membrane rupture (Fig. 6A). Furthermore, mocetinostat normalized the elevated iron and MDA levels observed in TAC hearts (Fig. 6B,C). Simultaneously, the TAC-induced decline in antioxidant defenses, including SOD and GSH, showed a statistically significant recovery following mocetinostat treatment. Furthermore, relative to the TAC group, mocetinostat intervention resulted in a statistically significant increase in GSSG levels and a marked elevation in the GSH/GSSG ratio (Fig. 6D–G).
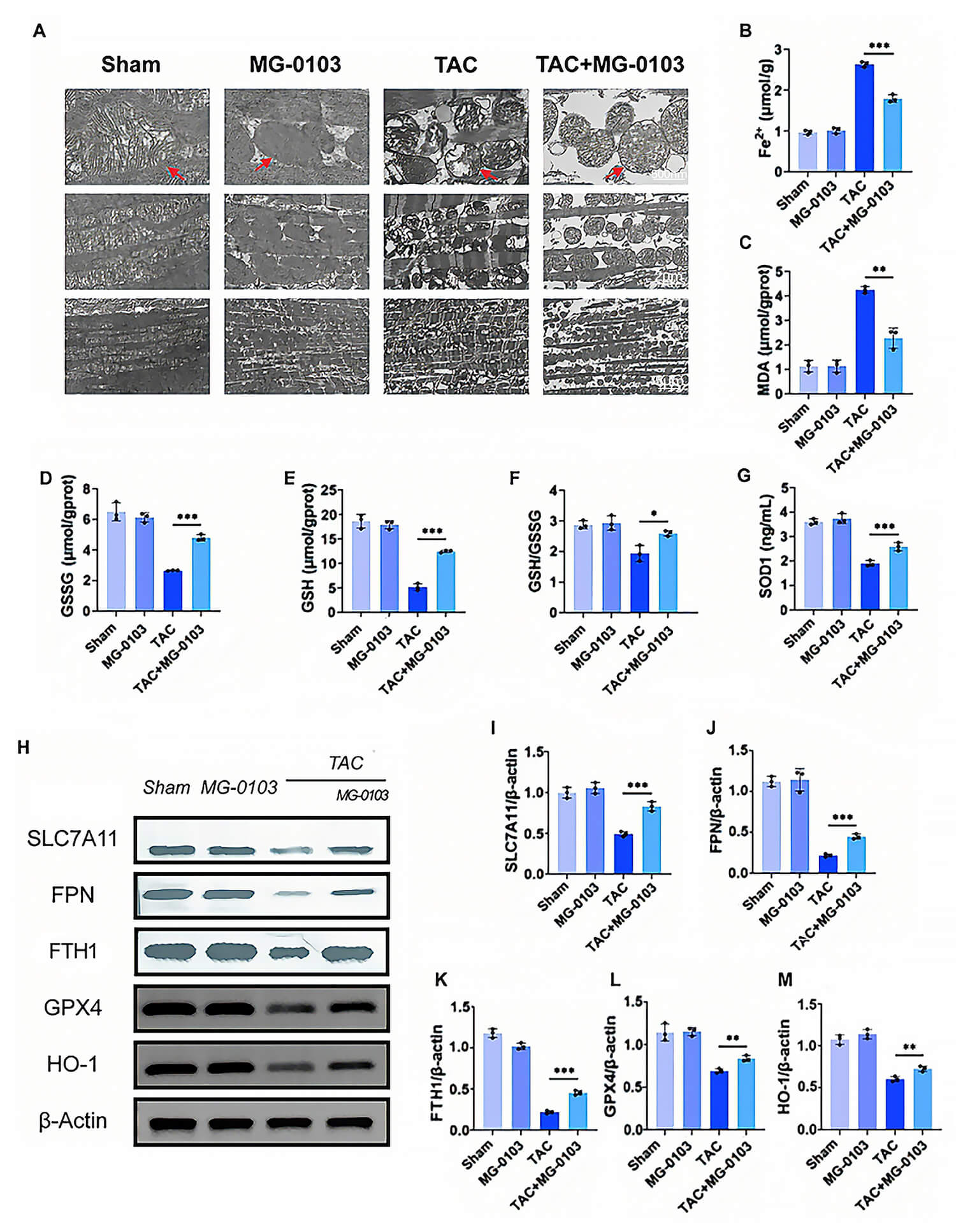 Fig. 6.
Fig. 6. Mocetinostat inhibits ferroptosis in hypertrophied myocardial tissue. (A) Ultrastructural morphology of myocardial tissue mitochondria under transmission electron microscopy. Red arrows indicate representative mitochondria with either typical normal morphology or typical ferroptotic morphology. Scale bars: 500 nm (top), 2 µm (middle), 5 µm (bottom). (B,C) Quantitative analysis of Fe2+ (B) and MDA (C) levels in murine myocardial tissue. (D–F) Quantification of GSH (D), GSSG (E), and GSH/GSSG ratio (F) in murine myocardial tissue. (G) Quantitative determination of SOD activity in murine myocardial tissue. (H–M) Western blotting and quantitative analysis of SLC7A11, FPN, FTH1, GPX4, and HO-1 expression in myocardial tissue. *p < 0.05, **p < 0.01, ***p < 0.001. N = 3 independent experiments. Data were compared via one-way ANOVA with Bonferroni post hoc tests.
Moreover, as revealed by Western blot analysis, downregulation of ferroptosis-associated proteins (SLC7A11, FPN, FTH1, GPX4, and HO-1) in TAC mice was markedly restored by mocetinostat therapy (Fig. 6H–M). Collectively, these data demonstrate that mocetinostat suppresses ferroptosis during the progression of pathological cardiac hypertrophy.
To further investigate the specific effects of the HDAC1 inhibitor mocetinostat on the Nrf2 pathway, Western blot analysis was performed on primary cardiomyocytes from each group. The results showed a substantial decrease in total Nrf2 levels and a moderate decrease in nuclear Nrf2 levels in these cells. After treatment with mocetinostat, it was found that these changes were reversed. As previously described, the expression and activity of Nrf2 are directly regulated by Keap1 [22]. Consequently, to determine whether the changes in Nrf2 were associated with Keap1, the expression levels of Keap1 in each group of cells were analyzed by Western blotting. However, we found no change in total Keap1 expression levels between the experimental groups (Fig. 7A–E).
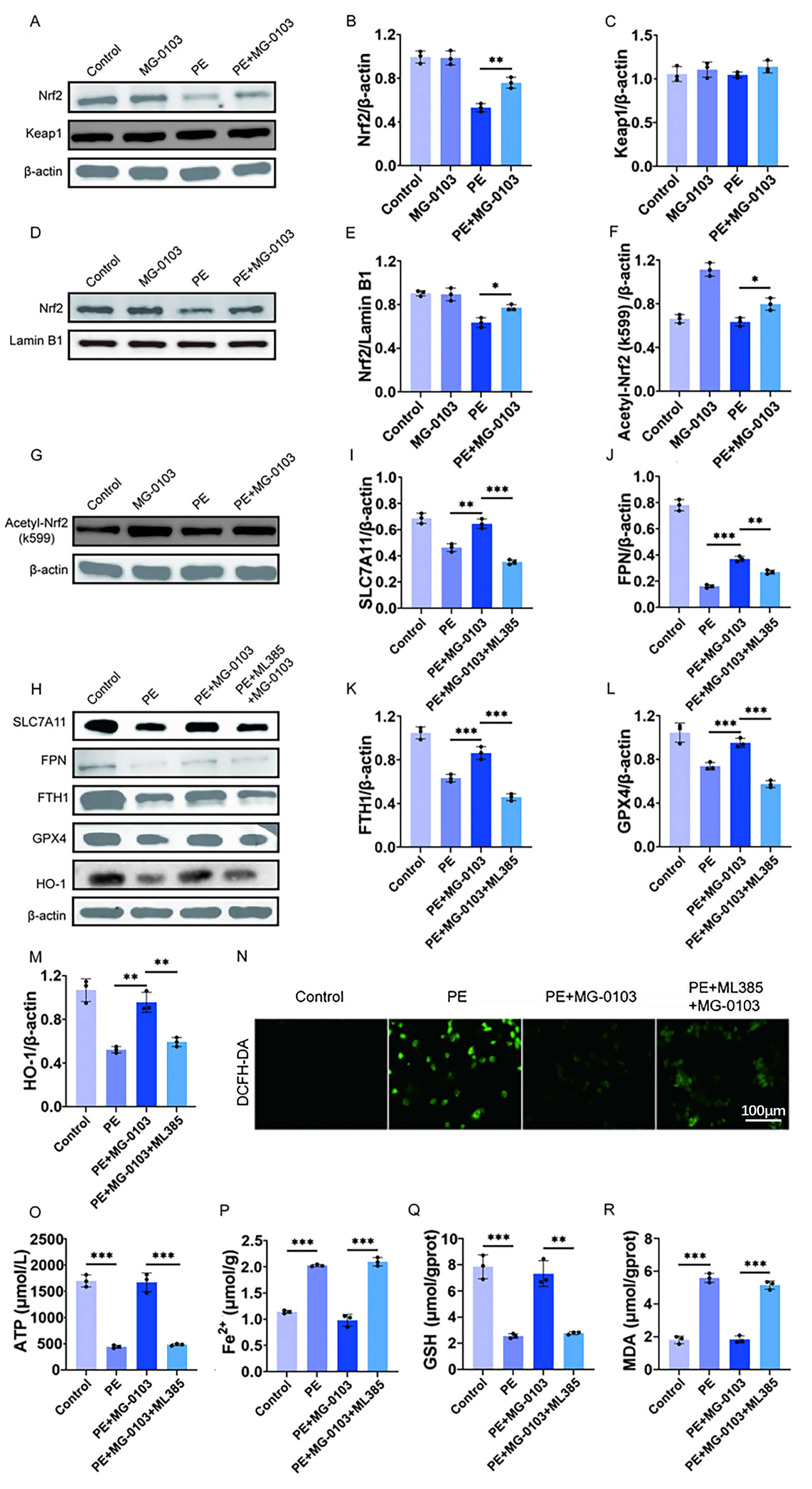 Fig. 7.
Fig. 7. Mocetinostat activates the Nrf2 pathway to exert anti-ferroptotic and cardioprotective effects. (A–E) Representative Western blotting images and corresponding densitometric analyses illustrating total Nrf2, nuclear Nrf2, and Keap1 protein levels in primary cardiomyocytes. (F,G) Western blot images and quantitative evaluation of acetylated Nrf2 (Ac-Nrf2) expression in primary cardiomyocytes. (H–M) Western blotting images and quantitative evaluation of expression levels of SLC7A11, FPN, FTH1, GPX4, HO-1 in H9c2 cells. (N) Measurement of Intracellular ROS levels in primary cardiomyocytes using the DCFH-DA fluorescence probe. Scale bar: 100 μm. (O) ATP concentration in H9c2 cells determined by bioluminescence assay. (P–R) Quantitative analysis of Fe2+, GSH, and MDA levels in H9c2 cells using commercial assay kits. *p < 0.05, **p < 0.01, ***p < 0.001. N = 3 independent experiments. Data were compared via one-way ANOVA with Bonferroni post hoc tests. Nrf2, nuclear factor erythroid 2-related factor 2.
Previous work has shown that acetylation stabilizes Nrf2 and also promotes its nuclear localization [29]. We hypothesized that mocetinostat induces Nrf2 nuclear localization by acetylating it. Consequently, we examined the acetylation status of Nrf2 on Lys599 by Western blotting and found an increase in acetylated Nrf2 protein levels in mocetinostat-treated samples (Fig. 7F,G).
To validate the functional role of Nrf2 as a regulatory mediator of mocetinostat’s anti-ferroptotic effects, the Nrf2 inhibitor ML385 was employed prior to PE treatment to specifically block its binding to antioxidant response elements (AREs) in the nucleus. It was found that PE treatment reduced key ferroptosis suppressor protein levels in cells, whereas combination therapy with mocetinostat increased these protein levels compared to PE treatment alone. However, this protective effect was abrogated by ML385-mediated Nrf2 inhibition (Fig. 7H–M). Similarly, the PE + mocetinostat group showed reduced ROS levels, which was reversed following Nrf2 inhibition (Fig. 7N). Bioluminescence assay of ATP in H9c2 cells further revealed that PE exposure markedly decreased ATP production, whereas co-treatment with mocetinostat restored ATP levels, an effect that was reversed by Nrf2 inhibition (Fig. 7O). Furthermore, measurements of intracellular Fe2+, GSH, and MDA concentrations showed that Nrf2 inhibition sufficiently reversed the anti-ferroptotic effects of mocetinostat (Fig. 7P–R). In summary, these data suggested that mocetinostat exerted its anti-ferroptotic effect by enhancing Nrf2 acetylation and promoting its nuclear translocation.
The heart is the vital organ responsible for systemic blood supply, yet it remains susceptible to diverse pathological stimuli. Pathological cardiac hypertrophy is an initial compensatory response to such stress, characterized by structural remodeling to maintain cardiac output. This condition typically initiates with diastolic impairment and progresses through interstitial fibrosis and cardiomyocyte death to maladaptive ventricular remodeling. As disease severity increases, ventricular dilation ensues, further compromising diastolic and systolic function and culminating in heart failure. Consequently, identifying effective interventions to prevent or treat the onset and progression of hypertrophy is critical. This study demonstrates that the HDAC1 inhibitor mocetinostat exerts therapeutic benefits against cardiac dysfunction, ventricular remodeling, and fibrosis in TAC-induced mice. Consistent with these in vivo findings, mocetinostat effectively attenuated PE-induced hypertrophic responses in cardiomyocytes in vitro. Consequently, the findings suggest that the HDAC1 inhibitor mocetinostat is a promising novel therapeutic agent for the management of pathological cardiac hypertrophy.
Ferroptosis, an iron-dependent lipid peroxidation-driven cell death, is critical in cardiovascular disease pathogenesis [32,33]. This process is driven by disruptions in amino acid metabolism, iron homeostasis, and mitochondrial function [34]. At the molecular level, excessive Fe2+ catalyzes the Fenton reaction with hydrogen peroxide to generate hydroxyl radicals, which trigger ROS accumulation [35], and subsequent lipid peroxidation [36]. Pathological conditions [32], such as ferritinophagy, accelerate this process by releasing excessive Fe2+ [12]. Emerging research highlights ferroptosis as a key driver in myocardial ischemia/reperfusion injury, DOX-induced cardiomyopathy, coronary atherosclerosis, abdominal aortic aneurysm [37] and cardiac hypertrophy [37,38].
Given its high metabolic demand, the heart possesses the body’s highest mitochondrial density [39]. Mitochondrial lipid peroxidation triggers dysfunction, increasing ROS and impairing respiratory capacity, which drives the progression of ferroptosis [40,41,42,43]. Consequently, mitochondria-targeted antioxidants that maintain homeostasis can mitigate ferroptosis-associated myocardial injury [44]. Although cellular defenses like GPX4, FTH1, HO-1, FPN, and SOD [45] normally sequester Fe2+ and neutralize ROS, these mechanisms are overwhelmed during exacerbated ferroptosis. Specifically, GPX4 inactivation and GSH depletion accelerate cell death. This study found that TAC-induced cardiac hypertrophy and PE-stimulated cardiomyocytes exhibit key hallmarks of ferroptosis, namely elevated MDA and Fe2+ levels, excessive ROS, mitochondrial ultrastructural damage (cristae reduction and membrane rupture) [46,47], and marked downregulation of ferroptosis suppressors (GSH, GPX4, SLC7A11, SOD, FTH1, FPN, and HO-1). These results establish that ferroptosis drives cardiac hypertrophy, indicating that inhibiting this iron-dependent cell death is a viable therapeutic strategy to treat or prevent pathological remodeling.
HDACs are involved in numerous cellular signaling pathways and have been implicated in the pathogenesis of various diseases. Numerous studies have revealed that protein acetylation plays a complex and critical regulatory role in various cardiovascular diseases, including pathological cardiac hypertrophy [48,49]. In particular, Class I HDACs have been shown to promote the progression of cardiac pathology. A key study using RNA interference identified HDAC1 and HDAC2 as critical drivers of pressure overload-induced hypertrophy, demonstrating that the class I/II HDAC inhibitor trichostatin A can reverse this pathology in the TAC model [50]. Additionally, a study found that mocetinostat treatment exerts therapeutic effects by regulating the renin–angiotensin system, thereby reversing TAC-induced cardiac remodeling, cardiomyocyte hypertrophy, and fibrosis in rats [20]. These findings suggest that class I HDAC inhibitors show therapeutic potential for mitigating pathological cardiac hypertrophy. Previously, we identified HDAC1 as a crucial driver of ferroptosis within the pathological microenvironment of heart failure and highlighted mocetinostat as a promising therapeutic agent [15]. Building on these findings, we examined mocetinostat’s effects on ferroptosis in a model of pathological myocardial hypertrophy. As expected, elevated HDAC1 expression was observed in the hypertrophic cardiac model. To elucidate the regulatory role of mocetinostat in cardiac hypertrophy, we employed both an in vitro model of PE-stimulated cardiomyocytes and an in vivo mouse model of pressure overload-induced myocardial hypertrophy.
The findings demonstrated that mocetinostat intervention ameliorated myocardial hypertrophy and improved cardiac function. Analyses of iron levels, MDA, and lipid peroxidation staining revealed that mocetinostat promoted cell survival through its antioxidant activity. Furthermore, mocetinostat treatment increased the average MMP in cardiomyocytes, improved mitochondrial ultrastructural abnormalities, and partially restored mitochondrial function. Importantly, mocetinostat upregulated key ferroptosis-related markers (GSH, SOD, SLC7A11, GPX4, FTH1, FPN, and HO-1) in both in vitro and in vivo cardiomyocyte models, thereby partially reversing the ferroptosis process.
During ferroptosis, Fenton reaction-generated oxygen free radicals disrupt cellular homeostasis, activating the Nrf2 transcription factor [51]. Oxidative modification of Keap1 cysteine thiols through oxidation or alkylation disrupts formation of the Keap1-Nrf2 complex, triggering Nrf2 release, as well as its nuclear translocation. In the nucleus, Nrf2 binds to AREs to initiate gene transcription [52] such as GPX4 and SOD, thereby maintaining redox balance and mitochondrial function. Beyond oxidative activation, post-translational modifications, specifically phosphorylation and acetylation, critically regulate Nrf2 [53]. In HepG2 cells, acetylation promotes Nrf2 nuclear localization and basic region leucine zipper complex formation to activate antioxidant transcription, whereas deacetylation favors cytoplasmic retention and transcriptional termination [54]. Based on these findings, it is hypothesized that the class I HDAC inhibitor mocetinostat mitigates cardiomyocyte ferroptosis by enhancing Nrf2 acetylation and nuclear translocation. This is supported by evidence showing that mocetinostat treatment in pathological myocardial hypertrophy models upregulated Nrf2 expression and increased nuclear Ac-Nrf2 levels. These cardioprotective effects were reversed by the Nrf2 inhibitor ML385. Collectively, these data suggest that mocetinostat exerts cardioprotective effects by inhibiting pathological myocardial hypertrophy in both in vitro and in vivo models. Therefore, this study provides the first evidence that the cardioprotective effect of mocetinostat against pathological hypertrophy is mediated, at least partially, by the Nrf2 pathway activation, specifically by upregulating Nrf2 expression, enhancing its acetylation, promoting its nuclear translocation, and subsequently activating antioxidant pathways that inhibit ferroptosis. Furthermore, consistent with neuroscience research indicating that HDAC1/2 inhibition alleviates neuronal ferroptosis via the Nrf2/HO-1 pathway [29], this study suggests that the HDAC1-Nrf2 regulatory axis constitutes a conserved therapeutic target across diverse tissues and pathologies.
It is noteworthy that mocetinostat exhibited a biphasic dose-response relationship in our in vitro assay, which acted as a protective agent at 0.1 µM but exhibited mild cytotoxicity at concentrations ≥0.25 µM. This concentration-dependent effect is consistent with previous reports of HDAC inhibitors, where low-dose HDAC inhibition confers cytoprotection while high-dose inhibition may induce cell death or growth arrest [55]. These findings highlight the importance of dose optimization to ensure the safe and effective therapeutic application of HDAC inhibitors in managing cardiovascular diseases.
There are several limitations in this study. First, anesthesia-associated risks in TAC-induced mice limited cardiac assessment to a single imaging session at 6 weeks post-surgery, failing to capture earlier, crucial stages. Second, the absence of “Fer-1 only” and “ML385 only” control groups restricted our ability to assess the baseline effects of these inhibitors; however, this does not invalidate the primary conclusion. ML385 reversed the protective effects of mocetinostat, confirming Nrf2 pathway specificity. Fer-1, a well-established ferroptosis inhibitor, exerted comparable protective effects, serving as a positive control. Notably, both inhibitors are widely used as standard pharmacological tools, further supporting the validity of our approach [56,57]. Third, although the authors observed Nrf2 upregulation in vitro following mocetinostat treatment, likely linked to increased H3K27 and histone acetylation at the Nrf2 promoter [58], the precise molecular mechanisms in the context of myocardial hypertrophy require further in vivo validation. Fourth, several methodological limitations should be noted regarding the mechanistic validation. Our mechanistic inferences rely primarily on the Nrf2 inhibitor ML385. However, without complementary genetic silencing approaches (e.g., small interfering RNA-mediated Nrf2 knockdown), off-target effects cannot be completely excluded. Future studies employing genetic loss-of-function models will help confirm the specificity of the Nrf2 pathway in mediating the cardioprotective effects of mocetinostat. Additionally, although our data demonstrate that mocetinostat enhances Nrf2 acetylation and nuclear translocation, we have not provided direct evidence of a physical interaction between HDAC1 and Nrf2. Co-immunoprecipitation experiments are necessary to determine whether HDAC1 directly binds and deacetylates Nrf2, or if these effects occur indirectly. Moreover, we did not perform a comprehensive in vivo dose-response analysis for mocetinostat. Based on literature indicating cardioprotective benefits, a 10 mg/kg dose was selected; preliminary studies confirmed this dosage partially reduced ventricular hypertrophy with no appreciable toxicity. Nevertheless, we acknowledge that future studies should include systematic dose-response evaluations—covering both lower and higher ranges—to define the optimal therapeutic window. Finally, this study confirms that mocetinostat alleviates cardiomyocyte ferroptosis, at least partly through the Nrf2 pathway; however, the role of other mechanisms, such as ferritinophagy, warrants future investigation.
Overall, our findings indicate that the HDAC1 inhibitor mocetinostat modulates Nrf2 expression and acetylation, driving its nuclear translocation to suppress cardiomyocyte ferroptosis and mitigate pathological myocardial hypertrophy. These data provide a reference for cardioprotection and suggest novel therapeutic strategies for stress-induced hypertrophy. Furthermore, this study suggests that HDAC1 may serve as both a therapeutic target and a biomarker for tracking the progression of pathological hypertrophy to heart failure.
ACSL4, Acyl-CoA Synthetase Long-chain Family Member 4; Actal, Actin Alpha 1; ANP, Atrial Natriuretic Peptide; ARE, Antioxidant Response Element; ATP, Adenosine Triphosphate; β-MHC, β-Myosin Heavy Chain; BNP, Brain Natriuretic Peptide; COLlal, Collagen Type I Alpha 1; CTGF, Connective Tissue Growth Factor; FPN, Ferroportin; FTH1, Ferritin Heavy Chain 1; GPX4, Glutathione peroxidase 4; GSH, Glutathione; GSSG, Glutathione oxidized; HDAC1, Histone Deacetylase 1; HO-1, Heme Oxygenase 1; Keap1, Kelch-like ECH-Associated Protein 1; MDA, Malondialdehyde; NCOA4, Nuclear Receptor Coactivator 4; NQO1, NAD(P)H Oxidoreductase 1; Nrf2, Nuclear Factor-E2-Related Factor 2; OXPHOS, Oxidative Phosphorylation; PE, Phenylephr ine Hydrochloride; ROS, Reactive Oxygen Species; SLC7A11, Solute Carrier Family 7 Member 11.
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
JL, HS and JZ was responsible for the conceptualization and investigation of the study. JS, PX and CW performed the main experiments. CC, PG and XW contributed to collection and assembly of data. QG, AX and YL performed statistical analysis of all data. QL contributed to conception, supervision, administration, and validation of this project. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All experiments involving animals were approved by the Animal Ethics Committee of Nantong University (Ethic Approval Number: S20220310-010) and in compliance with institutional Guide for the Care and Use of Laboratory Animals.The experiments were conducted in strict accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, 8th edition, 2011) and were in compliance with all applicable Chinese animal welfare legislation and institutional guidelines.The study is reported in compliance with the ARRIVE guidelines 2.0.
We thank Ms. Yiling Su, as a member of Prof. Qi Lu’s team, for her contribution to the animal ethics application. We also thank the administrative staff of the Laboratory Animal Center of Nantong University Medical School for their professional guidance and assistance.
This research was funded by the Affiliated Hospital of Nantong University Research Physician Development Fund, grant number YJXYY202204-YSB59, and Postgraduate Research & Practice Innovation Program of Jiangsu Province in 2023, grant number SJCX23_1791.
The authors declare no conflicts of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/Pharmazie51659.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.