











1 Jiangsu Key Laboratory of Marine Biological Resources and Environment, Jiangsu Key Laboratory of Marine Pharmaceutical Compound Screening, School of Pharmacy, Jiangsu Ocean University, 222005 Lianyungang, Jiangsu, China
2 Medicine Center, Guangxi University of Science and Technology, 545006 Liuzhou, Guangxi, China
3 Grand Medical Nutrition Science (Wuhan) Co., LTD., 430040 Wuhan, Hubei, China
†These authors contributed equally.
Abstract
Melanocortin-4 receptor (MC4R) is a key receptor in the hypothalamus regulating appetite and energy metabolism. Research has indicated that MC4R agonists have demonstrated potential for weight loss; its antagonists may stimulate appetite and weight gain, showing potential therapeutic applications in patients with anorexia or cachexia or those requiring weight restoration. However, the signaling mechanism, clinical applications, and safety profile of MC4R antagonists still require in-depth investigation. The purpose of this review is to comprehensively summarize the research progress of MC4R antagonists, analyze their mechanism of action and clinical applications, evaluate their safety profile, and explore future research directions. Future studies should focus on developing safer and more effective MC4R antagonists and exploring the therapeutic potential of MC4R in other diseases.
Keywords
- MC4R antagonist
- appetite
- cachexia
- wasting diseases
- mechanism of action
Weight disorders include obesity, anorexia, cachexia, etc., which cause abnormal changes in body weight and appetite. Obese patients experience continuous accumulation of body fat due to energy intake exceeding expenditure, which may be associated with excessive appetite and dysregulation of energy -metabolism, resulting in sustained weight gain [1, 2, 3]. Anorexia patients experience a significant decline in appetite due to psychological or physiological factors, consuming far less than their body’s requirements, leading to rapid weight loss [4]. Cachexia, often secondary to severe chronic diseases such as cancer or AIDS, is characterized not only by reduced appetite but also by substantial loss of muscle and fat tissue, causing pronounced weight decline [5, 6]. These diseases exert significant impacts on patients’ physical and mental health, making it of paramount importance to thoroughly investigate their pathogenesis and identify effective intervention measures.
The melanocortin receptors belongs to a subfamily of the G protein-coupled receptor (GPCR) superfamily with therapeutic potential, comprising MC1R, MC2R, MC3R, MC4R, and MC5R [7]. Melanocortin receptors (MC1R-MC5R) are located on various cell types and distributed across different body systems. MC1R is expressed in skin melanocytes and plays a crucial role in determining skin and hair pigmentation; it is also expressed in leukocytes, potentially mediating anti-inflammatory properties. MC2R, which refers to the adrenocorticotropic hormone (ACTH) receptor, mediating the effect of ACTH on steroid secretion. MC3R is distributed in multiple regions of the central nervous system and peripheral tissues, participating in the regulation of energy homeostasis. MC4R is primarily expressed in the central nervous system and plays a critical regulatory role in the processes of food intake and energy metabolism. MC5R is found in peripheral tissues and primarily contributes to exocrine functions [7, 8].
Melanocortin-4 receptor (MC4R) is widely distributed in the central nervous system (such as the hypothalamic arcuate nucleus and paraventricular nucleus) and peripheral tissues of mammals, and is primarily involved in regulating energy balance, appetite, and body weight homeostasis. Studies have shown that abnormalities in the MC4R gene are a major cause of severe obesity [9, 10]. Gene mutations often manifest as complete or partial loss of function, impairing receptor signal transduction and subsequently leading to metabolic abnormalities such as obesity, elevated leptin levels, hyperinsulinemia, and insulin resistance [11]. The statement indicates that the functional level of MC4R plays a pivotal role in the body’s energy metabolism and weight regulation. Under normal functional conditions, activation of MC4R suppresses appetite and increases energy expenditure (e.g., raising basal metabolic rate and thermogenic capacity). By modulating the distribution and metabolism of adipose tissue, it reduces fat accumulation. MC4R agonists have shown highly effective in appetite suppression and weight loss, but long-term use may trigger compensatory metabolic adaptations and cannot meet the needs of catabolic conditions such as anorexia nervosa or cachexia.
Recent studies have shown that, beyond its classic role in energy metabolism,
MC4R also plays a crucial neuroprotective role, primarily through
anti-inflammatory mechanisms [12]. Activation of MC4R can inhibit signaling
pathways such as NF-
Given that MC4R plays a crucial role in both metabolic diseases and neurological disorders, precise regulation of its function is of paramount importance. MC4R antagonists dynamically regulate energy balance through reversible inhibition of MC4R, and are applicable for the treatment of anorexia, cachexia, anxiety, depression, and metabolic disorders [14, 15]. The development of MC4R antagonists has gained attention due to their higher selectivity and specificity, as well as their potential therapeutic value in treating cachectic diseases. This article primarily outlines the current research and development status of MC4R antagonists, aiming to provide insights for subsequent studies.
Melanocortins are a class of hormones and proteins with diverse biological
functions, first discovered in 1916 [16]. They exert their effects by interacting
with G protein-coupled melanocortin receptors, activating multiple intracellular
signaling pathways. In humans, melanocortins primarily function through
regulating energy balance, immune responses, and pigmentation. In 1993, MC4R was
cloned using degenerate PCR, but its function remained unknown [17]. Subsequent
studies have shown that MC4R may be involved in regulating energy homeostasis. In
1997, a series of groundbreaking studies on mice validated this hypothesis. In
1998, human genetic studies discovered that MC4R gene mutations could cause
monogenic obesity [17], revealing the critical role of MC4R in human weight
regulation. In the early 2000s, researchers began to explore the molecular
mechanism of MC4R. For example, in 2001, studies demonstrated that MC4R gene
mutations lead to receptor internalization and functional inactivation [18].
Additionally, studies in 2003 revealed the role of MC4R in energy homeostasis and
its binding properties with
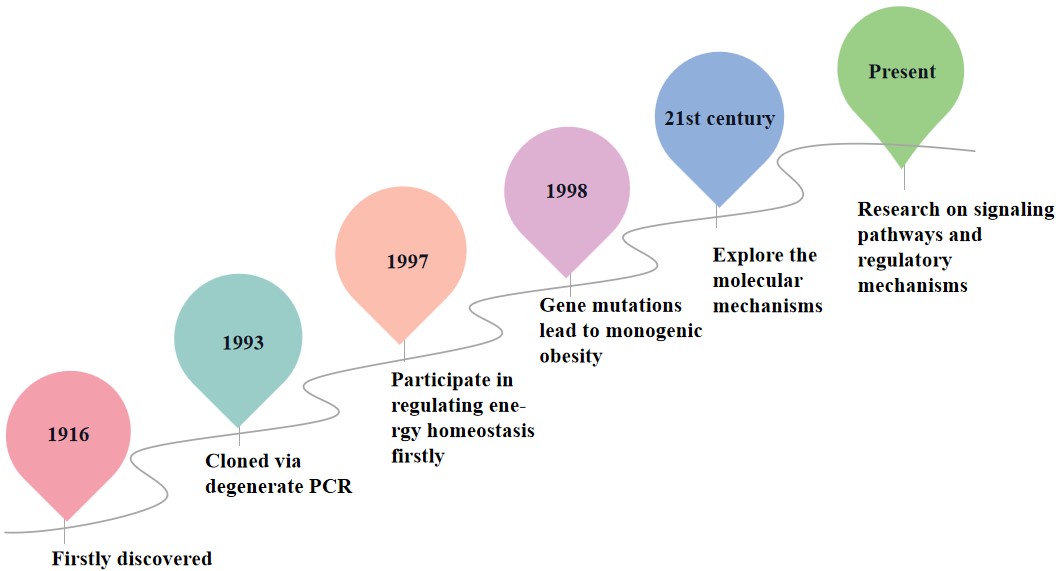 Fig. 1.
Fig. 1.
The discovery process of MC4R. MC4R, Melanocortin-4 receptor.
The MC4R protein, composed of 332 amino acids, is encoded by a gene located at chromosome 18q21.3, which contains only one exon [21]. MC4R is composed of an extracellular N-terminus, seven transmembrane segments, three intracellular loops, three extracellular loops, and a cytoplasmic tail [22, 23, 24], as shown in Fig. 2. This structural configuration enables it to regulate signal transduction by binding to G proteins. In 2020, Yu et al. [25] resolved the crystal structure of MC4R bound to the antagonist SHU9119 [26], this structure revealed the three-dimensional conformation of MC4R and its binding mode with SHU9119. The study demonstrated that MC4R is a seven-transmembrane helical protein (TM1–TM7) containing a binding pocket for SHU9119 and a divalent calcium ion (Ca2+) cofactor [26].
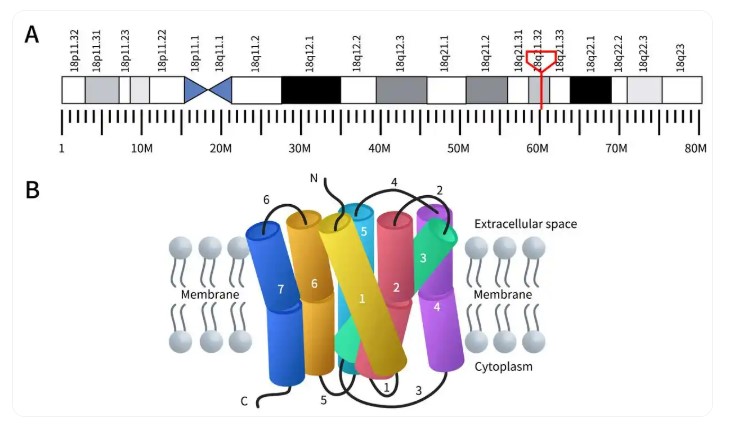 Fig. 2.
Fig. 2.
Gene location and protein structure of MC4R. (A) The genomic location of MC4R. The red box indicates the location of the gene encoding the MC4R protein. (B) Its protein structure. (The genomic location data derived from WIKIPWDIA.)
MC4R is a key receptor regulating energy balance and appetite. Its active sites
are primarily located in the transmembrane domains (especially TM3 and TM6),
which interact with various agonists and antagonists to trigger or inhibit signal
transduction, thereby influencing energy metabolism and appetite regulation. For
example, the endogenous ligand
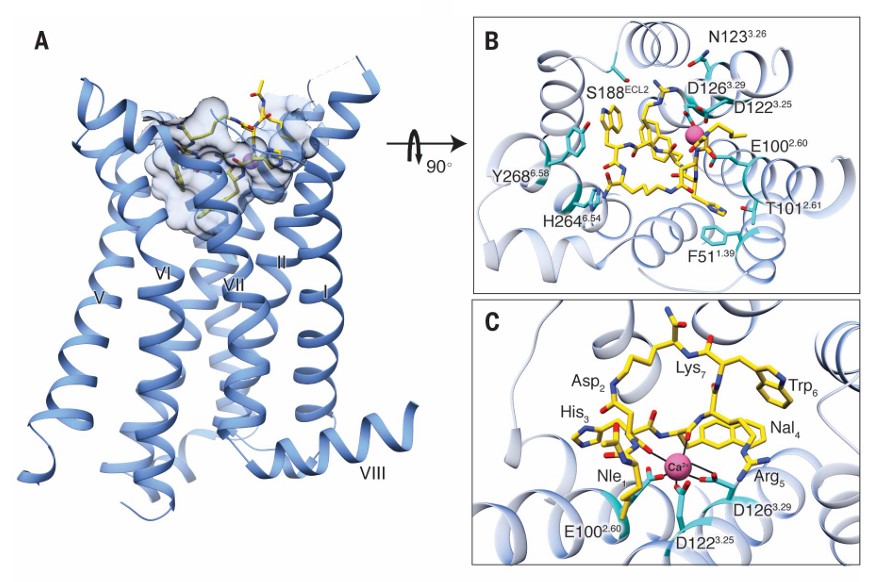 Fig. 3.
Fig. 3.
Structure of MC4R bound with SHU9119 [25]. (A) Side view of the crystal structure of the MC4R-SHU9119 complex. (B) Structure viewed from the extracellular side, illustrating the interaction network among MC4R, SHU9119, and metal ions. (C) Expanded view of the metal ion-binding site in the protein.
SHU9119 prevents the transition of L1333.36 from the inactive
state to the active state through steric hindrance. L1333.36 is located in
the third transmembrane region (TM6) of MC4R, which acts as a key regulator for
the activation of Class A GPCRs. It interacts with W2586.48 to influence the
displacement of TM6. In the inactive state, L1333.36 maintains a CH-
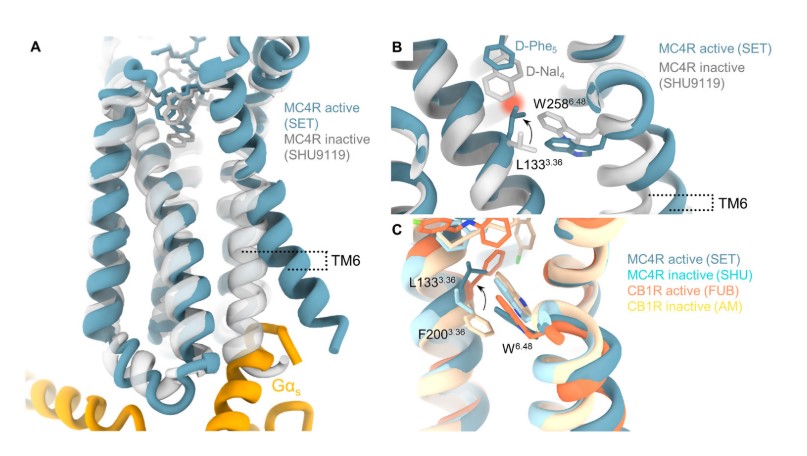 Fig. 4.
Fig. 4.
MC4R activation switch [28]. (A) Superimposition of the MC4R active complex with the antagonist-bound receptor. (B) The D-Nal4 fragment in SHU9119 stabilizes the inactive switch conformation. (C) Superimposition of the MC4R activation switch (active—light blue, inactive—sky blue) with the classical “toggle switch” of CB1R (active—pink, inactive—orange conformation).
In addition, under certain circumstances, Ca2+ ions also participate in the ligand binding process. When MC4R binds to the agonist setmelanotide, the carbonyl oxygen atom forms a coordination bond with Ca2+, thereby enhancing binding stability [28, 29]. This binding pattern suggests that Ca2+ plays a pivotal role in the ligand-binding process, possibly by stabilizing the receptor conformation or facilitating interactions between the ligand and receptor. The structure of the SHU9119-MC4R complex (DB ID: 6W25) reveals the binding mode of the antagonist SHU9119 and further demonstrates the critical role of Ca2+ ions within the binding pocket [28]. In this complex, SHU9119 binds to amino acid residues F184, Y268, and F284 through hydrophobic interactions, while the carbonyl oxygen atom forms hydrogen bonds with Ca2+, thus enhancing the binding affinity between the ligand and the receptor [28].
The Ca2+-mediated fine-tuning mechanism ensures that MC4R can sensitively
respond to fluctuations in upstream signals, thereby establishing its structural
basis as a central hub for energy balance. The most representative regulatory
pathway is the leptin-
| Pathway | Main downstream molecules | Effect on obesity/energy metabolism |
| G-protein | G protein alpha s (G |
PKA phosphorylates Melanocortin-4 receptor (MC4R), strengthening its anorectic (appetite-suppressing) effect. |
| Mediates MC4R’s G-protein-independent signaling, contributing to long-term regulation of energy balance. | ||
| Ca2+ pathway | Phospholipase C beta (PLC |
Alters neuronal excitability, thereby modulating feeding impulses. |
| PI3K/Akt | Phosphoinositide 3-kinase (PI3K) |
Interacts with leptin signaling and regulates hypothalamic energy sensing. |
The primary signaling pathways involved in MC4R include the G protein,
The complexity of the MC4R signaling pathway is not only reflected in its
diverse physiological functions and regulatory mechanisms but also in its close
association with G protein-coupled receptors. When delving into the complexity of
the MC4R signaling pathway, we must focus on one of its core mechanisms, the G
protein-coupled signaling pathway. MC4R activates the cAMP-PKA signaling pathway
through direct coupling with G proteins, which constitutes its primary signal
transduction mechanism (see Table 2, Ref. [32, 33, 34]). This mechanism plays a critical role in regulating
appetite suppression and energy balance within the hypothalamus [35].
Specifically, after MC4R binds to the G protein, the
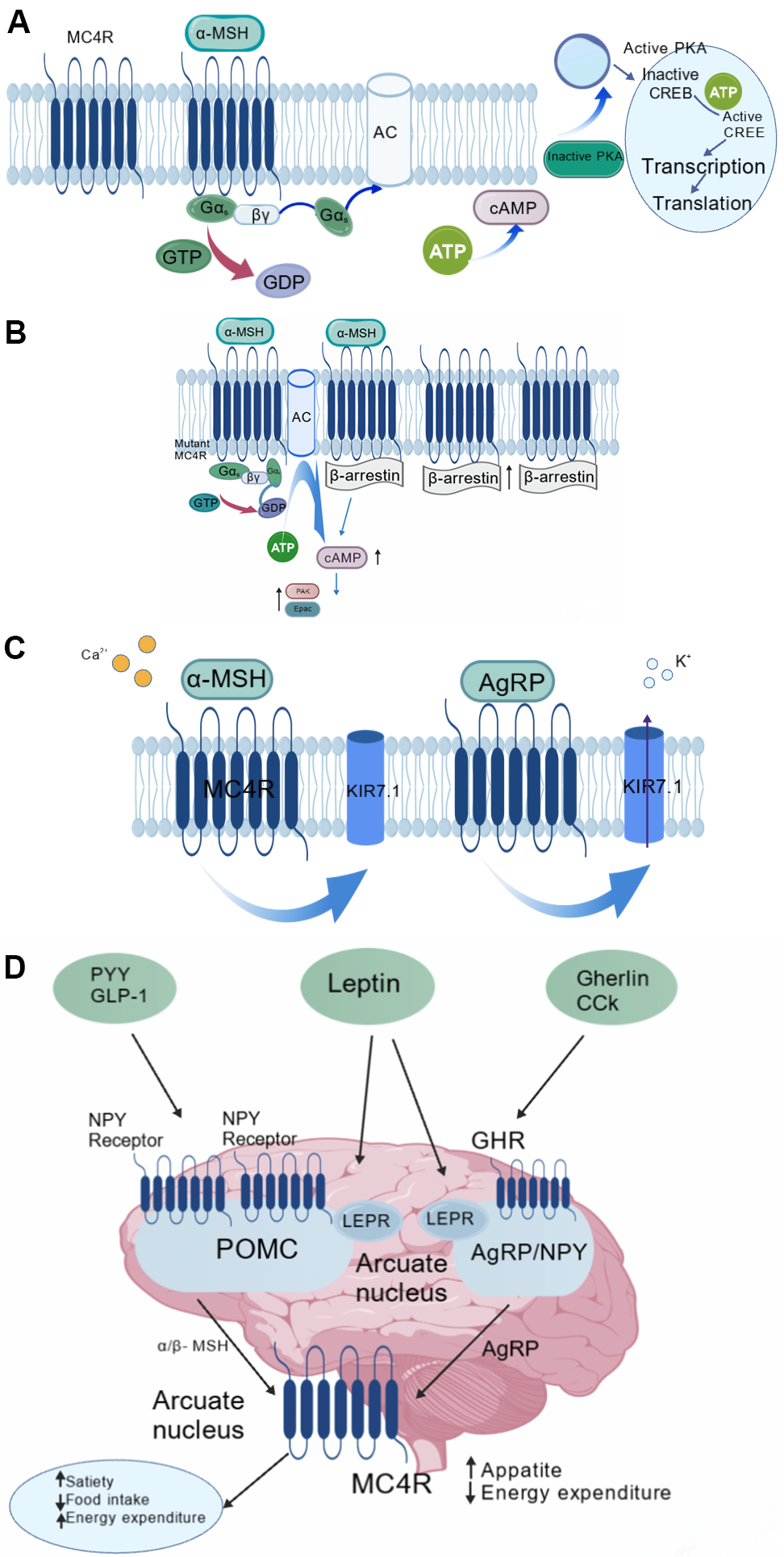 Fig. 5.
Fig. 5.
Signaling pathways of MC4R [34, 37]. (A) G protein-coupled
signaling pathway. (B)
| Features | G protein-coupled signaling | Non-G protein-dependent signaling |
| Core mediators | G protein subtypes such as G |
|
| Signal initiation conditions | After ligand binding, it induces conformational changes in the receptor and activates G proteins | After the receptor is phosphorylated by GRK, it recruits |
| Main pathways | G |
|
| Signal initiation speed | Fast (in seconds), such as rapid accumulation of cAMP | Slow (in minutes to hours), such as ERK phosphorylation regulating gene transcription |
| Signal duration | Transient (depending on cAMP degradation) | Persistent (continuous activation of endosomal signals) |
| Main effects | Acute appetite suppression and metabolic regulation | Regulation of gene expression and receptor recycling |
| Case associations | Inactivating mutations in MC4R lead to obesity | Signal imbalance is associated with insulin resistance |
In addition to the classical G protein signaling pathway, the MC4R signaling pathway also involves non-G protein-dependent pathways, adding flexibility and diversity to its signal transduction and further enriching its complex signaling network.
Studies show that after MC4R is activated by ligands, G protein-coupled receptor
kinases (GRKs) phosphorylate its C-terminal domain and recruit
MC4R also activates the Ca2+ signaling pathway, demonstrating its
functional complexity [35]. Ca2+ can cooperate with
Leptin–Melanocortin Pathway is a key system for regulating energy balance and
appetite, functioning through specific neurons and receptors in the hypothalamus.
Under satiety conditions, adipose tissue secretes leptin, which activates the
leptin receptor (LEPR) in the arcuate nucleus. After leptin binds to its
receptor, it stimulates pro-opiomelanocortin (POMC) neurons to secrete
As a member of the GPCR family, MC4R not only transmits signals through the
classical G
Under the theoretical framework of “pathway-selective inhibition”, the biased
signaling of MC4R plays a critical role. Biased signaling refers to the
phenomenon where specific ligands or receptor variants preferentially activate
one of several downstream signaling pathways of the receptor [39]. Human genetic
studies have confirmed that gain-of-function mutants, such as V103I and I251L,
exhibit a characteristic
Molecular mechanism studies have shown that enhanced
Additionally, studies have reported that certain agonists preferentially
activate the Gq/11 pathway rather than the Gs pathway. These ligands
significantly reduce food intake by enhancing meal-related satiety, while
avoiding full activation of the Gs-cAMP pathway, which leads to a significant
reduction in cardiovascular side effects (e.g., blood pressure elevation) in
preclinical models [41, 42]. This suggests that activation of the
In conclusion, from the molecular to the animal level, the V103I and I251L
mutants provide comprehensive evidence that biased signaling mediated by the
To achieve this precise design goal, future research methods must also be innovated: for example, single-cell analysis can be used to resolve the dynamics of MC4R signaling pathways across different cell types in spatial and temporal dimensions [43]; additionally, label-free assays and other non-invasive techniques can capture whole-cell responses to ligand stimulation, identifying unique biased signaling fingerprints [44]. These advancements will enable rational design of highly selective antagonists that target specific signaling branches, effectively controlling body weight while maximizing therapeutic safety.
MC4R plays a critical role in regulating appetite and metabolism. Its dysfunction is associated with diseases such as obesity, cachexia, and anorexia, making it a crucial target for drug development. This article primarily analyzes the research status of MC4R antagonists from two stages: preclinical and clinical.
Before discussing specific MC4R antagonists, it is essential to clarify an
important pharmacological subdivision: the distinction between neutral
antagonists and inverse agonists. Although both can block the action of
endogenous agonists (e.g.,
GPCRs generally exhibit a certain level of constitutive activity (i.e., basal
activity), and MC4R is no exception [45]. Inverse agonists can actively suppress
this basal activity, lowering receptor signaling below baseline levels; in
contrast, neutral antagonists merely block the binding of exogenous agonists
(e.g.,
In summary, clearly distinguishing inverse agonists from neutral antagonists provides key insights for developing MC4R-targeted drugs that address different pathophysiological mechanisms. In recent years, numerous MC4R ligands with well-defined pharmacological properties have been reported, and their diversity is summarized in Table 3 (Ref. [16, 47, 48, 49, 50, 51]).
| Compound | Type | Mode of action | Key activity data (IC50/Ki) |
| AGRP (86-132) [47] | Endogenous peptide | Inverse agonist (reduces basal cAMP) | IC50 |
| SHU 9119 [51] | Synthetic peptide | Neutral antagonist (exhibits inverse-agonist activity in specific neurons and cellular contexts) | IC50 |
| MCL0020 [48] | Small molecule | Inverse agonist (suppresses basal signaling) | IC50 |
| ML00253764 [48] | Small molecule | Inverse agonist (suppresses basal signaling) | / |
| PF 07258669 [47] | Small molecule | Neutral antagonist (does not affect basal activity) | IC50 |
| TCMCB-07 [49] | Peptide | Neutral antagonist | IC50 |
| GSC000580 [50] | Small molecule | Neutral antagonist | IC50 |
| Ipsen 5i [16] | Small molecule | Inverse agonist (suppresses basal signaling) | Ki |
The primary objective of early studies was to validate the feasibility of MC4R as a therapeutic target rather than pursuing subtype selectivity, therefore, researchers have prioritized the development of compounds with antagonistic activity, while placing relatively lower requirements on selectivity. During this phase, researchers developed a series of non-selective MC4R antagonists (e.g., SHU9119).
SHU9119, as an antagonist in early studies, exhibits inhibitory effects
on both melanocortin receptors (MC3R and MC4R), thereby leading to off-target
effects. Research has shown that SHU9119 was initially designed as a
non-selective antagonist capable of acting on both MC3R and MC4R, demonstrating
significant pharmacological effects in obesity models and acute inflammation
models in mice. The primary mechanism of action is primarily through competitive
binding to MC3R and MC4R, blocking the activation of endogenous ligands such as
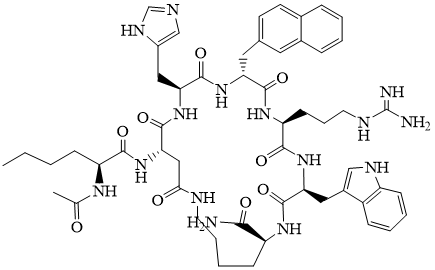 Fig. 6.
Fig. 6.
The structure of SHU9119 [51].
Early compounds demonstrated significant antagonistic activity against MC4R, but exhibited cross-reactivity with other melanocortin receptors (e.g., MC1R, MC3R). Through studies on early-stage compounds, researchers not only validated the feasibility of MC4R as a drug target, but also provided critical structural foundations and mechanistic insights for subsequent MC4R-targeted drug design.
Although non-selective antagonists have successfully validated the therapeutic potential of MC4R, their cross-reactive inhibitory activity against subtypes such as MC1R and MC3R leads to off-target effects (e.g., metabolic disorders, anxiety-like behaviors), severely limiting clinical translation. The root of this issue lies in the high sequence homology of the melanocortin receptor family (MC1R-MC5R) [56, 57], particularly in the transmembrane regions of MC3R and MC4R, which exhibit particularly high homology, making it challenging for traditional phenotypic screening-based compounds to achieve selective binding. The research focus has shifted to the development of highly selective MC4R antagonists based on this, aiming to circumvent interference from MC3R and other subtypes through rational design, while optimizing pharmacokinetic properties (such as brain permeability and metabolic stability) to meet the long-term therapeutic requirements for chronic disease treatment.
Supported by structural biology, Structure-Based Drug Design (SBDD) has become a critical strategy for developing highly selective MC4R antagonists. By leveraging the crystallographic data of MC4R, researchers can precisely identify and optimize the interactions between small molecules and the receptor. Specifically, these strategies include molecular docking, virtual screening, and structural optimization strategies, which are used to design and optimize antagonists with high selectivity and good pharmacokinetic properties. The application of these methods not only accelerated the discovery of novel antagonists but also provided crucial insights into understanding the structure-function relationship of MC4R.
HS014 (Fig. 7A), as a synthetic cyclic MSH (melanocyte-stimulating
hormone) analog, is a selective MC4R antagonist. It suppresses MC4R, thereby
reducing the appetite-suppressing effect induced by
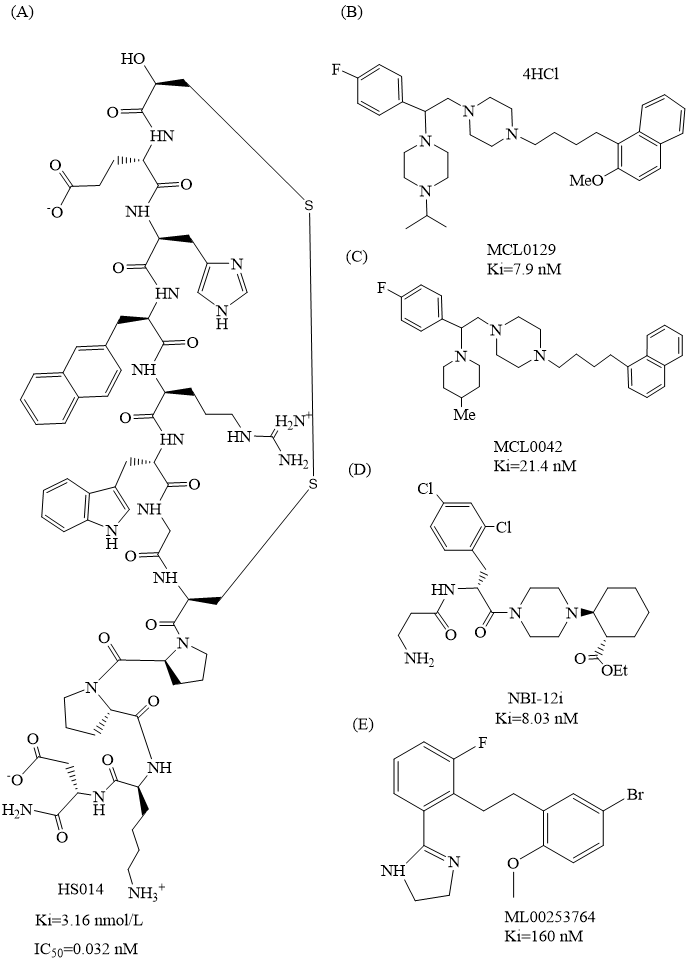 Fig. 7.
Fig. 7.
Structures of highly selective MC4R antagonists. Panels (A–E) show the chemical structures of the five compounds HS014, MCL0129, MCL0042, NBI-12i, and ML00253764, along with their corresponding affinity values.
HT1-0 is a linear peptide extracted from conotoxin, it exhibits the property of competitive antagonism on G protein-coupled receptors. Specifically, it acts as an MC4R antagonist with relatively lower binding affinity, but requires the combination of hydrophobic groups and negatively charged residues to bind to MC4R. This demonstrates that HT1-0 is a unique high-affinity ligand, representing the first MC4R antagonist derived from snake venom peptides, which provides a new direction for the development of novel MC4R antagonists [60].
Highly selective MC4R antagonists can also be achieved through non-peptidic compounds. Studies have identified MCL0129 and MCL0042 as potent and selective MC4R antagonists, which function through MC4R signaling pathways and exhibit unique pharmacological properties. In animal models, they demonstrate anxiolytic and antidepressant effects.
MCL0129 (Fig. 7B) is a novel MC4R antagonist developed by Taisho
Pharmaceutical Co., Ltd. (Tokyo, Japan), currently at the preclinical stage as
its highest development phase. MCL0129 exerts its effects by inhibiting
the binding of
MCL0042 (Fig. 7C) is a novel compound developed by Taisho Pharmaceutical Co., Ltd. Currently, the drug’s highest development stage is preclinical, intended for the treatment of anxiety and depression. MCL0042 is a non-peptide MC4R antagonist that exerts its effects by antagonizing the MC4 receptor and inhibiting the serum transporter [61]. MCL0042 demonstrates significant efficacy in animal models of anxiety and depression. For example, in swimming stress-induced animal models, MCL0042 administration reduces the time spent with arms extended by swimming-stressed rats in the elevated plus maze task [62], demonstrating its anxiolytic potential; in the olfactory bulbectomy (OBX)-induced rat model, OBX rats exhibited a significant increase in locomotor activity. Administration of MCL0042 significantly attenuated the locomotor activity in OBX rats [62], suggesting that MCL0042 possesses antidepressant-like potential.
NBI-12i (Fig. 7D) is a small-molecule MC4R antagonist characterized by high affinity and selectivity, it exhibits nanomolar affinity for the MC4R receptor (Ki = 8.03 nM) with 30 to 200-fold selectivity over other melanocortin receptor subtypes [63], and can penetrate the central nervous system following peripheral administration, by blocking MC4R, it significantly ameliorated the cachexia state in uremic mice, including increased food intake, body weight, and body composition, while reducing basal metabolic rate. Its mechanism of action may involve the regulation of appetite signals in the central nervous system and energy metabolism in brown adipose tissue. This study provides an important theoretical basis for developing novel anti-cachectic drugs targeting MC4R [64].
ML00253764 (Fig. 7E) (developed by Millennium Pharmaceuticals, Inc., Cambridge, MA, USA) is an MC4R antagonist that, besides its potential applications in obesity research, has increasingly attracted attention for its role in oncology. This compound exhibits low affinity for MC4R (Ki = 160 nM) and displays inverse agonist activity in the Gs-cAMP pathway, selectively inhibiting both wild-type and various constitutively active human MC4R forms [16]. Notably, its pharmacological effects show marked species specificity: in humans and mouse models it primarily acts as an antagonist/inverse agonist, whereas in fish MC4R (e.g., grass carp and zebrafish) it functions as an agonist, an effect that can be competitively blocked by other ligands [48, 64]. In tumor biology, functional expression of MC4R has been confirmed in several malignancies. For instance, in glioblastoma (GBM), targeted inhibition of MC4R blocks the ERK1/2 and Akt signaling pathways, producing anti-proliferative and pro-apoptotic effects; moreover, ML00253764 combined with temozolomide shows remarkable synergistic efficacy in preclinical studies [65]. Similarly, in melanoma models, MC4R antagonism suppresses ERK1/2 phosphorylation and down-regulates BCL-XL, thereby inhibiting tumor growth. The combination of ML00253764 with the BRAF inhibitor vemurafenib demonstrates strong synergistic activity both in vitro and in vivo, with preclinical safety assessments revealing no significant toxicity and indicating a favorable safety profile [66]. In summary, ML00253764 is a pharmacologically complex MC4R modulator that not only serves as a crucial tool for elucidating the diverse functions of the melanocortin system but also holds significant translational value in cancer therapy, especially within combination treatment strategies.
Studies have shown that highly selective MC4R antagonists demonstrate potential therapeutic value in treating obesity, depression, and anxiety disorders. Although highly selective MC4R antagonists hold great theoretical potential, they still face several challenges in practical applications. The current comprehensive evaluation of various MC4R subtypes remains insufficient, and the biological activity and selectivity of some compounds still require further validation. Furthermore, how to balance the selectivity and efficacy of antagonists to avoid adverse reactions remains an important research direction [67]. In summary, significant progress has been made in the design and development of highly selective MC4R antagonists, but further research is still required to overcome existing challenges and fully realize their therapeutic potential in disease treatment.
In research on MC4R antagonists, certain compounds have garnered notable attention due to their unique pharmacological effects and promising clinical application prospects. These compounds not only demonstrate significant efficacy in regulating appetite and energy metabolism, but also hold potential value in the treatment of various diseases. The following section will focus on elaborating the pharmacological effects and clinical development progress of two representative MC4R antagonists in clinical stages (e.g., PF-07258669 and TCMCB07).
PF-07258669 (Fig. 8C) (Ref. [47]) is an innovatively designed MC4R antagonist. Developed by Pfizer as a small-molecule drug, it aims to treat appetite loss-related disorders by targeting the MC4R signaling pathway. Currently, the drug’s highest development stage is Phase I clinical trial, intended for the treatment of nutritional deficiency. The design of PF-07258669 employs a spirocyclic structure, which not only optimizes the pharmacological efficacy of the MC4 receptor antagonist but also improves ADME (absorption, distribution, metabolism, and excretion) properties, while avoiding the endogenous hormone-activating activity (hERG) observed in earlier series. For example, by optimizing the spirocyclic structure, the antagonist PF-07258669 was ultimately obtained, which exhibits potent MC4R antagonist activity (IC50 = 13 nM, Ki = 0.46 nM) and excellent metabolic stability; and exhibits a lower MDR efflux ratio (MDR efflux ratio of 4.7), which is beneficial for brain penetration; it does not produce hERG-active metabolites, significantly reducing the associated safety risks [47]. Additionally, PF-07258669 demonstrated potent efficacy in animal models, particularly exhibiting significant appetite stimulation and weight gain effects in aged rat models. For example, after orally administering PF-07258669 at different doses of 0.3, 1, 3, and 10 mg/kg twice daily to rats for 20 days, compared to the control group, the cumulative food intake in each dose group increased by 23%, 26%, 40%, and 111%, respectively, the body weight increased by 2%, 5%, 9%, and 22% respectively [47], as shown in Fig. 8A,B (Ref. [47]). From a clinical development perspective, PF-07258669 is currently undergoing Phase 1 clinical trials, aimed at evaluating its safety, pharmacokinetics, and pharmacodynamics risks in healthy adult subjects [47]; this indicates that the safety and effectiveness of the drug in humans are being further validated, and it may potentially become a candidate drug for treating cachexia-related diseases in the future.
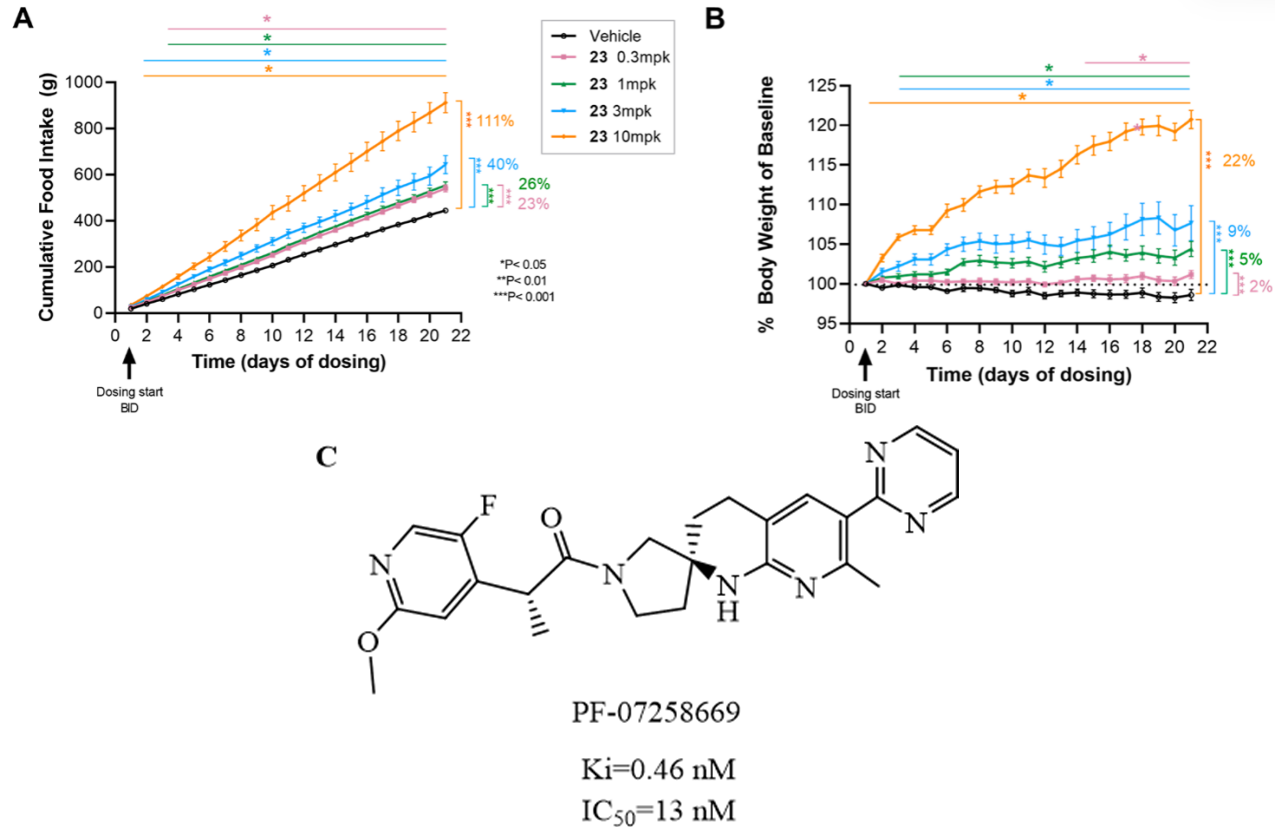 Fig. 8.
Fig. 8.
Effects of different doses of PF-07258669 on food intake and
body weight in rats and the structure of PF-07258669 [47]. (A) Cumulative food
intake since the baseline of self-pretreatment. (*p
Bile acid transport peptides are an emerging class of peptide therapeutics that have attracted attention because they can be actively transported by multispecific bile acid transporters and have the potential to cross the blood–brain barrier. However, the generally rapid hepatic clearance of these peptides severely limits their druggability. TCMCB07 (Endevica Bio Inc., Northbrook, IL, USA) represents a breakthrough case in this context. As a member of the bile acid peptide family, it incorporates a clever modification of the C-terminal amino-acid sequence that slows hepatic clearance [68]. This key modification not only addresses the core bottleneck of this peptide class but also provides a platform approach that can be applied to other similar peptides, opening a new pathway for developing drug-like peptides with desirable pharmacokinetic properties.
TCMCB07 (Fig. 9, Ref. [69]) is a cyclic nonapeptide MC4R antagonist developed by Endevica Bio Inc. that is currently in Phase II clinical trials for the treatment of cachexia. Its mechanism of action involves inhibition of central MC4R signaling, which stimulates appetite and improves energy balance. Additionally, it has been shown to reduce the expression of inflammatory genes in the hypothalamus [70]. Evidence in inflammatory models: For example, in a lipopolysaccharide (LPS)-induced systemic inflammation model, LPS triggers a severe cytokine storm that leads to multi-organ damage [71]. Studies have demonstrated that, regardless of administration route—intraperitoneal, subcutaneous, or oral—TCMCB07 significantly improves 24-hour food intake and body-weight changes in LPS-treated rats, confirming its efficacy under acute inflammatory conditions. Effects in cachexia disease models: In a cancer-associated cachexia model, subcutaneous injection of TCMCB07 markedly suppresses the expression of several hypothalamic inflammatory genes (such as Il1b, Il1r1, Il6, and Selp), consistent with its anti-inflammatory action. In a chronic kidney disease (CKD) cachexia model, although typical inflammatory gene expression changes are not pronounced, TCMCB07 treatment significantly attenuates the up-regulation of the Agrp gene, which may represent a key mechanism by which it improves appetite through a different pathway [70]. These preclinical studies, which demonstrate improvements in feeding, body weight, and the regulation of neuroinflammation and appetite signaling, collectively reveal the therapeutic potential of TCMCB07 and provide a theoretical basis for further investigation in human patients.
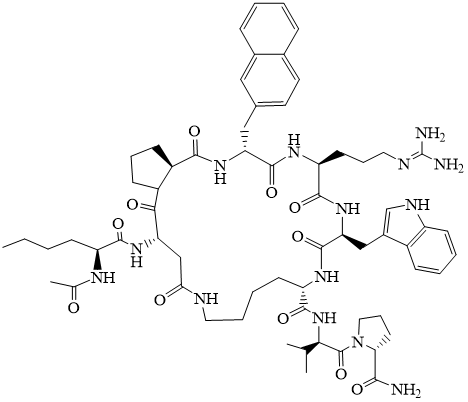 Fig. 9.
Fig. 9.
Structure of TCMCB07 [69].
Research on MC4R antagonists has achieved significant progress in structural elucidation, selectivity optimization, and preclinical evaluation, revealing the pivotal role of MC4R in energy metabolism and appetite regulation, while providing a structural foundation for the design of novel antagonists. However, there are still challenges such as enhancing subtype-specific selectivity, optimizing pharmacokinetic properties (including blood–brain barrier permeability and metabolic stability), and achieving personalized treatment strategies. Future research should focus on optimizing the subtype selectivity of antagonists through structural biology and computational chemistry, exploring novel strategies to enhance pharmacokinetic performance, and developing precision treatment regimens integrated with individualized medicine to promote breakthroughs in clinical applications.
Melanocortin receptors are key regulators of energy balance, and the tissue distribution and functional differences among their multiple subtypes pose challenges for the development of subtype-selective antagonists. Current research primarily focuses on the development of subtype-specific ligands, rational design based on cryo-EM structures, and the development of allosteric modulators and dual-target antagonists.
Subtype-specific ligands achieve specific binding to target subtypes through screening and optimization, thereby reducing side effects and enhancing therapeutic efficacy. For example, certain ligands can achieve higher selectivity by binding to non-conserved regions [72]. However, due to the high structural homology of MC4R with other subtypes, traditional drug design approaches often rely on low-resolution models and empirical screening methods, making it particularly challenging to accurately identify and target the unique structural features of MC4R. The advancements in cryo-electron microscopy (cryo-EM) technology have provided revolutionary tools for the structural elucidation of MC4R. Its high-resolution three-dimensional structure has revealed key binding pockets and allosteric sites, enabling researchers to perform rational design at the atomic level [73, 74]. Based on these structural insights, researchers have initiated the exploration of developing allosteric modulators and dual-target antagonists to further enhance the selectivity and pharmacological efficacy of MC4R antagonists. Additionally, combining allosteric modulators with traditional antagonists can further enhance the drug’s selectivity and therapeutic efficacy [74]. These strategies have been successfully applied to the drug development of other GPCRs, providing new opportunities for the innovative design of MC4R antagonists [75, 76, 77].
Despite progress in basic research and drug design, the clinical translation of MC4R antagonists still faces challenges, including optimization of blood–brain barrier penetration, personalized therapy, and biomarker research.
MC4R is highly expressed in the central nervous system, and developing drugs capable of penetrating the blood–brain barrier is crucial. Traditional small molecules and peptide compounds have limitations in penetrating the blood–brain barrier, resulting in insufficient drug concentration in the brain and compromising therapeutic efficacy [78]. By optimizing the chemical structure of drug molecules (e.g., introducing lipophilic groups), their ability to cross the blood–brain barrier can be enhanced. Furthermore, due to individual variations in MC4R subtype expression, it is particularly important to develop individualized treatment regimens based on genotype and receptor expression levels. Biomarker research aids in predicting therapeutic outcomes and adverse reactions, thereby promoting precision medicine advancements.
In summary, research on MC4R antagonists should focus on enhancing subtype selectivity, optimizing pharmacokinetic properties, and addressing key challenges in translational medicine to overcome the limitations of existing drugs and provide novel therapeutic solutions for the treatment of obesity, cachexia, metabolic disorders, and related diseases.
MC4R, as a key receptor in the melanocortin system, plays a crucial role in regulating energy balance, body weight control, and appetite. MC4R antagonists can improve appetite decline and weight loss in patients with anorexia or cachexia by blocking its activation. In early studies, non-selective MC4R antagonists such as SHU9119 were widely used to investigate the physiological roles of MC4R, including its effects on feeding behavior and energy homeostasis, which validated the feasibility of targeting MC4R. However, due to the activity of SHU9119 on other melanocortin receptors, off-target effects have limited its clinical application. To overcome the limitations of non-selective antagonists, researchers have developed selective MC4R antagonists. For example, compounds such as HS014, MCL0129, and MCL0042 have been demonstrated to be selective for MC4R and exhibit potential therapeutic value in treating obesity, depression, and anxiety disorders. Recently, studies have found that PF-07258669 and TCMCB07 demonstrate broad therapeutic potential in the treatment of cachexia and related diseases.
However, Cachexia is characterized not only by loss of appetite and weight loss, but also by significant muscle wasting and emotional disorders such as anxiety and depression. Recent studies have shown that MC3R plays an important role in the development of cachexia. Unlike MC4R, which primarily regulates appetite, MC3R agonists can both stimulate feeding and alleviate negative emotions such as anxiety during treatment; conversely, MC3R antagonists suppress feeding [79]. This suggests that MC3R may be a novel therapeutic target. Therefore, an ideal cachexia treatment strategy should act on both MC4R and MC3R: using an MC4R antagonist to increase appetite while modulating MC3R signaling to improve anxiety, depression, and other symptoms.
Although MC4R antagonists exhibit potential in treating conditions such as appetite regulation, they still face challenges including insufficient selectivity, poor blood–brain barrier penetration, low metabolic stability, unclear dosing and administration regimens, and a lack of effective biomarkers to guide personalized treatment. Future research should focus on developing safer, more efficient, and personalized MC4R antagonists while exploring individualized treatment strategies to achieve more precise disease intervention.
This review systematically dissects the central role of MC4R in the regulation of energy balance and argues for the therapeutic potential of MC4R antagonists in alleviating disease-related anorexia, such as that seen in cachexia. At the same time, we recognize that the pathophysiology of cachexia is far more complex than merely reduced appetite; it also involves independent pathways mediated by MC3R that drive muscle wasting. To overcome the current challenges of MC4R-targeted therapy—such as selectivity and blood–brain barrier penetration—and to achieve comprehensive and effective management of cachexia, the MC4R field needs to focus on structure-based drug design, develop biomarker-guided personalized strategies, and actively explore combinatorial treatments targeting both MC4R and MC3R. Through these efforts, a novel, integrated therapeutic solution for cachexia may become attainable.
JS was responsible for the overall conceptualization of the review, literature search and drafting of the initial manuscript. JX was responsible for the overall conception and design of this review and conducted multiple rounds of critical revisions of the key content. CH contributed to the review’s design and the preparation of figures and tables. LH was primarily responsible for the creation of the figures and diagrams, including the key structural and mechanistic illustrations in this review. YC, as the corresponding author, oversaw the overall conceptualization of the review, the final manuscript approval, responses to reviewer comments, and communication with the editorial office. All authors have read and approved the final manuscript, ensuring the authenticity and integrity of the research content. All authors contributed to editorial changes in the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
Despite Ling Huang being employed by Grand Medical Nutrition Science (Wuhan) Co., LTD., the judgments in data interpretation and writing were not influenced by this relationship. The remaining authors declare no financial conflicts of interest that could have influenced the results of this study.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.