



 , Meiyun Xu 2, Mansa Li 2, Manyu Xu 1,2, Xi Han 1,*
, Meiyun Xu 2, Mansa Li 2, Manyu Xu 1,2, Xi Han 1,* , Qiongying Hu 2,*
, Qiongying Hu 2,*1 School of Basic Medical Sciences, Jiamusi University, 154007 Jiamusi, Heilongjiang, China
2 Department of Basic Medicine, School of Medicine, Taizhou University, 318000 Taizhou, Zhejiang, China
Abstract
Ferroptosis is a regulated form of cell death characterized by iron-dependent lipid peroxidation. Intracellular iron promotes the generation of reactive oxygen species via the Fenton reaction, leading to lipid peroxide accumulation, membrane damage, and subsequent cell death. Increasing evidence indicates that ferroptosis plays a critical role in cancer progression and therapeutic response. Recent studies have identified a diverse range of ferroptosis-inducing compounds, including natural products, semi-synthetic derivatives, and synthetic small molecules, which exert antitumor effects by targeting key regulatory nodes such as iron metabolism, system Xc-, and glutathione peroxidase 4. However, the clinical translation of ferroptosis-based strategies remains limited by insufficient tumor selectivity, systemic toxicity, the lack of reliable biomarkers, and adaptive resistance mechanisms. In this review, we summarize the molecular regulatory networks governing ferroptosis, critically evaluate representative ferroptosis-inducing compounds and the associated mechanisms of action, and discuss current translational challenges and unresolved controversies. Furthermore, an integrative framework is proposed to define ferroptosis as a context-dependent metabolic vulnerability in tumors, thereby providing a conceptual basis for the rational development of ferroptosis-based anticancer therapies.
Keywords
- ferroptosis
- lipid peroxidation
- iron metabolism
- glutathione peroxidase 4
- solute carrier family 7 member 11
- antineoplastic agents
Ferroptosis is a distinct form of regulated cell death driven by iron-dependent lipid peroxidation and oxidative membrane damage. Since its first description in 2012, ferroptosis has been recognized as a unique death modality that is mechanistically and morphologically different from apoptosis, necrosis, and autophagy [1]. Increasing evidence indicates that ferroptosis plays a critical role in diverse physiological and pathological processes, including development, immune regulation, neurodegeneration, and particularly tumor progression [2, 3]. In cancer, ferroptosis has emerged as an attractive therapeutic vulnerability. Therapy-resistant and mesenchymal-like tumor cells frequently exhibit elevated iron demand, altered lipid metabolism, and enhanced oxidative stress, rendering them selectively sensitive to ferroptotic perturbations [4, 5]. However, the regulatory networks governing ferroptosis in tumors are highly complex and context dependent, and the clinical translation of ferroptosis-based therapies remains limited by insufficient tumor selectivity and systemic toxicity [6, 7].
In recent years, substantial progress has been made in elucidating the molecular mechanisms underlying ferroptosis, particularly in the areas of iron homeostasis, lipid peroxidation, and antioxidant defense systems [2, 3]. Meanwhile, a growing number of natural products and synthetic small molecules have been identified as potent ferroptosis inducers, providing new opportunities for anticancer drug discovery [8, 9]. In this review, we summarize the current understanding of ferroptosis regulation in tumor cells, systematically discuss representative ferroptosis-inducing compounds and their molecular targets, and highlight the major challenges and future directions for the clinical translation of ferroptosis-based cancer therapy.
Ferroptosis is an iron-dependent, regulated form of cell death characterized by excessive lipid peroxidation and reactive oxygen species (ROS) accumulation, distinguishing it from apoptosis, necrosis, and autophagy [10, 11]. Since its discovery, ferroptosis has been recognized as a key contributor to the pathogenesis and treatment response of multiple diseases, including cancer, neurodegenerative disorders, ischemia–reperfusion injury, cardiovascular diseases, kidney injury, and liver diseases [1, 10, 11]. The unique biochemical features of ferroptosis, including iron accumulation, glutathione depletion, and lipid peroxidation, provide both vulnerabilities and therapeutic opportunities, depending on the disease context [11, 12]. The major molecular mechanisms and disease associations of ferroptosis are summarized in Fig. 1.
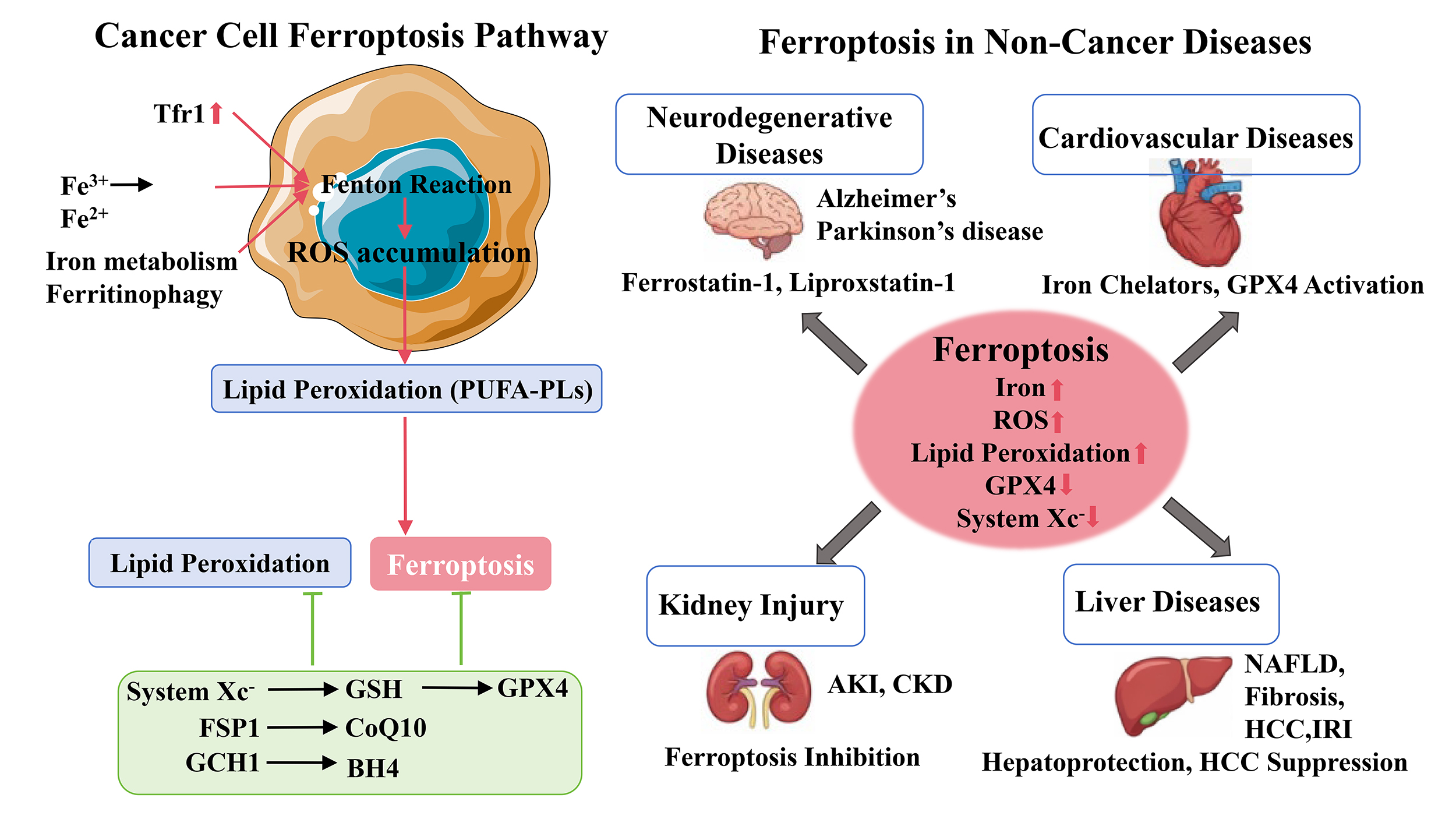 Fig. 1.
Fig. 1.
Molecular mechanisms regulating ferroptosis and its involvement in human diseases. Ferroptosis is an iron-dependent form of regulated cell death characterized by lipid peroxidation and ROS accumulation. Dysregulated iron metabolism increases intracellular iron levels and promotes ROS generation through the Fenton reaction, leading to lipid peroxidation of polyunsaturated phospholipids and ferroptotic cell death. Antioxidant defense systems, including the System Xc–-GSH-GPX4 axis, the FSP1-CoQ10 pathway, and the GCH1-BH4 pathway, inhibit lipid peroxidation and suppress ferroptosis. Ferroptosis is associated with various diseases, including neurodegenerative diseases, cardiovascular diseases, kidney injury, and liver diseases. Red arrows indicate promotion, green lines indicate inhibition, and black arrows indicate disease associations. ROS, reactive oxygen species; GPX4, glutathione peroxidase 4; Fe2+, ferrous iron; GSH, glutathione; Tfr1, transferrin receptor 1; CoQ10, Coenzyme Q10. Created using Microsoft PowerPoint.
Cancer cells often exhibit altered iron metabolism, increased ROS production, and dysregulated lipid metabolism, all of which sensitize them to ferroptosis [2, 13]. Induction of ferroptosis can inhibit tumor growth, particularly in apoptosis-resistant cancers, and enhance the efficacy of conventional therapies including chemotherapy, radiotherapy, and immunotherapy [14]. Mechanistically, the cystine/glutamate antiporter system Xc– and glutathione peroxidase 4 (GPX4) are central regulators of ferroptosis; inhibition of either promotes lipid ROS accumulation and ferroptotic cell death [2, 10].
Tumor suppressor proteins, notably p53, modulate ferroptosis by repressing SLC7A11, a key subunit of system Xc–, thereby enhancing lipid peroxidation and ferroptotic susceptibility [2]. Oncogenic pathways such as RAS–RAF–MEK and NRF2 also influence ferroptosis sensitivity by regulating iron homeostasis, lipid metabolism, and antioxidant responses [2, 14]. Furthermore, ferroptosis induction can overcome drug resistance in cisplatin-resistant ovarian cancer and sorafenib-resistant hepatocellular carcinoma [13, 14]. Collectively, these findings indicate that ferroptosis offers a selective therapeutic strategy against malignant cells while sparing normal tissues [2, 13, 14].
Beyond cancer, ferroptosis contributes to multiple organ injuries and degenerative diseases, where iron dysregulation, ROS accumulation, and lipid peroxidation are central pathological features [1, 10]. In neurodegenerative disorders such as Alzheimer’s disease and Parkinson’s disease, iron accumulation and lipid ROS drive neuronal death [15]. Pharmacological inhibition of ferroptosis with agents such as ferrostatin-1 or liproxstatin-1 mitigates neurodegeneration and preserves neuronal function in experimental models [15, 16]. Ferroptosis is also a critical mediator in ischemia–reperfusion injury (IRI) affecting the heart, liver, kidney, and brain. Reperfusion generates excessive ROS, promotes iron accumulation, and induces lipid peroxidation, triggering ferroptotic cell death and exacerbating tissue damage [17]. Interventions targeting ferroptosis via iron chelators, lipid ROS scavengers, or GPX4 activation significantly reduce tissue injury and improve organ function [17, 18]. In cardiovascular diseases, ferroptosis contributes to myocardial infarction, heart failure, and atherosclerosis [18, 19]. Iron overload and lipid peroxidation induce cardiomyocyte death and compromise cardiac function, whereas ferroptosis inhibition alleviates myocardial injury and preserves cardiac function [18]. Similarly, ferroptosis mediates acute kidney injury (AKI) and chronic kidney disease (CKD) through iron-dependent tubular epithelial cell death, and its pharmacological inhibition demonstrates renoprotective effects [20]. Liver diseases, including non-alcoholic fatty liver disease, liver fibrosis, hepatocellular carcinoma, and hepatic ischemia-reperfusion injury, also involve ferroptosis. Iron overload and lipid peroxidation exacerbate hepatocyte injury, whereas inhibition of ferroptosis mitigates liver damage [21]. Conversely, ferroptosis induction can suppress tumor progression in hepatocellular carcinoma, reflecting its context-dependent dual role [14, 21].
Ferroptosis is a distinct form of regulated cell death driven by iron-dependent lipid peroxidation and oxidative damage to cellular membranes, which differentiates it from apoptosis, necrosis, and other cell death modalities [1, 10]. Morphologically, ferroptotic cells are characterized by condensed mitochondria with reduced or absent cristae and increased membrane density, while nuclear morphology remains largely preserved, indicating the absence of chromatin condensation typical of apoptosis [1, 5].
At the biochemical level, ferroptosis arises from an imbalance between reactive oxygen species (ROS) production and cellular antioxidant capacity. Intracellular labile iron catalyzes ROS generation via the Fenton reaction, initiating the peroxidation of polyunsaturated fatty acid (PUFA)-containing phospholipids in cellular membranes [2, 10]. The accumulation of lipid peroxides disrupts membrane integrity and ultimately leads to cell death. Lipid remodeling enzymes, including acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3), facilitate the incorporation of PUFAs into membrane phospholipids, thereby increasing susceptibility to ferroptosis [2, 22].
Cellular defense against ferroptosis is primarily mediated by antioxidant systems that limit lipid peroxidation. The cystine/glutamate antiporter system Xc– maintains intracellular cysteine levels required for glutathione (GSH) synthesis, while glutathione peroxidase 4 (GPX4) reduces lipid hydroperoxides to non-toxic lipid alcohols [2, 10]. Inhibition of system Xc– or GPX4 leads to rapid accumulation of lipid peroxides and ferroptotic cell death. Additional GPX4-independent pathways, including the ferroptosis suppressor protein 1 (FSP1)-coenzyme Q10 (CoQ10) axis and the GTP cyclohydrolase 1 (GCH1)-tetrahydrobiopterin (BH4) pathway, provide complementary protection against oxidative membrane damage and contribute to ferroptosis resistance [22, 23].
Ferroptosis is mechanistically, morphologically, and biochemically distinct from other forms of regulated cell death, including apoptosis, necrosis, necroptosis, pyroptosis, and autophagy-dependent cell death. Apoptosis is characterized by caspase activation, chromatin condensation, and apoptotic body formation, whereas ferroptosis occurs independently of caspase activation and does not involve nuclear fragmentation. Instead, ferroptotic cells exhibit mitochondrial shrinkage, increased mitochondrial membrane density, and reduced cristae without significant changes in nuclear morphology [24, 25]. Necrosis is traditionally considered an uncontrolled form of cell death associated with cell swelling, plasma membrane rupture, and inflammatory responses; in contrast, plasma membrane rupture in ferroptosis occurs secondary to lipid peroxidation–induced membrane damage rather than osmotic imbalance. Necroptosis and pyroptosis are regulated necrotic cell death pathways mediated by RIPK1/RIPK3/MLKL signaling and gasdermin pore formation, respectively, both of which involve membrane permeabilization through protein-mediated pore formation rather than oxidative lipid damage, further distinguishing ferroptosis from these pathways [25].
Autophagy exhibits a complex relationship with ferroptosis rather than a strictly independent pathway. Selective autophagy processes, particularly ferritinophagy, can promote ferroptosis by degrading ferritin and increasing intracellular labile iron levels, thereby enhancing lipid peroxidation and ferroptotic cell death [26, 27]. This functional interaction indicates that ferroptosis is not only a distinct form of regulated cell death but also part of a broader network of metabolically regulated cell death pathways. These mechanistic and morphological differences highlight ferroptosis as a unique iron-dependent oxidative cell death pathway and support its potential as a therapeutic target distinct from apoptosis-targeted anticancer strategies.
Ferroptosis is regulated by the coordinated interplay of iron metabolism, lipid peroxidation, and antioxidant defense systems, forming a highly integrated network that determines tumor cell susceptibility. The precise balance among these regulatory nodes is context-dependent, influenced by cellular metabolic state and the tumor microenvironment. A comprehensive overview of the ferroptosis regulatory network is schematically summarized in Fig. 2 (Ref. [2, 28, 29]), providing a visual framework for understanding the subsequent mechanistic sections.
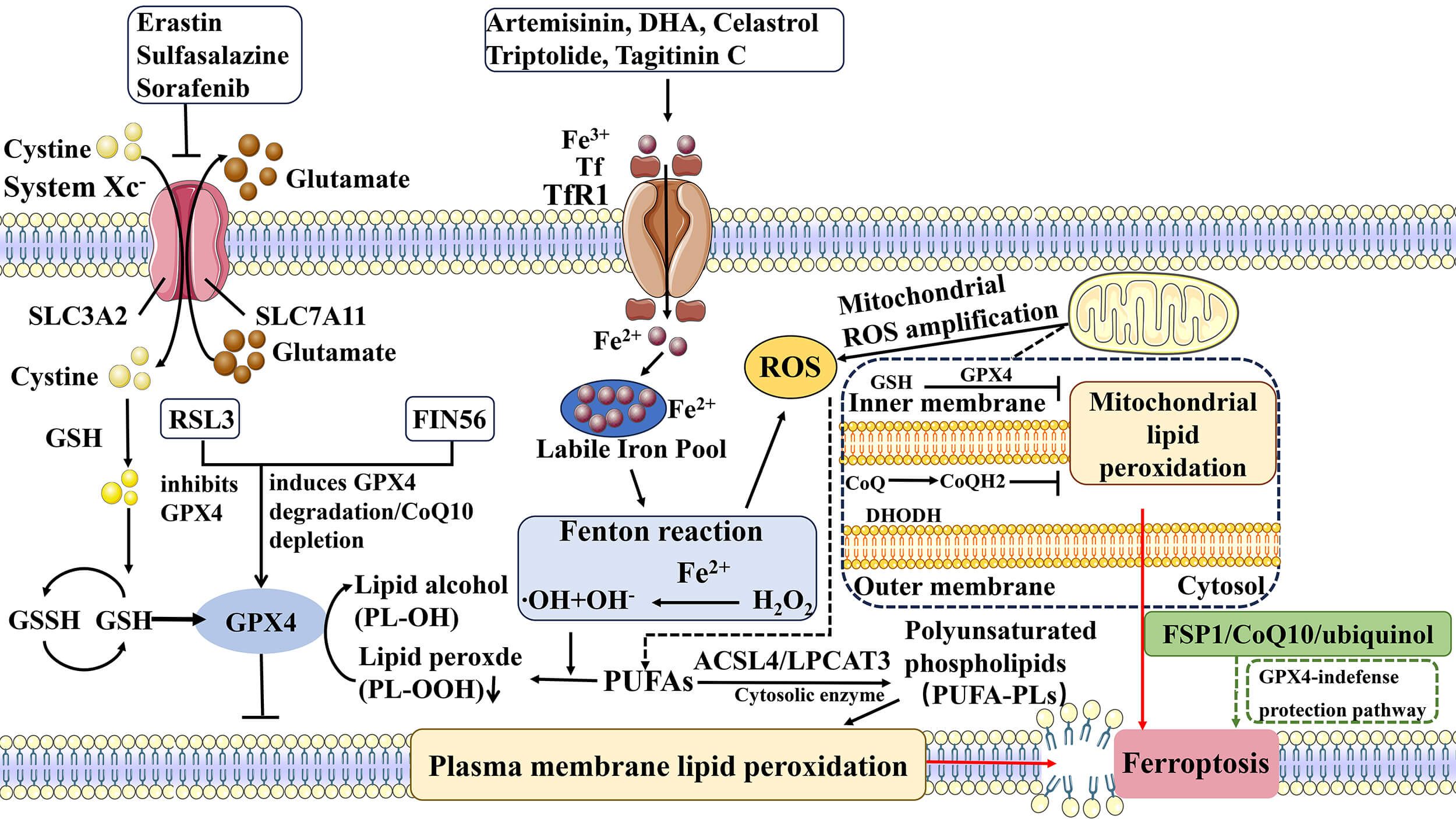 Fig. 2.
Fig. 2.
Regulatory network and pharmacological modulation of
ferroptosis [2, 28, 29]. Ferroptosis is an iron-dependent form of regulated cell death
characterized by lipid peroxidation and reactive oxygen species accumulation. The
process is mainly regulated by iron metabolism, the System Xc–-GSH-GPX4
antioxidant axis, lipid peroxidation pathways, and the FSP1-CoQ10 system.
Iron-dependent ROS generation and phospholipid peroxidation play central roles in
ferroptosis, while inhibition of System Xc– or GPX4 leads to lipid peroxide
accumulation. Various synthetic compounds and natural products modulate
ferroptosis by targeting these regulatory pathways. The symbol “
Iron metabolism is a critical determinant of ferroptosis sensitivity in tumor cells, as ferroptosis is characterized by iron-dependent accumulation of lipid peroxides and oxidative membrane damage [30]. Cellular iron is primarily imported through transferrin receptor 1 (TFR1)-mediated endocytosis and stored in ferritin, while ferroportin mediates iron export to maintain intracellular iron homeostasis [31]. Disruption of iron homeostasis leads to expansion of the labile iron pool, which catalyzes the Fenton reaction and promotes reactive oxygen species (ROS) generation, thereby enhancing lipid peroxidation and ferroptotic cell death [31, 32]. Tumor cells frequently exhibit altered iron metabolism, including increased iron uptake and reduced iron export, which contributes to their elevated sensitivity to ferroptosis and highlights iron metabolism as a potential therapeutic target in cancer treatment [32, 33].
Ferritinophagy, a selective form of autophagy mediated by nuclear receptor coactivator 4 (NCOA4), regulates intracellular iron availability by degrading ferritin and releasing stored iron into the cytosolic labile iron pool [34]. Increased ferritinophagy elevates intracellular Fe2+ levels and enhances ROS production, thereby promoting lipid peroxidation and ferroptosis [34, 35]. The interaction between autophagy and iron metabolism forms an important regulatory mechanism controlling ferroptosis sensitivity in tumor cells [35]. Proteins involved in iron uptake, storage, export, and ferritinophagy, including TFR1, ferritin, ferroportin, and NCOA4, are considered potential molecular targets for ferroptosis-based anticancer therapy [32, 33].
Lipid peroxidation represents the core execution process of ferroptosis and mainly involves oxidation of polyunsaturated fatty acid (PUFA)-containing phospholipids in cellular membranes [30, 36]. The incorporation of PUFAs into membrane phospholipids is regulated by acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3), which determine cellular sensitivity to ferroptosis by shaping membrane lipid composition [36]. These PUFA-phospholipids undergo oxidation through iron-dependent non-enzymatic reactions or lipoxygenase-mediated enzymatic reactions, resulting in accumulation of lipid hydroperoxides and membrane damage [31, 36]. Suppression of ACSL4 or inhibition of lipid peroxidation significantly reduces ferroptosis sensitivity, indicating that lipid remodeling enzymes and lipid peroxidation pathways represent key regulatory nodes and pharmacological targets in ferroptosis-related cancer therapy [32, 36].
Tumor cells employ multiple antioxidant systems to counteract lipid peroxidation and maintain redox homeostasis, thereby preventing ferroptotic cell death [30]. The canonical System Xc–-GSH-GPX4 axis represents the central defense pathway against ferroptosis, in which cystine is imported through System Xc–, reduced to cysteine, and used for glutathione (GSH) synthesis, while glutathione peroxidase 4 (GPX4) utilizes GSH to reduce phospholipid hydroperoxides into non-toxic lipid alcohols [30, 37]. Pharmacological inhibition of System Xc– by erastin or direct inhibition of GPX4 by RSL3 results in the accumulation of lipid peroxides and ultimately triggers ferroptotic cell death [13, 37]. Loss of GPX4 activity has been demonstrated to be sufficient to induce ferroptosis in various cancer cell types, highlighting its essential role in maintaining lipid peroxide homeostasis and cell survival [37].
In addition to the GPX4-dependent pathway, GPX4-independent antioxidant systems have been identified that suppress ferroptosis through parallel redox mechanisms [23, 30]. Ferroptosis suppressor protein 1 (FSP1), previously known as AIFM2, reduces coenzyme Q10 (CoQ10) to ubiquinol using NADPH, which functions as a lipophilic radical-trapping antioxidant that prevents lipid peroxidation propagation in cellular membranes [23]. Another ferroptosis defense mechanism involves the GTP cyclohydrolase 1 (GCH1)–tetrahydrobiopterin (BH4) pathway, where BH4 acts as a potent antioxidant and protects membrane phospholipids from peroxidation independently of GPX4 activity [5]. These parallel antioxidant systems enable tumor cells to survive under conditions of oxidative stress and contribute to ferroptosis resistance in the tumor microenvironment [30, 38].
More recently, dihydroorotate dehydrogenase (DHODH), a mitochondrial enzyme involved in pyrimidine synthesis, has been identified as a mitochondrial ferroptosis defense factor that reduces ubiquinone to ubiquinol within mitochondria and suppresses lipid peroxidation in mitochondrial membranes [39]. The coexistence of GPX4, FSP1, GCH1, and DHODH antioxidant systems indicates that ferroptosis resistance is regulated by multiple layers of redox control located in different cellular compartments [30, 39]. Targeting these antioxidant defense pathways, either individually or in combination, has emerged as an important strategy for pharmacologically inducing ferroptosis in tumor cells and improving the efficacy of anticancer therapies [13, 23].
Ferroptosis is regulated by multiple signaling pathways that control iron metabolism, lipid peroxidation, and antioxidant defense systems in tumor cells [30]. The tumor suppressor p53 is one of the most extensively studied regulators of ferroptosis and can promote ferroptosis by repressing the expression of SLC7A11, a key component of the cystine/glutamate antiporter system Xc–, thereby reducing cystine uptake and glutathione synthesis [12]. In addition, p53 has been reported to enhance ferroptosis through regulation of lipid metabolism and reactive oxygen species accumulation, indicating that p53 participates in ferroptosis through both metabolic and transcriptional mechanisms [12, 13].
The nuclear factor erythroid 2-related factor 2 (NRF2) signaling pathway plays a central role in protecting cells against ferroptosis by regulating antioxidant responses and iron metabolism-related genes [30, 40]. NRF2 activation induces the expression of genes involved in glutathione synthesis, GPX4 activity, ferritin expression, and NADPH regeneration, thereby suppressing lipid peroxidation and ferroptosis [40]. Aberrant activation of NRF2 signaling in tumor cells contributes to ferroptosis resistance and has been associated with tumor progression and drug resistance [40, 41].
Several additional signaling pathways have also been implicated in ferroptosis regulation. The MAPK signaling pathway has been shown to influence ferroptosis sensitivity by regulating oxidative stress and lipid metabolism [5]. The Hippo-YAP pathway can modulate ferroptosis through regulation of cell density-dependent lipid metabolism and iron uptake [42]. The AMPK signaling pathway links cellular energy stress to ferroptosis by regulating lipid biosynthesis and redox homeostasis [43]. These signaling pathways collectively form a complex regulatory network that determines ferroptosis sensitivity in tumor cells and provides multiple potential targets for therapeutic intervention [5, 30].
Identification of molecular targets involved in ferroptosis has facilitated the development of ferroptosis-inducing compounds for cancer therapy [30]. Key molecular targets include system Xc–, GPX4, FSP1, DHODH, ACSL4, and iron metabolism-related proteins such as transferrin receptor and ferritin [13, 30]. Inhibitors of system Xc–, such as erastin, block cystine uptake and glutathione synthesis, whereas GPX4 inhibitors such as RSL3 directly inactivate lipid peroxide detoxification pathways, both leading to ferroptotic cell death [13, 44]. Targeting FSP1 and DHODH has also emerged as a strategy to overcome GPX4-independent ferroptosis resistance mechanisms in tumor cells [39].
Screening methods for ferroptosis-inducing compounds typically involve cell viability assays combined with ferroptosis rescue experiments using ferroptosis inhibitors such as ferrostatin-1 or liproxstatin-1 [30]. Lipid peroxidation detection using C11-BODIPY fluorescence probes, measurement of intracellular iron levels, and glutathione depletion assays are commonly used to confirm ferroptosis induction [45]. High-throughput screening approaches based on chemical libraries and CRISPR genetic screens have further accelerated the discovery of novel ferroptosis-related targets and compounds [46]. These screening strategies have contributed to the identification of numerous ferroptosis-inducing agents, including natural products, small molecules, and drug repurposing candidates, which are currently being investigated for anticancer therapy [30, 46].
Although iron metabolism, lipid peroxidation, and antioxidant defense systems are often described as independent regulatory modules, increasing evidence indicates that these processes are highly interconnected and collectively determine cellular susceptibility to ferroptosis. Iron overload promotes reactive oxygen species generation through the Fenton reaction, while lipid peroxidation provides the direct execution mechanism of ferroptotic cell death. At the same time, antioxidant systems such as the System Xc–-GSH-GPX4 axis and the FSP1-CoQ10 pathway function as critical defense mechanisms that limit lipid peroxide accumulation and maintain redox homeostasis [2, 47]. These findings indicate that ferroptosis is not controlled by a single pathway but rather by a complex metabolic network integrating iron metabolism, lipid metabolism, and redox regulation.
The sensitivity of tumor cells to ferroptosis is closely associated with their metabolic state and genetic background. Tumor cells with high iron demand, increased polyunsaturated lipid synthesis, or impaired antioxidant capacity are more vulnerable to ferroptosis. Metabolic reprogramming in cancer cells, including altered glutamine metabolism, lipid metabolism, and mitochondrial function, has been shown to significantly influence ferroptosis sensitivity [14, 48]. In addition, the tumor microenvironment also plays an important role in ferroptosis regulation. Hypoxia, nutrient deprivation, and immune cell interactions can modulate iron metabolism and lipid peroxidation, thereby affecting ferroptosis susceptibility in tumor cells [49]. Despite the therapeutic potential of ferroptosis induction in cancer treatment, tumor cells may develop resistance to ferroptosis through upregulation of antioxidant pathways such as GPX4, FSP1, and GCH1-mediated pathways. Increased expression of SLC7A11, enhanced NADPH production, and activation of lipid repair mechanisms have also been reported to contribute to ferroptosis resistance [11, 50]. These resistance mechanisms may limit the effectiveness of ferroptosis-inducing therapies when used alone.
Targeting ferroptosis in combination with other cancer therapies has attracted increasing attention. Ferroptosis induction has been reported to enhance the efficacy of chemotherapy, radiotherapy, and immunotherapy in various tumor models. Combination strategies may overcome drug resistance and improve therapeutic outcomes by simultaneously disrupting redox balance and tumor survival pathways [51]. Further studies are needed to better understand the interaction between ferroptosis and tumor metabolism, as well as the tumor microenvironment, which may provide new strategies for ferroptosis-based cancer therapy.
Accumulating evidence demonstrates that chemically diverse compounds can induce ferroptosis in tumor cells by disrupting iron homeostasis, promoting lipid peroxidation, or disabling lipid-directed antioxidant systems. Based on their origin and chemical characteristics, these agents can be broadly classified into natural/semi-synthetic compounds and fully synthetic small molecules. An overview of representative ferroptosis-inducing compounds and their regulatory mechanisms is schematically illustrated in Fig. 3.
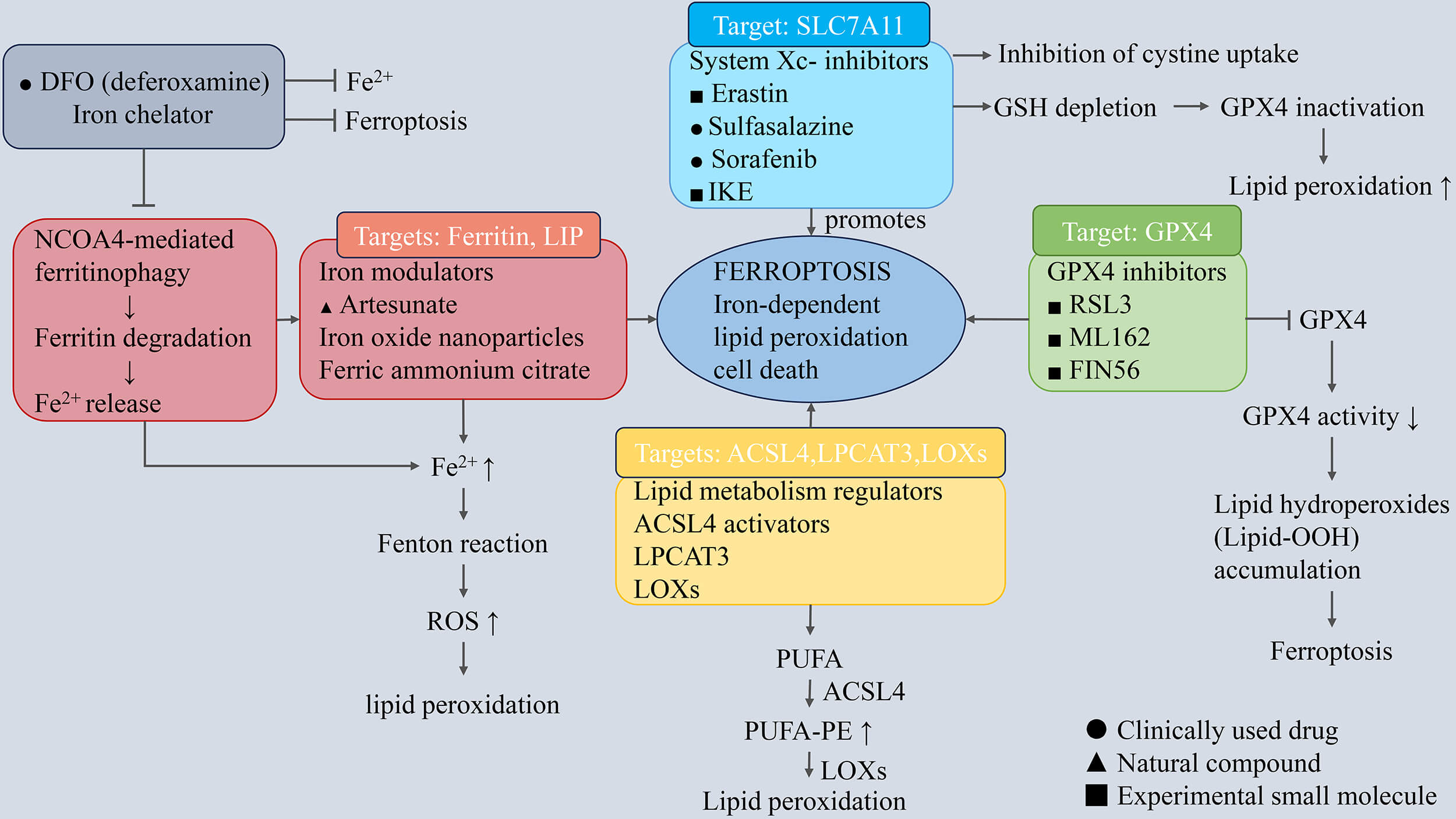 Fig. 3.
Fig. 3.
Molecular mechanisms of ferroptosis and ferroptosis-modulating
compounds. Ferroptosis is an iron-dependent form of regulated cell death
characterized by lipid peroxidation. The process is mainly regulated by iron
metabolism, the System Xc–/GSH/GPX4 antioxidant system, and PUFA-dependent lipid
peroxidation pathways. Iron accumulation promotes reactive oxygen species
generation via the Fenton reaction, while inhibition of System Xc– results in
glutathione depletion and GPX4 inactivation, leading to lipid peroxide
accumulation. Enzymes involved in lipid metabolism, including ACSL4, LPCAT3, and
LOXs, promote phospholipid peroxidation. Ferroptosis-modulating compounds
regulate ferroptosis by targeting different pathways, including System Xc–
inhibitors, GPX4 inhibitors, iron metabolism modulators, and lipid peroxidation
inducers. The symbols “
Natural products represent an important source of ferroptosis inducers due to their ability to simultaneously modulate redox homeostasis, iron metabolism, and lipid remodeling. Compared with synthetic agents that often target single molecular nodes, many natural compounds exhibit multi-target regulatory effects and selectively exploit metabolic vulnerabilities of tumor cells [9, 52].
Artemisinin and its semi-synthetic derivatives, including artesunate and dihydroartemisinin, primarily induce ferroptosis through iron-dependent mechanisms. These compounds promote NCOA4-mediated ferritinophagy, enhance intracellular iron availability, and expand the labile iron pool, thereby amplifying Fenton reaction-driven ROS generation and lipid peroxidation [53]. In parallel, artemisinin derivatives suppress the System Xc––GSH–GPX4 axis, further weakening antioxidant capacity and reinforcing ferroptotic signaling [9]. Compared with classical System Xc– inhibitors, artemisinin derivatives exhibit a stronger dependence on iron mobilization rather than direct GPX4 inactivation.
Triptolide induces ferroptosis by coordinately disrupting iron homeostasis and antioxidant defense systems. In cervical and lung cancer models, triptolide suppresses NRF2-dependent cytoprotective pathways and downregulates SLC7A11 and GPX4 expression, leading to excessive ROS accumulation and lipid peroxidation [54, 55]. Although triptolide exhibits potent antitumor activity, its narrow therapeutic window and systemic toxicity remain major challenges for clinical translation.
Tagitinin C induces ferroptosis mainly through activation of endoplasmic reticulum stress-associated signaling pathways. Activation of the PERK–NRF2–HO-1 axis promotes iron release and lipid peroxidation, culminating in ferroptotic cell death in colorectal cancer cells [56]. The specificity of Tagitinin C-induced ferroptosis is supported by selective rescue with ferroptosis inhibitors but not apoptosis or necroptosis inhibitors, indicating a context-dependent ferroptotic mechanism.
Fully synthetic ferroptosis inducers are characterized by higher target specificity and predictable pharmacological properties. These compounds have been widely used as chemical probes to dissect ferroptosis signaling networks and, in some cases, as lead structures for translational development.
Erastin is the prototypical synthetic ferroptosis inducer identified through
phenotype-based chemical screening [57]. By inhibiting the cystine/glutamate
antiporter System Xc–, erastin blocks cystine uptake, depletes intracellular
glutathione, and indirectly inactivates GPX4, resulting in lethal lipid peroxide
accumulation [58]. In addition, erastin induces endoplasmic reticulum stress and
activates the PERK–eIF2
Sulfasalazine, a clinically approved anti-inflammatory drug, also inhibits System Xc– but exhibits weaker ferroptosis-inducing potency than erastin. Nevertheless, its established clinical safety profile makes sulfasalazine an attractive candidate for drug repurposing. In multiple tumor models, sulfasalazine suppresses Solute Carrier Family 7 Member 11 (SLC7A11) expression, enhances lipid peroxidation, and induces ferroptosis at relatively high concentrations [60, 61].
Sorafenib, a multikinase inhibitor approved for hepatocellular carcinoma, has also been reported to function as a System Xc– inhibitor in specific cellular contexts. By suppressing cystine uptake and impairing antioxidant capacity, sorafenib induces ferroptosis in various cancer types [62]. Notably, sorafenib-induced ferroptosis is highly context-dependent and modulated by stress-responsive signaling pathways such as endoplasmic reticulum stress and ATF2 activation [14, 63].
RSL3 is a canonical covalent inhibitor of GPX4 that irreversibly inactivates
GPX4 through covalent modification of its active site [64]. This direct mode of
action renders ferroptosis induction independent of glutathione depletion.
Downstream signaling pathways, such as NF-
FIN56 represents a distinct class of synthetic ferroptosis inducers that promote post-translational degradation of GPX4 rather than inhibiting its enzymatic activity. In addition, FIN56 suppresses coenzyme Q10 biosynthesis through modulation of the mevalonate pathway, thereby weakening lipid antioxidant defenses [8, 67]. Autophagy-dependent ferritin degradation further contributes to iron accumulation and lipid peroxidation, establishing FIN56 as a multifaceted ferroptosis inducer with potent activity in several tumor models [68].
Fully synthetic ferroptosis inducers predominantly act through precise disruption of key antioxidant defense nodes, particularly System Xc– and GPX4. Their high specificity and mechanistic clarity make them valuable tools for ferroptosis research and attractive starting points for drug development. However, challenges related to tumor selectivity, systemic toxicity, and adaptive resistance remain significant obstacles for clinical translation, emphasizing the need for combination strategies and biomarker-guided patient stratification. The representative ferroptosis-inducing compounds, their molecular targets, and tumor models are summarized in Table 1 (Ref. [8, 9, 53, 54, 55, 56, 59, 60, 61, 62, 67, 69, 70]).
| Compound | Chemical structure | Category | Tumor cell line(s) | Primary ferroptosis mechanism | Reference |
| Artemisinin (ART) | 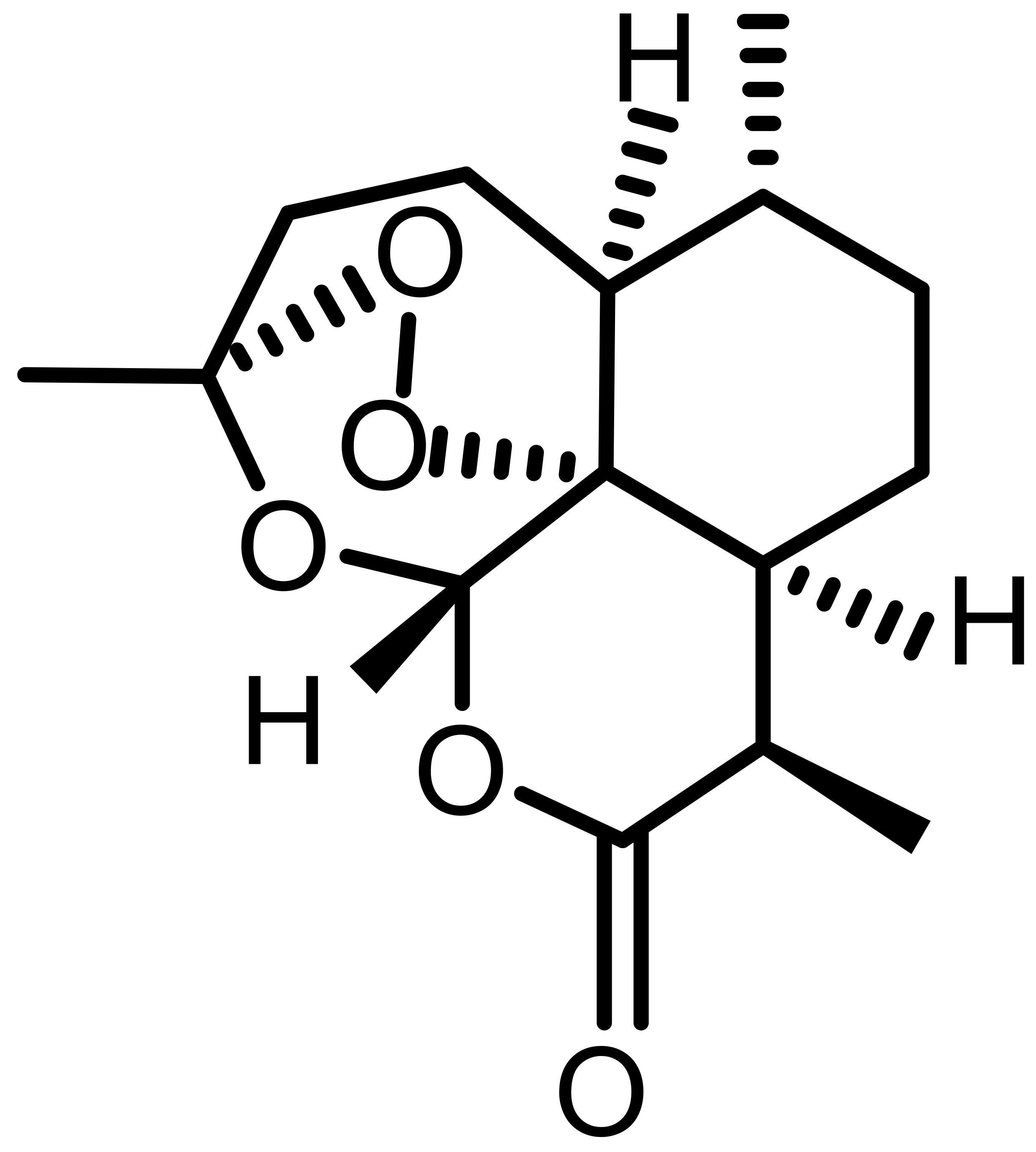 |
Natural | KTCTL-26, CRC, glioma | Induces ferritinophagy; increases LIP; ER stress activation | [9] |
| Triptolide (TPL) | 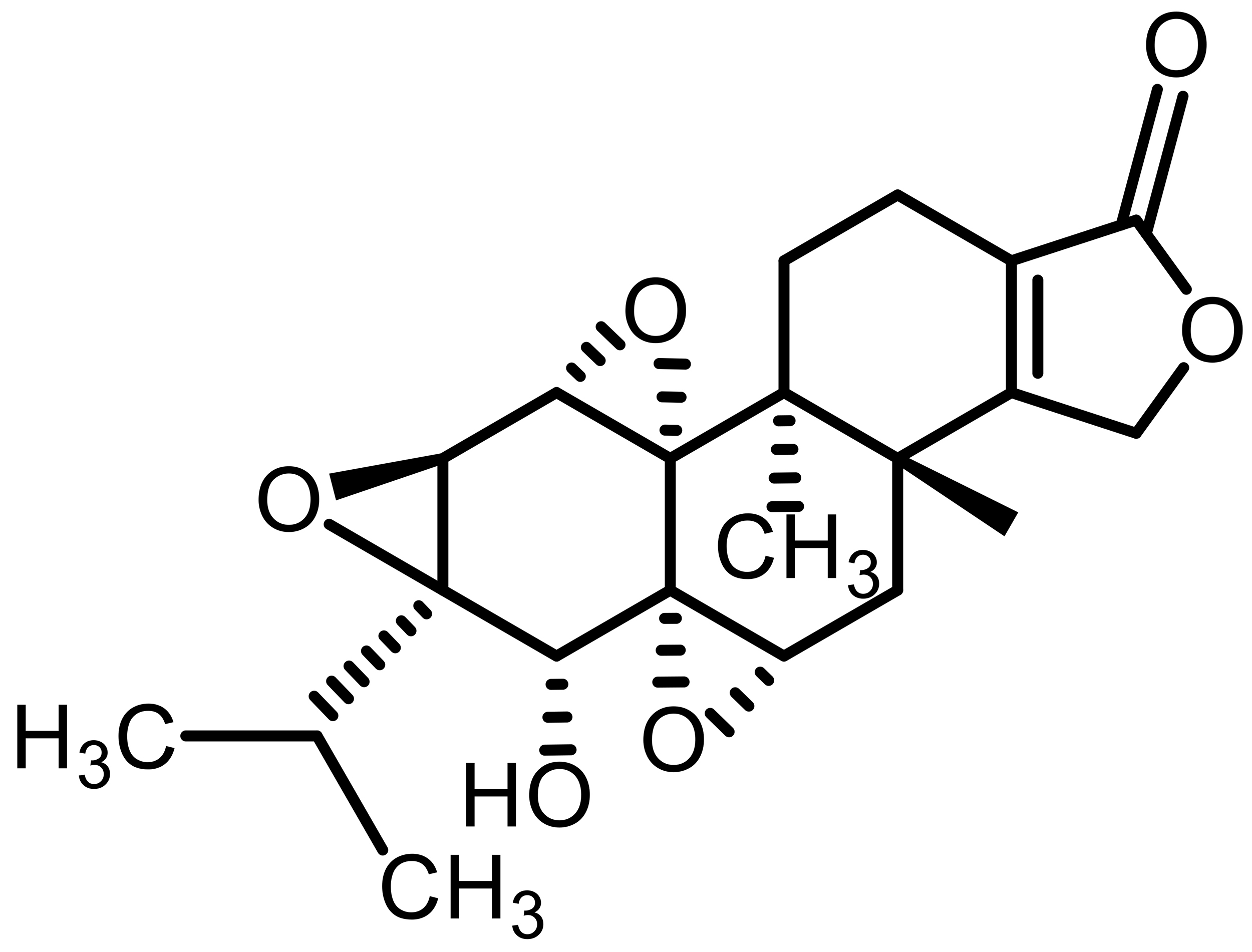 |
Natural | A549 lung cancer | Inhibits NRF2 signaling; downregulates GPX4/SLC7A11 | [54, 55] |
| Tagitinin C (TC) | 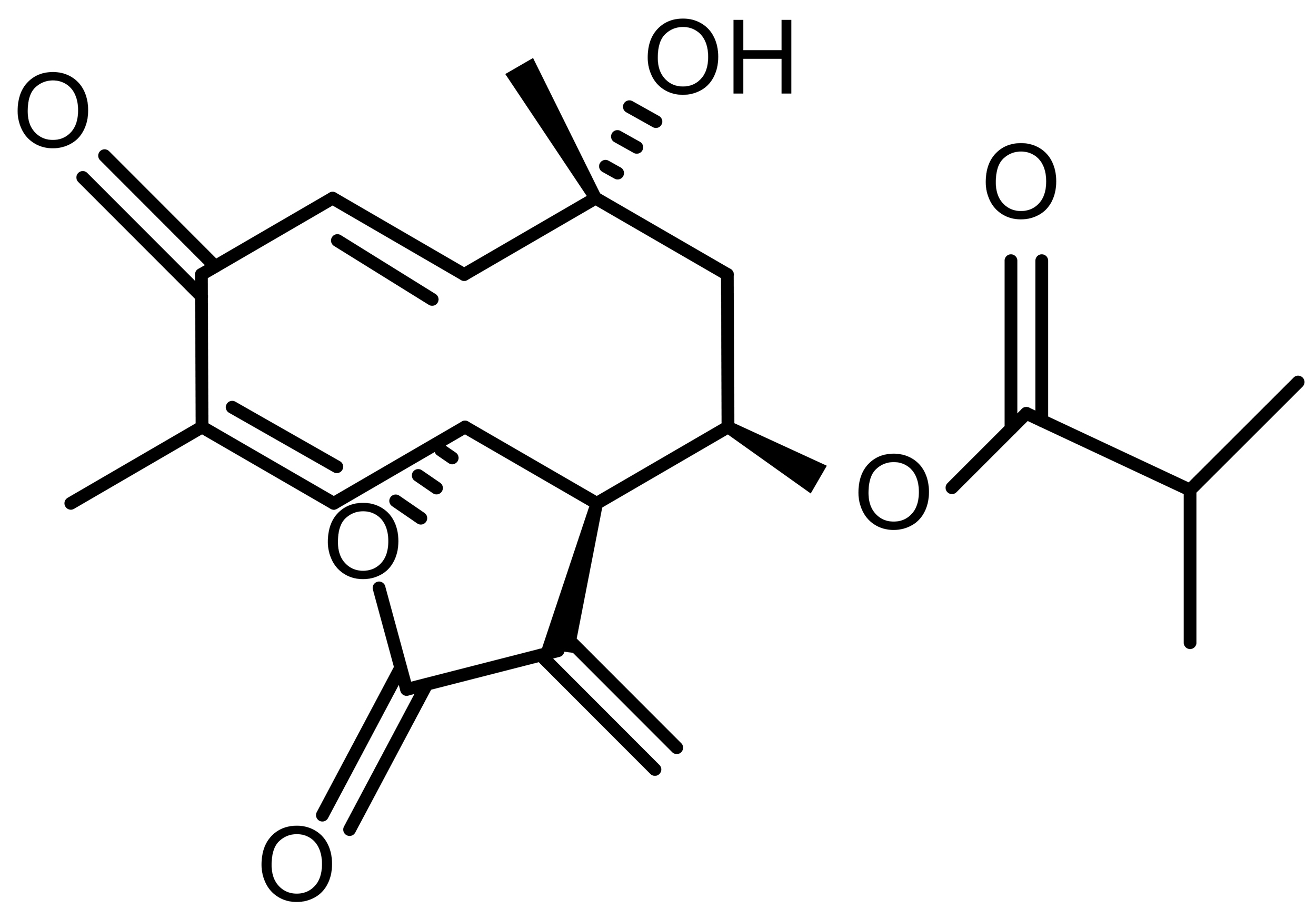 |
Natural | HCT116 colorectal cancer | PERK–NRF2–HO-1 activation; iron-dependent lipid ROS | [56] |
| Artesunate (ATS) | 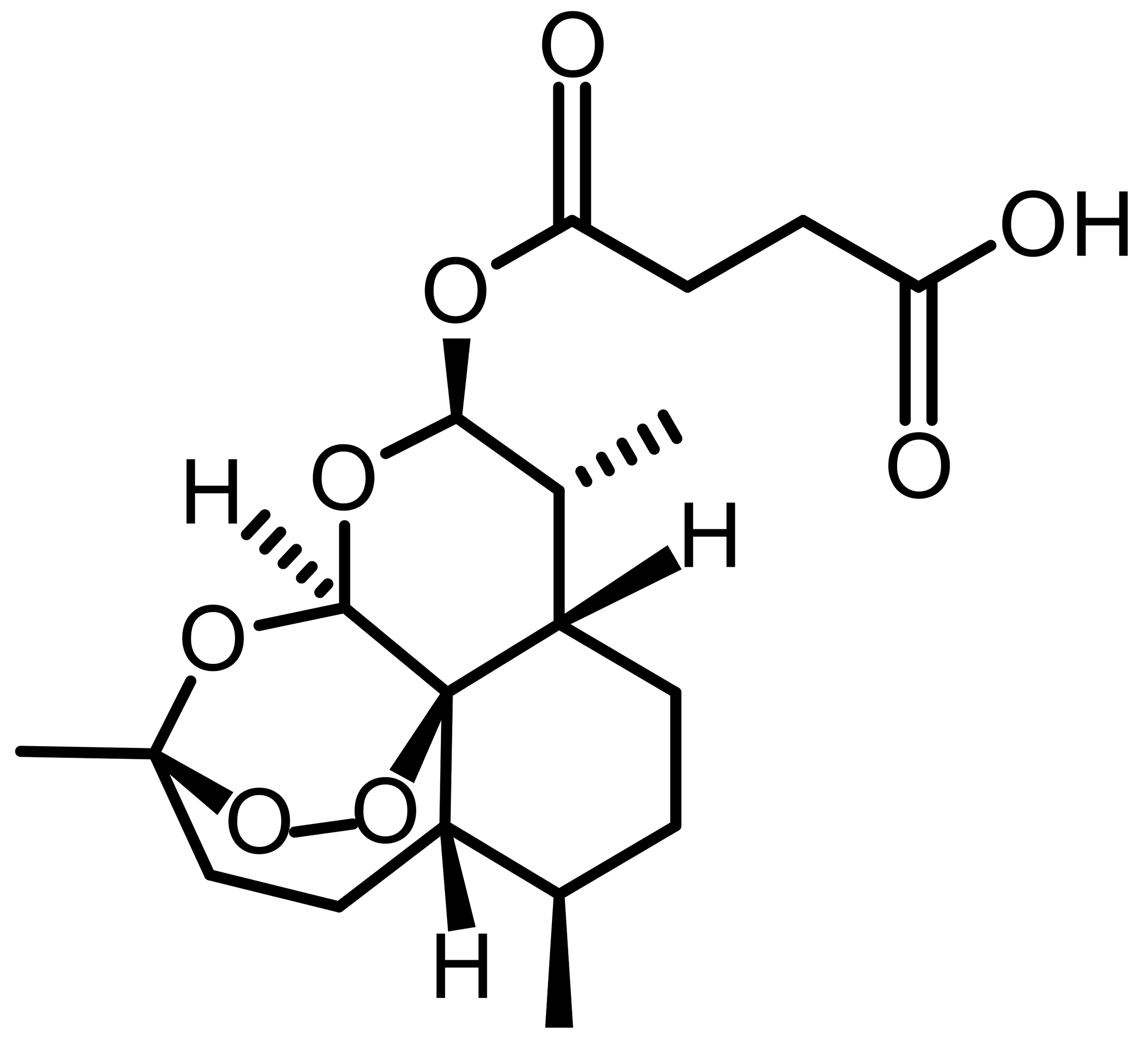 |
Semi-synthetic | MCF-7, MDA-MB-231 | Iron-dependent ROS generation; GPX4 suppression | [53] |
| Dihydroartemisinin (DHA) | 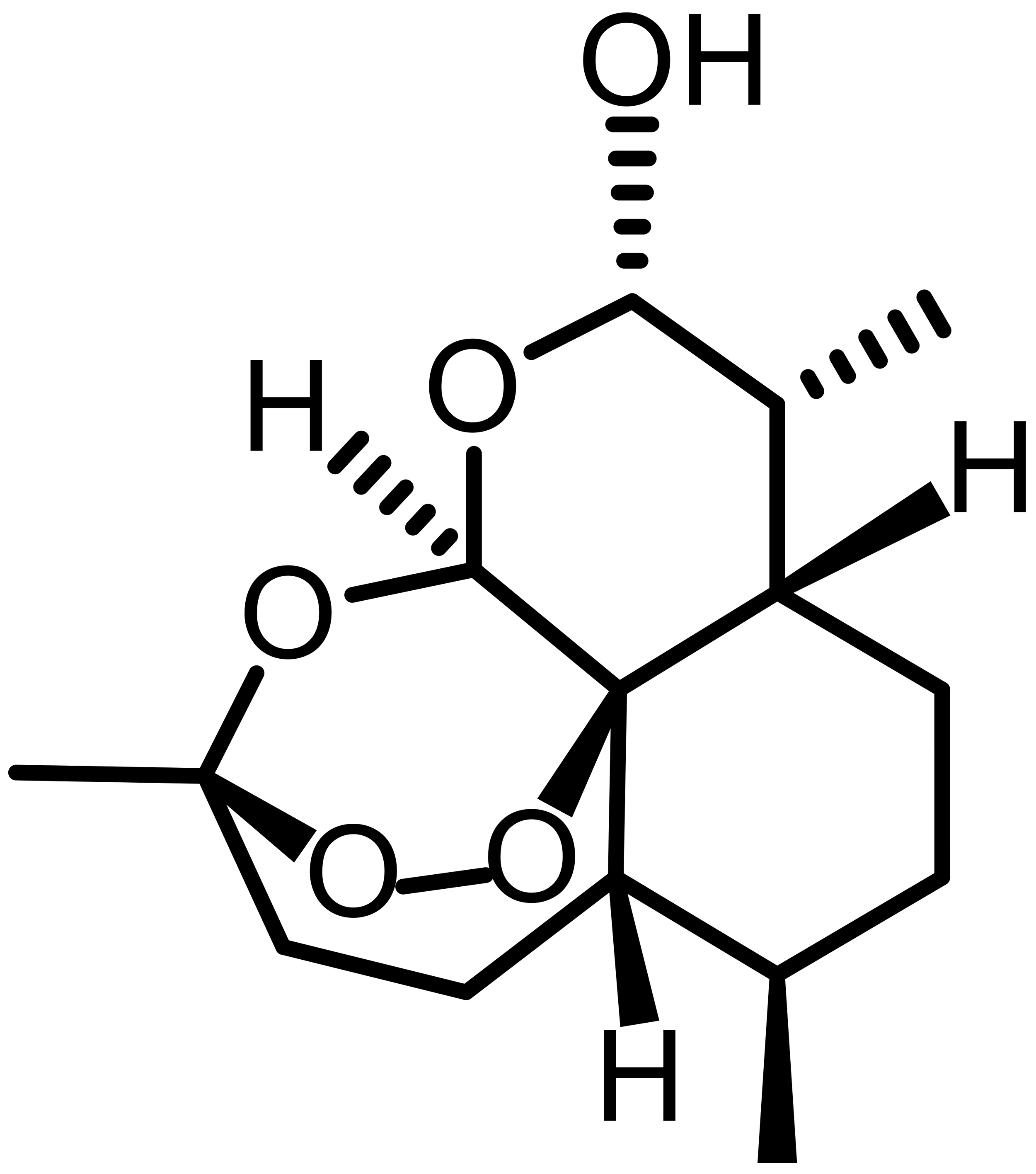 |
Semi-synthetic | HL-60, THP-1 | Ferritinophagy; iron accumulation; lipid peroxidation | [53] |
| Erastin | 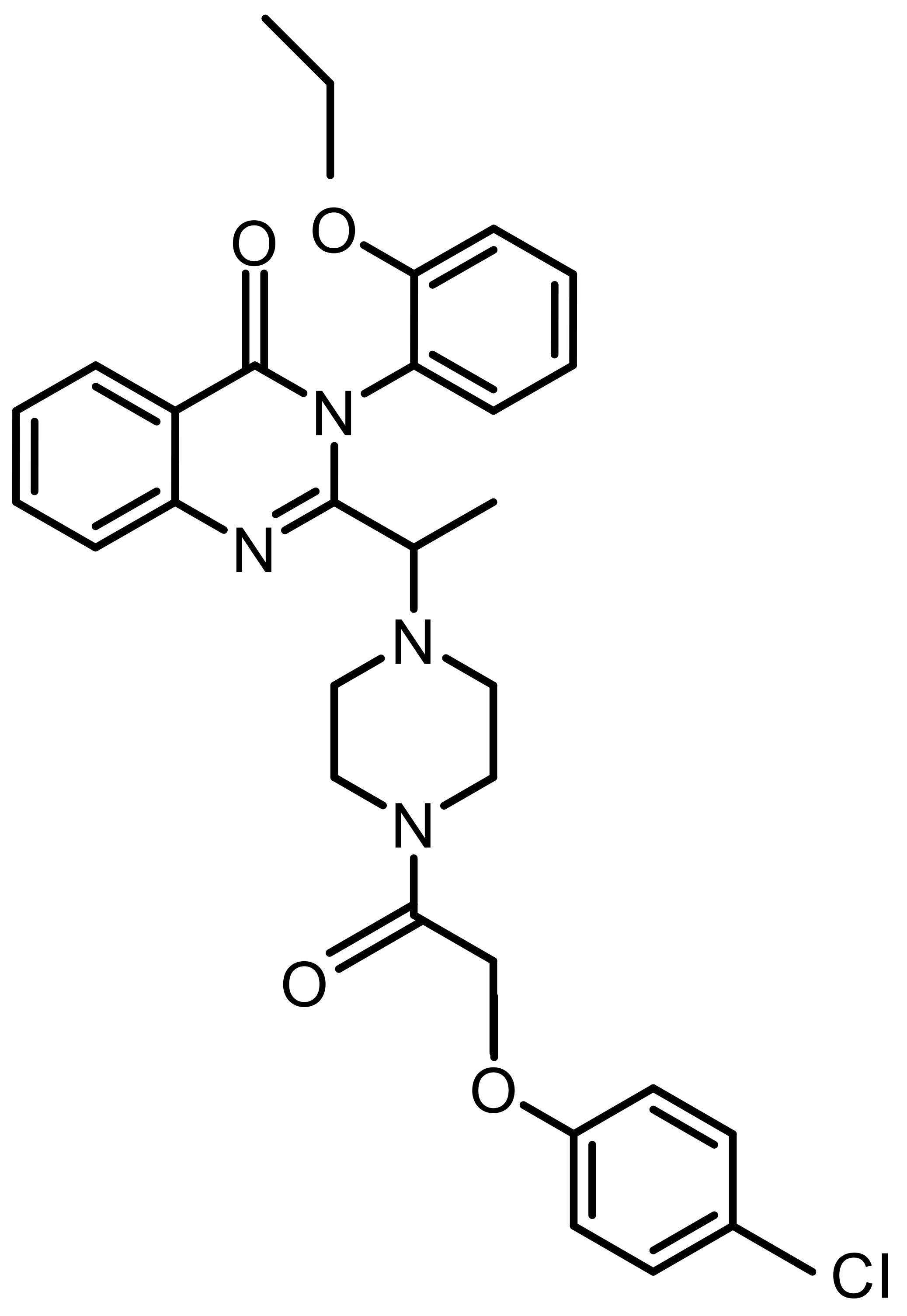 |
Synthetic | HeLa, SiHa | System Xc– inhibition; GSH depletion | [59, 69] |
| Sorafenib | 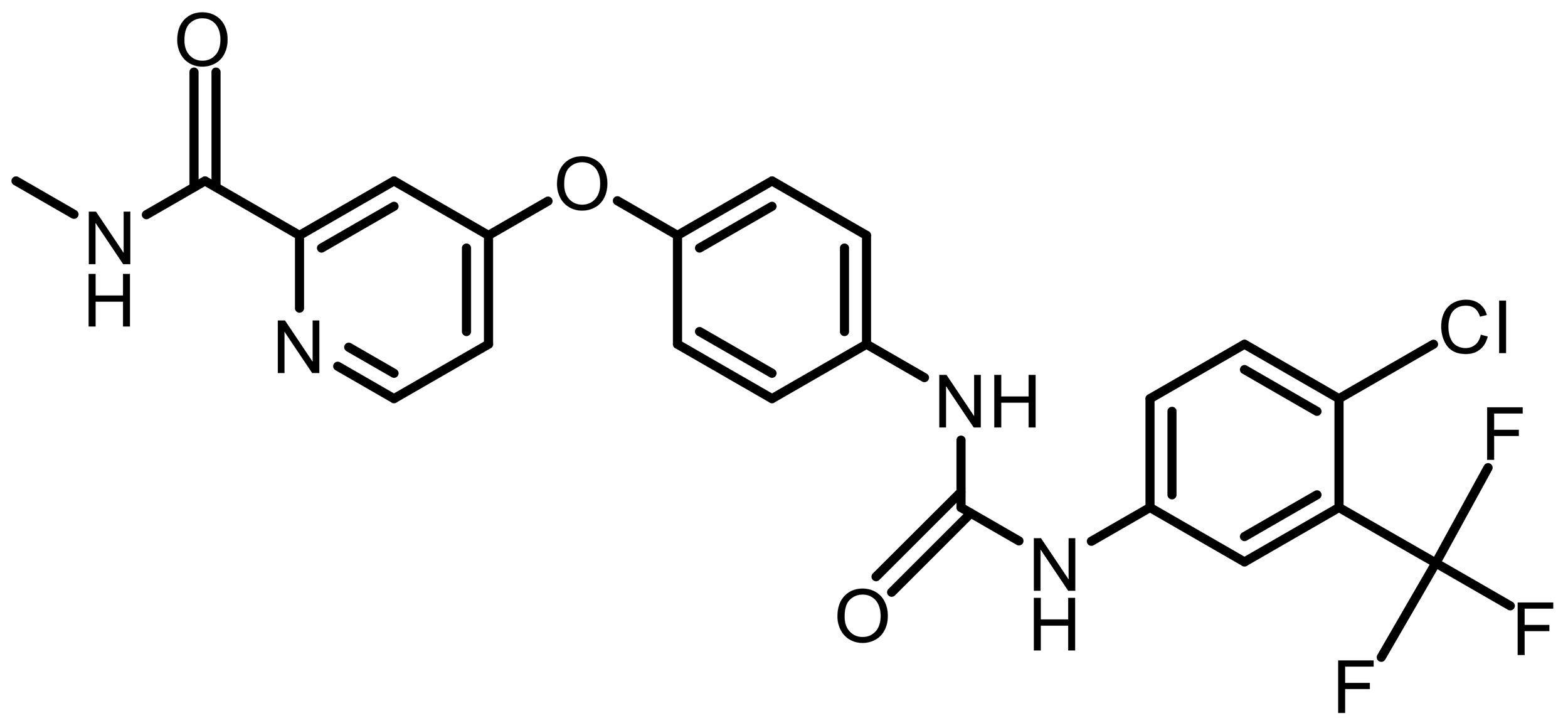 |
Synthetic | Hepatocellular carcinoma | System Xc– inhibition; ferroptosis induction | [62] |
| Sulfasalazine (SAS) | 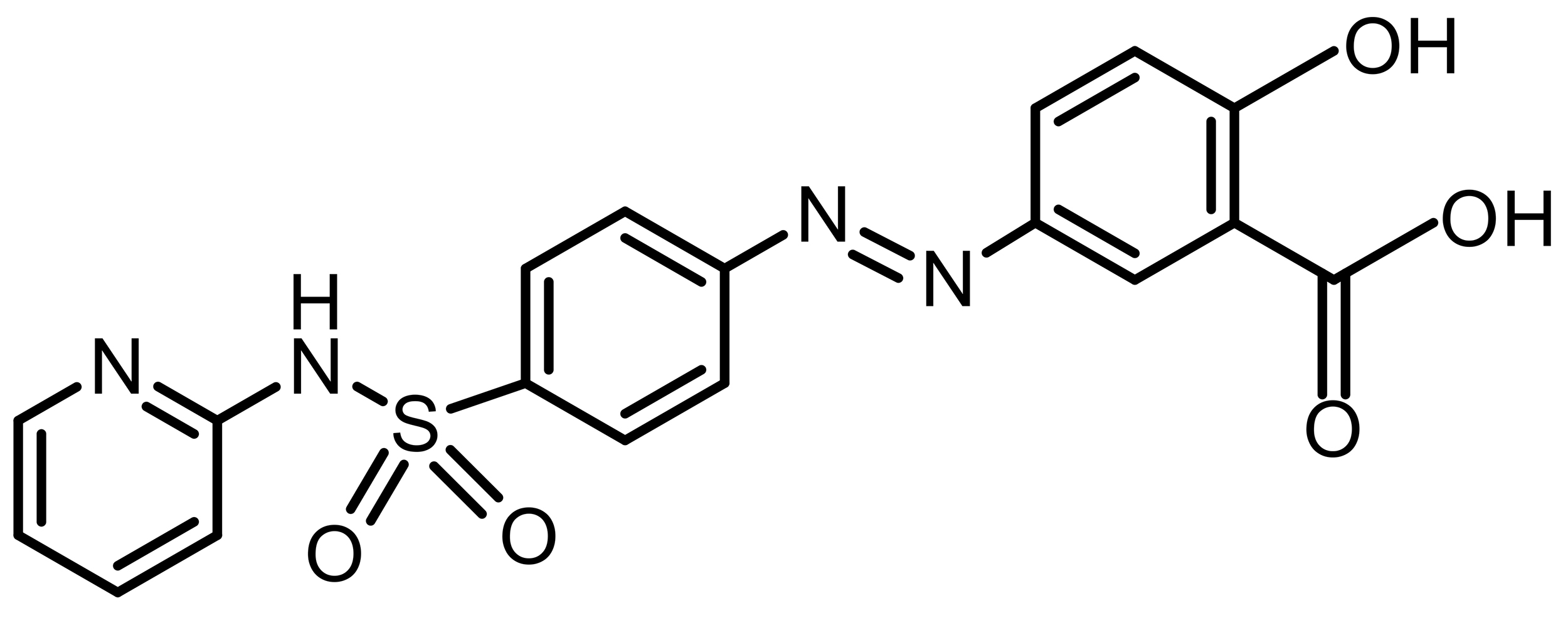 |
Synthetic | TE-1 esophageal cancer | System Xc– inhibition; SLC7A11 suppression | [60, 61] |
| FIN56 | 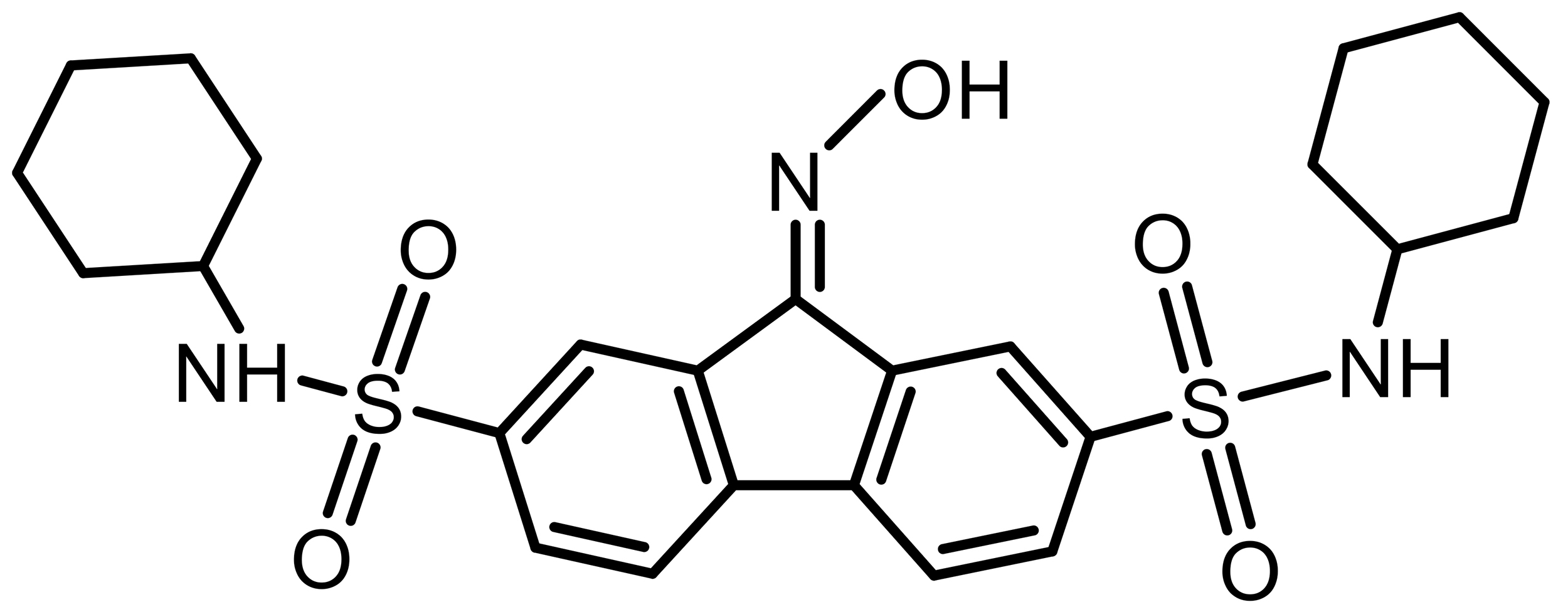 |
Synthetic | Breast cancer, LN229 | Promotes GPX4 degradation; CoQ10 depletion | [8, 67] |
| RAS-selective lethal 3 (RSL3) | 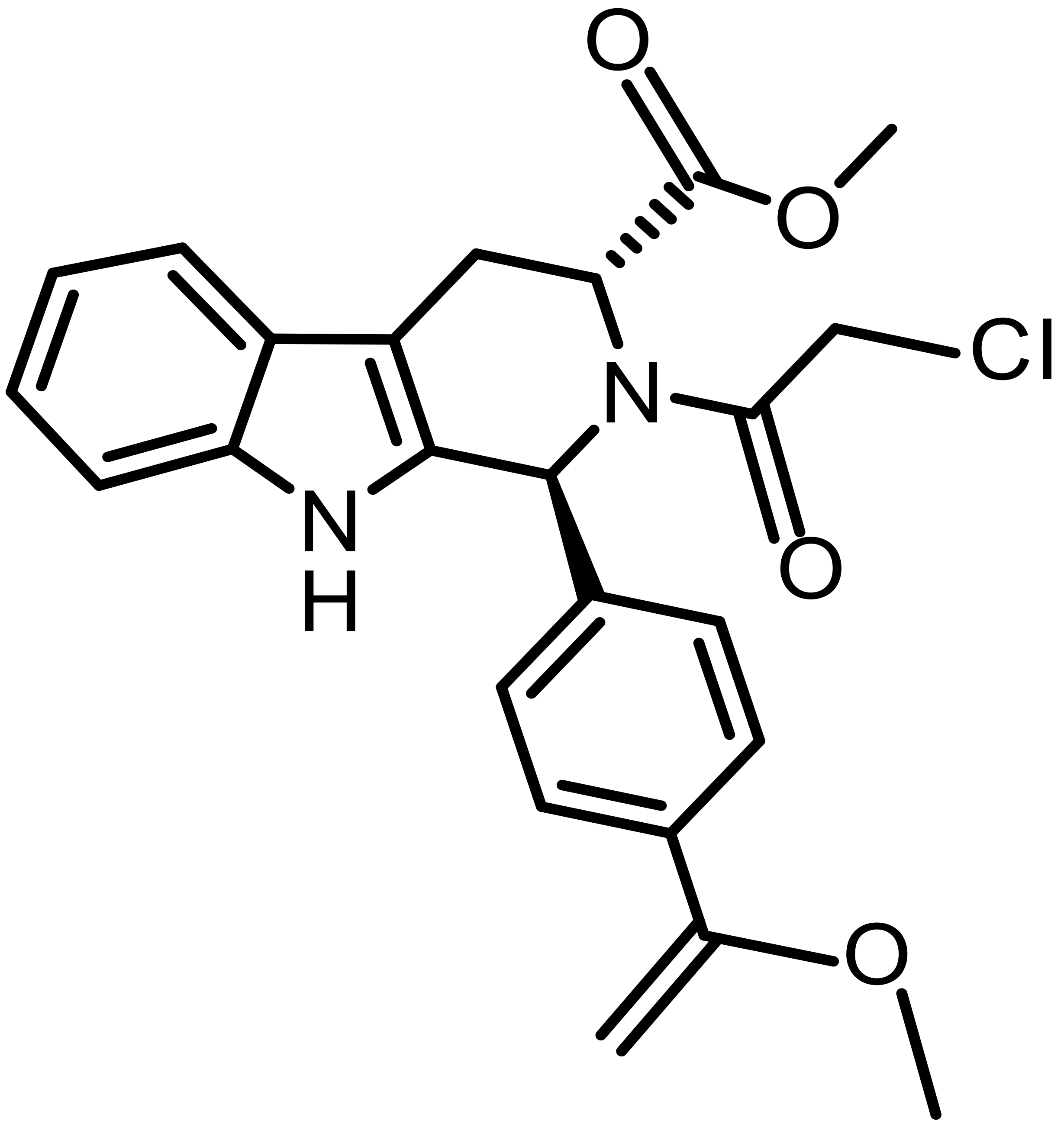 |
Synthetic | U87, U251 glioma | Direct GPX4 inhibition | [70] |
Recent studies have identified a variety of ferroptosis-inducing compounds targeting system Xc–, GPX4, lipid metabolism, and iron homeostasis; however, most of these compounds remain at the experimental stage and have not yet progressed to clinical application. One major limitation is the pharmacokinetic profile of many ferroptosis inducers, as poor solubility, low bioavailability, and rapid metabolic clearance significantly restrict their in vivo efficacy [2]. In addition, ferroptosis is not a tumor-specific process, and excessive lipid peroxidation may also cause damage in normal tissues, particularly in the liver, kidney, and nervous system, which raises concerns regarding systemic toxicity and therapeutic safety [5]. Another important issue is the lack of tumor specificity. Many ferroptosis-inducing agents target general antioxidant systems such as GPX4 or glutathione metabolism, which are also essential for normal cell survival. Strategies including nanoparticle delivery systems, liposomes, and prodrug design have been proposed to improve tumor targeting and reduce off-target effects [8].
Accumulating evidence suggests that ferroptosis induction may be more effective when combined with other anticancer therapies rather than used alone. Ferroptosis has been reported to enhance the efficacy of chemotherapy, radiotherapy, and immunotherapy by increasing oxidative stress and altering tumor cell metabolic vulnerability [49]. Ferroptotic cells can also release damage-associated molecular patterns and lipid peroxidation products that influence the tumor immune microenvironment, indicating potential synergy between ferroptosis induction and immune checkpoint blockade therapy [14]. Another important challenge is ferroptosis resistance. Tumor cells can develop resistance through upregulation of SLC7A11, activation of the FSP1–CoQ10 pathway, increased NADPH production, and lipid repair pathways, which may reduce the effectiveness of ferroptosis-inducing compounds [2]. Understanding the mechanisms of ferroptosis sensitivity and resistance, identifying predictive biomarkers, and developing combination therapeutic strategies may represent important directions for future research and may promote the translation of ferroptosis-based therapy from basic research to clinical oncology.
Ferroptosis has emerged as a potential therapeutic vulnerability in cancer; however, its clinical translation remains limited by several pharmacological and biological challenges. Iron accumulation and lipid peroxidation are not tumor-specific processes, and normal tissues with high metabolic activity may also be susceptible to oxidative damage, raising concerns regarding systemic toxicity and therapeutic safety [2]. In addition, many ferroptosis-inducing compounds exhibit suboptimal pharmacokinetic properties, including poor solubility, rapid metabolism, and narrow therapeutic windows, which restrict their in vivo efficacy [10]. Tumor selectivity also remains a major obstacle, since inhibition of GPX4 or glutathione metabolism affects fundamental antioxidant systems required for normal cell survival. Advanced drug delivery strategies, such as nanoparticle carriers, liposomes, and tumor-activated prodrugs, may improve intratumoral drug accumulation while reducing off-target effects and systemic toxicity [6].
Another major challenge is the identification of reliable biomarkers for predicting ferroptosis sensitivity and therapeutic response. Several ferroptosis-related molecules, including SLC7A11, GPX4, ACSL4, and FSP1, have been proposed as potential biomarkers; however, their predictive value appears to be tumor-type dependent and has not yet been validated in clinical settings [5]. Tumor heterogeneity, metabolic reprogramming, and microenvironmental factors further influence ferroptosis sensitivity. In addition, tumor cells can develop ferroptosis resistance through NRF2 activation, upregulation of FSP1, enhanced iron sequestration, lipid remodeling, and metabolic adaptation, which may limit the efficacy of ferroptosis-inducing therapies [5, 10]. These findings indicate that ferroptosis sensitivity is determined by integrated metabolic and genetic contexts rather than a single molecular target.
From a translational research perspective, the development of ferroptosis-based cancer therapy requires a systematic strategy that integrates mechanistic research, biomarker identification, drug development, and clinical evaluation. Some clinically approved drugs, such as sorafenib and sulfasalazine, have been reported to induce ferroptosis under specific conditions, suggesting that drug repurposing may accelerate clinical translation [7]. Ferroptosis induction may also enhance the efficacy of chemotherapy, targeted therapy, and immunotherapy, indicating that combination therapy could represent a more realistic clinical strategy than single-agent ferroptosis induction. Future research should focus on integrated biomarker panels, patient stratification strategies, tumor-targeted drug delivery systems, and clinical trials evaluating combination therapies. Bridging the gap between mechanistic studies and clinical oncology will be essential for translating ferroptosis from experimental research into clinical cancer therapy [6, 7].
RYZ and XH conceived the study. RYZ and MeiX prepared the original draft and figures. MSL and ManX contributed to figure preparation. QYH contributed to the conceptualization of the study and provided critical guidance on the review framework. XH and QYH supervised the study. All authors contributed to manuscript revision. All authors have read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflicts of interest.
During the preparation of this manuscript, ChatGPT was used only for language editing and improving readability. The authors critically reviewed, revised, and approved the final manuscript and take full responsibility for the integrity and accuracy of the content.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.