





1 Division of Cardiology, Johns Hopkins University, Baltimore, MD 21287, USA
Abstract
Cardiac amyloidosis (CA) results from the deposition of amyloid fibrils in the myocardium and is mainly caused by two parent proteins: transthyretin (ATTR) and light chain immunoglobulin (AL). ATTR is further differentiated into wild-type (ATTRwt) and hereditary (ATTRv) forms, based on the presence or absence, respectively, of mutations in the TTR gene. Historically, CA has been overlooked in clinical practice, with many cases misdiagnosed or diagnosed at advanced stages due to overlapping features with other cardiomyopathies, such as hypertrophic cardiomyopathy. However, recent advancements in both diagnostic techniques and awareness have led to an increasing recognition of CA, particularly in patients with heart failure with preserved ejection fraction and other forms of restrictive cardiomyopathy. Moreover, the advent of multimodality imaging has significantly enhanced the diagnosis of CA. Imaging modalities such as echocardiography, cardiac magnetic resonance (CMR), and nuclear scintigraphy (using bone-seeking tracers such as 99mTc-pyrophosphate) play pivotal roles in identifying myocardial involvement early in the disease course. CMR imaging allows precise tissue characterization, identifying myocardial edema, fibrosis, and amyloid deposition; meanwhile, nuclear scintigraphy with 99mTc-pyrophosphate has emerged as a non-invasive, highly sensitive imaging technique for detecting ATTR infiltration in the heart; the diagnosis of AL requires histological confirmation. Following the advent of disease-modifying therapies, the need for early disease detection has become more critical to enhance survival rates and improve quality of life.
Keywords
- cardiac amyloidosis
- transthyretin cardiac amyloidosis
- light chain amyloidosis
- cardiac magnetic resonance
- echocardiography
- Tc-pyrophosphate scintigraphy
Cardiac amyloidosis (CA) is a multifaceted infiltrative condition marked by the
accumulation of amyloid fibrils—abnormally folded protein aggregates—within
the extracellular space of various tissues [1]. These fibrils, primarily composed
of
Clinically, CA presents as a restrictive form of cardiomyopathy and is broadly categorized into two main types: light chain (AL) amyloidosis and transthyretin (ATTR) amyloidosis. AL results from the deposition of fibrils derived from the N-terminal fragment of a monoclonal immunoglobulin light chain, which is produced by an abnormal population of plasma cells in the bone marrow [4]. On the other hand, ATTR is caused by the destabilization and misfolding of transthyretin (TTR)—a liver-derived protein responsible for transporting thyroxine and retinol-binding protein. ATTR is further classified into wild-type (ATTRwt), occurring without genetic mutations, and variant (ATTRv), which is associated with mutations in the TTR gene [5].
Despite the availability of modern diagnostic methods, CA remains significantly underrecognized due to its clinical resemblance to other cardiac conditions such as hypertrophic cardiomyopathy. Studies indicate that CA is present in 13% of patients with heart failure with preserved ejection fraction (HFpEF), 16% of those with age-related aortic stenosis, and 9% of patients previously diagnosed with hypertrophic cardiomyopathy [6, 7, 8]. Additionally, post-mortem analyses have shown myocardial amyloid deposits in up to 25% of individuals over the age of 80, highlighting a substantial burden of undiagnosed disease in elderly populations [9].
With growing awareness and improvements in imaging techniques and biomarker identification, there is a renewed emphasis on recognizing CA early in its course [10]. Early detection is particularly important, as emerging therapies that target the disease mechanism offer the potential to alter disease progression if implemented promptly [11]. This review will delve into the distinct pathophysiological and epidemiological aspects of ATTRwt, ATTRv, and AL. It will also examine early clinical indicators, the diagnostic value of multimodal imaging and endomyocardial biopsy, and current approaches to the management of CA.
ATTRv results from over 130 known mutations in the TTR gene, all of which are inherited in an autosomal dominant manner. The clinical presentation of ATTRv is highly variable, depending on the specific mutation, the type of amyloid fibrils formed, and the geographic region [12] (Table 1). The age of onset, primary manifestations (such as cardiomyopathy, neuropathy, or both), and disease progression can differ significantly between individuals with different mutations.
| Feature | AL | ATTRwt | ATTRv |
| Epidemiology | ~8–12 cases/million/year; median age ~60s; male |
Increasingly recognized in elderly men (typically |
Variable depending on mutation; autosomal dominant inheritance; some mutations more common in specific ethnic groups (e.g., V122I in African ancestry; T60A in Irish) |
| Organ Involvement | Multisystem: heart, kidneys, liver, peripheral/autonomic nerves, GI tract | Primarily cardiac | Variable: cardiac, neurologic, mixed phenotypes depending on mutation |
| Onset | Subacute, rapidly progressive | Slowly progressive | Variable; early onset in some mutations (e.g., Val30Met); late-onset in others |
| Common Cardiac Presentation | Rapidly progressive HF, often with hypotension, low voltage on ECG, severe diastolic dysfunction | Gradual onset of HFpEF, often with conduction disease or arrhythmia | Similar to ATTRwt, though some variants have more neurologic involvement |
| Red Flag Features | Macroglossia, periorbital purpura, nephrotic-range proteinuria, musculoskeletal involvement, significant weight loss | Carpal tunnel syndrome, Trigger finger, biceps tendon rupture, lumbar spinal stenosis | Family history, neuropathy, carpal tunnel syndrome (depending on mutation) |
| Diagnostic Biomarkers | Elevated free light chains (FLCs); abnormal serum/urine immunofixation | Normal FLCs; positive bone scintigraphy (Grade 2–3) | Genetic testing confirms TTR mutation; bone scintigraphy or biopsy-based diagnosis |
| Prognosis (Untreated) | Poor: median survival |
Better than AL: median survival ~3–5 years | Highly variable depending on genotype and phenotype |
| Treatment | Chemotherapy targeting plasma cells (e.g., daratumumab-based regimens), stem cell transplant | TTR stabilizers (e.g., tafamidis); supportive HF care | Gene silencers (e.g., patisiran, vutrisiran), TTR stabilizers; mutation-specific approach |
AL, light chain cardiac amyloidosis; ATTRwt, wild-type cardiac amyloidosis; ATTRv, variant or hereditary cardiac amyloidosis; HFpEF, heart failure with preserved ejection fraction; TTR, transthyretin; MGUS, monoclonal gammopathy of unknown significance; GI, gastrointestinal; HF, heart failure; ECG, electrocardiogram.
Although the exact prevalence of ATTRv remains uncertain, certain mutations are more commonly observed in specific populations. In the United States, the V122I mutation is the most prevalent, found in about 3.4% of African Americans [13]. This variant often presents similarly ATTRwt, predominantly affecting men and leading to late-onset cardiomyopathy with minimal neuropathy, typically manifesting around the age of 70.
Another notable mutation in the United States is T60A, which originates from Northern Ireland and typically presents with a mixed cardiomyopathy and neuropathy phenotype [14]. Globally, the V30M mutation is the most common form of ATTRv, with a broad range of phenotypic expressions [15]. In endemic regions, particularly in Sweden, Portugal, and Japan, V30M often leads to early-onset disease (before age 50), presenting primarily with neurological symptoms. In contrast, the late-onset form of V30M, more common in nonendemic areas, may present with both cardiac and severe neurological symptoms [16].
Additional mutations such as L111M are more prevalent in Denmark, while I68L is often seen in Italy [17]. These regional differences further illustrate the genetic diversity of ATTRv and its variable clinical manifestations.
Recent enhancements in non-invasive diagnostic methods have dramatically broadened our understanding of ATTRwt, particularly in older populations. Historically, ATTRwt accounted for under 3% of CA cases between 1987 and 2009, but this share rose to approximately 14% in 2010–2015 and further climbed to around 25% from 2016 to 2019 [18]. As non-invasive diagnostic criteria were validated, screening among high-risk groups revealed many covert cases, suggesting earlier prevalence estimates significantly underestimated disease burden.
The reported prevalence of ATTRwt varies across studies—largely due to differences in patient selection and diagnostic protocols. For example:
These discrepancies reflect variation in clinical settings, inclusion thresholds, and diagnostic tools. Nonetheless, they collectively highlight that proactive screening uncovers a much higher frequency of ATTRwt than previously believed.
ATTRwt predominantly affects elderly individuals—primarily men of Caucasian ethnicity [21]. In over 90% of cases, the heart is the main organ involved. The median age at diagnosis is around 75 years, and median survival from diagnosis is approximately 3.6 years [22].
AL remains the most prevalent CA subtype, accounting for about 55% of cases [23]. Around 2200 new AL cases are diagnosed annually in the U.S. [18], with the average age at diagnosis near 65, and a slight male predominance. The disease typically follows a more aggressive course than ATTR, due mainly to the harmful impact of amyloidforming light chains on the heart [24].
Though AL can be systemic, it predominantly affects the kidneys (74%) and the heart (60%) [25]. When the heart is involved, prognosis worsens dramatically—median survival drops to approximately six months for patients presenting with symptomatic heart failure who receive no treatment [18].
In CA, amyloid fibrils accumulate throughout the myocardial extracellular matrix, resulting in concentric thickening and biventricular remodeling. This phenomenon is often misinterpreted as left ventricular hypertrophy (LVH), though the process fundamentally differs from cellular hypertrophy. In ATTRwt, amyloid deposition tends to permeate the entire thickness of the ventricular walls (transmurally), whereas in AL, deposits are more frequently localized near the subendocardium [26].
This infiltration stiffens the ventricles, limiting compliance and increasing diastolic filling pressures. In early disease stages, left ventricular ejection fraction (LVEF) is usually preserved. However, as the ventricle fails to remodel and walls stiffen, stroke volume becomes fixed and limited, reducing cardiac output. This dynamic distinguishes CA from other forms of heart failure [27]. Patients also become heavily reliant on heart rate to maintain output, making them poorly tolerant of negative chronotropic medications like beta-blockers and calcium channel blockers [28, 29].
Amyloid involvement also disrupts atrial structure and function, promoting arrhythmias such as atrial fibrillation, increasing thromboembolic risk due to atrial mechanical impairment [30, 31, 32]. Conduction system anomalies—including atrioventricular block and HisPurkinje disruptions—are also common, potentially leading to symptomatic AV block. Ventricular arrhythmias and valvular thickening can further contribute to cardiac dysfunction [33, 34, 35].
CA often manifests as progressive heart failure, presenting with exertional dyspnea and signs of right-sided failure—such as peripheral edema, jugular venous distension, and, in severe cases, ascites or cardiogenic shock [36]. Patients may also experience angina in the absence of coronary artery obstruction, often attributable to microvascular dysfunction driven by amyloid in small intramyocardial vessels, leading to reduced coronary flow reserve [37, 38].
Syncope in CA may result from arrhythmias or autonomic dysfunction. Atrial fibrillation is the most common arrhythmia, although bradyarrhythmias and heart block are also regularly observed (Fig. 1). In older ATTRwt patients, lowflow, lowgradient aortic stenosis may be an initial clinical indicator [39].
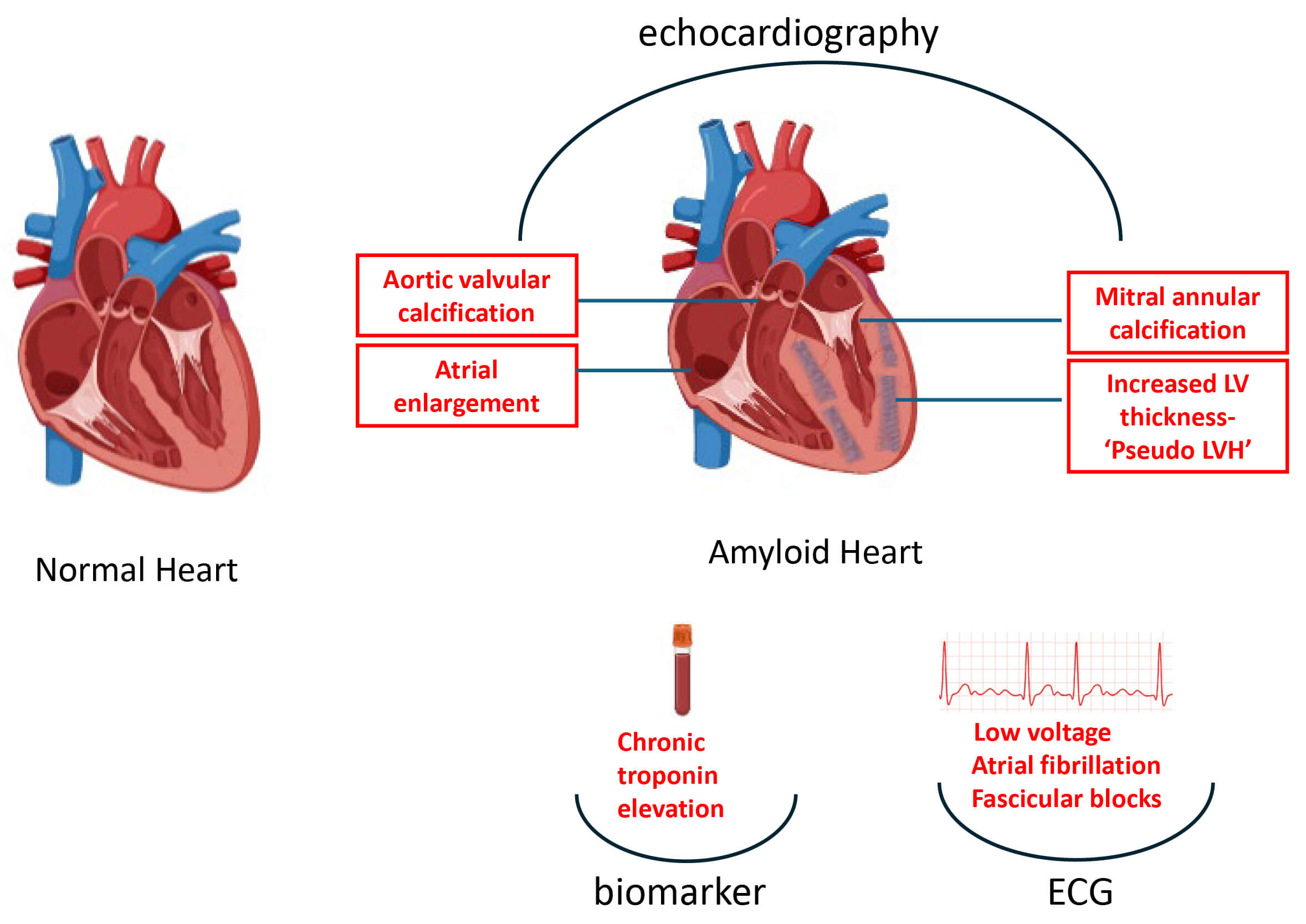 Fig. 1.
Fig. 1.
Pathological changes and associated biomarker, ECG, and echocardiographic findings in amyloid vs. normal heart. LVH, left ventricular hypertrophy; LV, left ventricle; ECG, electrocardiogram.
Early non-cardiac signs can serve as red flags for ATTRwt. Carpal tunnel syndrome, caused by amyloid deposits in the carpal tunnel and flexor retinaculum, is extremely common—present in about 50% of ATTRwt patients—and often precedes cardiac involvement by 5 to 9 years [40]. Among carpal tunnel surgery patients, detectable tenosynovial amyloid is found in 10–16%, even though only about 2% show signs of CA at that time [41]. Screening these individuals—especially non-obese elderly men—can reveal early ATTRwt cases with a high diagnostic yield [42].
Other musculoskeletal indicators include lumbar spinal stenosis (amyloid in ligamentum flavum; affecting over one-third of elderly undergoing surgery), spontaneous biceps tendon rupture (~33% of ATTRwt patients), and elevated rates of hip and knee joint replacements [43, 44, 45].
AL can involve nearly any organ outside the brain. Kidney involvement is most common, typically presenting with nephrotic syndrome and significant proteinuria due to glomerular amyloid deposition. In approximately 10% of cases, amyloid affects renal vessels and tubulointerstitium, causing renal impairment without heavy proteinuria [46]. Rarely, AL can present with acute kidney injury via amyloid cast nephropathy [47].
Hepatic involvement often results in hepatomegaly via infiltration or congestion. Autonomic neuropathy may lead to orthostatic hypotension, gastrointestinal dysmotility, and erectile dysfunction. Peripheral nerve involvement typically begins as distal sensory neuropathy and progresses to motor involvement. Softtissue signs—like macroglossia—are unique and diagnostic hallmarks of AL [46, 47].
Lowvoltage QRS (
Echocardiography is the frontline imaging modality for diagnosing CA and distinguishing it from hypertensive cardiomyopathy, hypertrophic cardiomyopathy, aortic stenosis, and Fabry disease. Classic findings include concentric LV wall thickening—though asymmetric thickening appears in ~23% of ATTRwt cases [56]—with wall thickness often exceeding 15 mm and greater in ATTRwt than AL [57]. A small subset of CA patients may display normal LV thickness [58]. Occasionally, dynamic LV outflow obstruction mimicking hypertrophic obstructive cardiomyopathy is seen [59]. Additional features include reduced LV cavity size, biatrial enlargement, interatrial septal thickening, and a granular “speckled” myocardial texture (Fig. 2). Diastolic dysfunction—evident through steep deceleration time, low mitral annular tissue Doppler velocity, and elevated E/e ratio—is a hallmark, whereas systolic function degrades gradually as disease progresses [60]. Tissue Doppler and speckle-tracking strain imaging, especially global longitudinal strain (GLS) with relative apical sparing (the “cherry-on-top” pattern), provide enhanced specificity in differentiating CA from other LVH etiologies; an apical-to-mid/basal GLS ratio around 1 is highly sensitive and specific [61, 62].
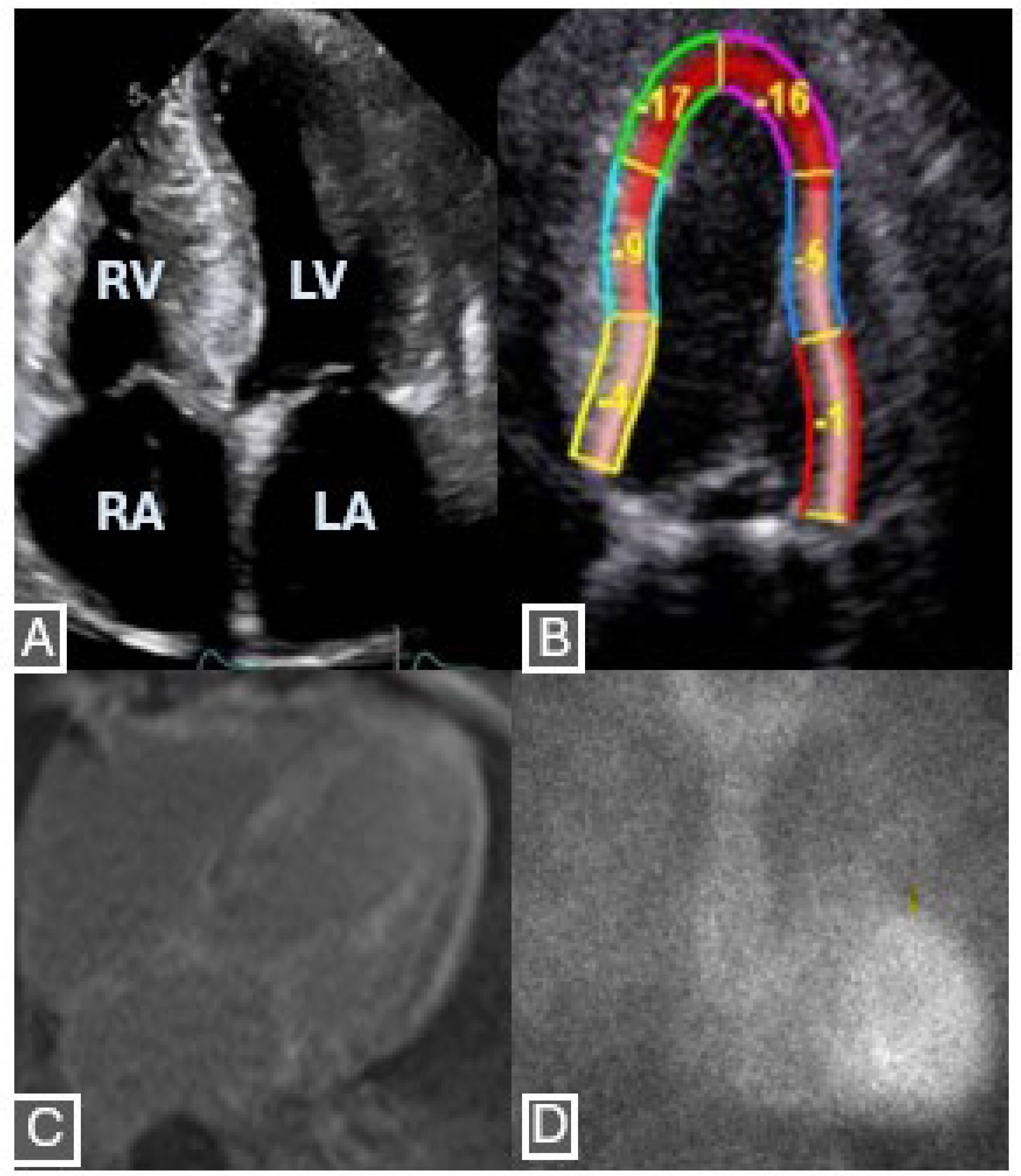 Fig. 2.
Fig. 2.
Features on echocardiogram, cardiac magnetic resonance and nuclear scintigraphy of a 59-year-old patient diagnosed with ATTRwt. (A) Restrictive physiology, characterized by enlarged atria and small-normal ventricular size, in addition to severe LV thickness. (B) Abnormal longitudinal strain with apical sparing on speckle tracking echocardiogram. (C) Diffuse transmural late gadolinium enhancement on cardiacmagnetic resonance. (D) Positive Tc-pyrophosphate scintigraphy with visual grade 3 on planar image. Retrospective use of fully anonymized archival images. RV, right ventricle; RA, right atrium; LA, left atrium.
CMR offers superior structural and tissue characterization in CA, though it
cannot distinguish ATTR from AL. Post-gadolinium imaging often reveals diffuse
subendocardial or transmural late gadolinium enhancement (LGE) due to expanded
extracellular space from amyloid deposition—a finding associated with advanced
disease and poorer outcomes [63, 64]. Use of contrast must be cautious in patients
with renal impairment (GFR
Bone-seeking radiotracers—such as 99mTc-PYP (United States), 99mTc-HMDP, and 99mTc-DPD (Europe)—offer a non-invasive alternative for diagnosing ATTR [73, 74, 75]. ATTR shows significantly more microcalcifications than AL, enhancing diagnostic specificity [76].
Scoring is based on tracer uptake relative to bone using a 0–3 grading scale (0
= none; 1 = less than bone; 2 = equal; 3 = greater than bone) [77]. In the
context of negative paraprotein testing, grade 2 or 3 uptake has 100%
specificity and PPV for ATTR in biopsy-confirmed cases [78]. Quantitative
assessment via heart-to-contralateral lung (H/CL) ratios
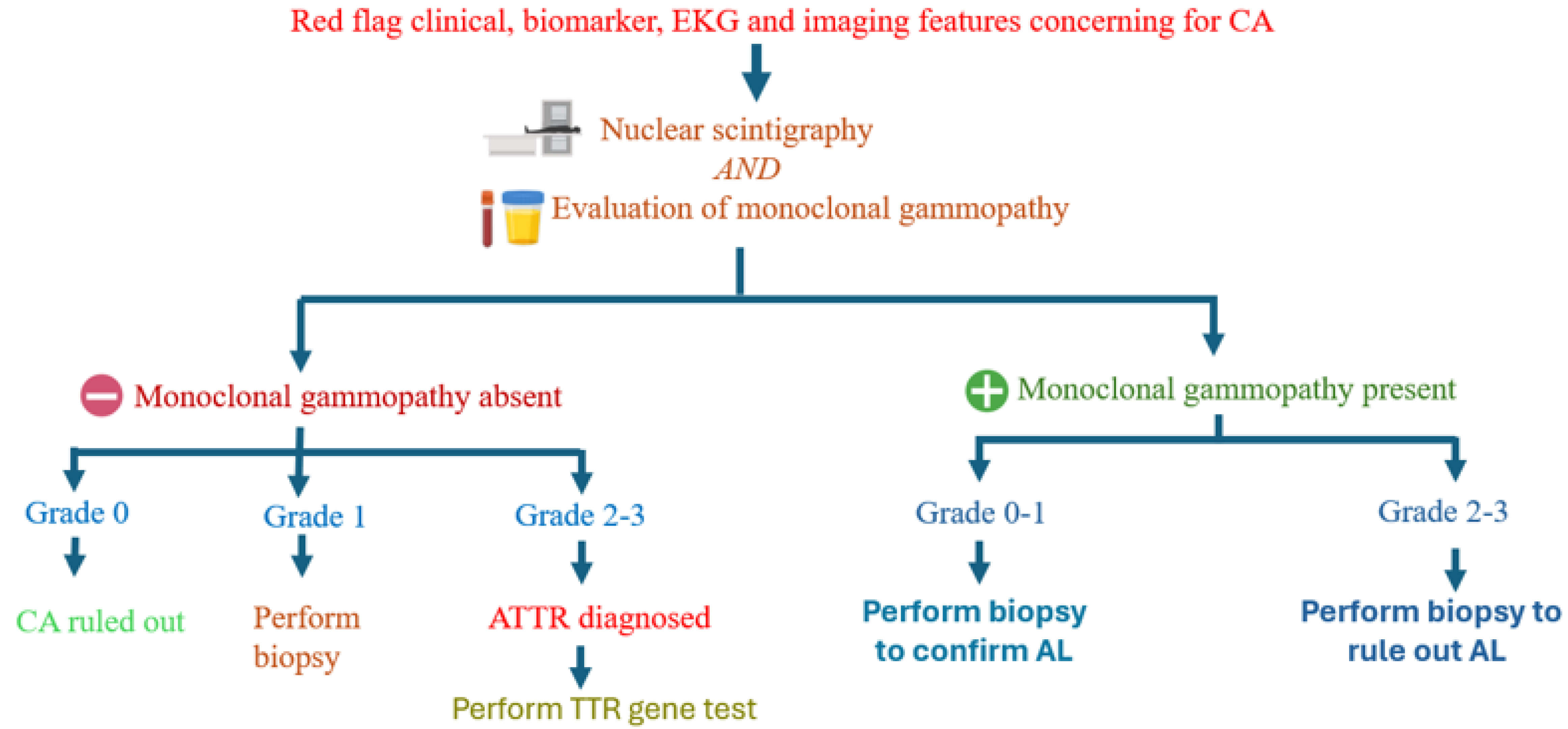 Fig. 3.
Fig. 3.
Flowchart outlining diagnostic process for ATTR and AL. AL, light chain immunoglobulin; CA, cardiac amyloidosis; TTR, transthyretin.
Biopsy is mandatory when a monoclonal light chain is detected, as AL must be
definitively diagnosed by histologic confirmation. Biopsy may also be needed in
suspected ATTR when non-invasive findings are inconclusive: low-grade tracer
uptake (
Endomyocardial biopsy—with Congo red–positive amyloid showing apple-green birefringence—is the gold standard, allowing amyloid typing via immunohistochemistry or mass spectrometry [82, 83]. While highly sensitive, it carries risks such as perforation, tamponade, and arrhythmia. Fat-pad aspiration offers a less invasive option but is poorly sensitive—particularly for ATTRwt—and often yields insufficient tissue [84].
Quantum dots (QDs) are nanoscale semiconductor particles known for their unique optical and electronic characteristics, including size-tunable fluorescence emission and high photostability, which make them promising tools for molecular imaging. In the context of CA, QDs can potentially be engineered to target amyloid fibrils through conjugation with amyloid-specific ligands. While QD-based imaging has been explored in other amyloid-related conditions such as Alzheimer’s disease [85], their application in CA remains largely unexplored. Further research is needed to evaluate their diagnostic potential in this setting, particularly in enhancing early detection and differentiation of amyloid subtypes.
The diagnosis of CA requires a comprehensive approach, integrating clinical suspicion, advanced imaging techniques, and histological confirmation. While non-invasive tools such as echocardiography, cardiac magnetic resonance imaging, and nuclear scintigraphy play pivotal roles in detecting and differentiating CA, tissue biopsy remains the gold standard for definitive diagnosis in AL, and also in ambiguous cases of ATTR. The challenge of differentiating between ATTR and AL highlights the importance of a multifaceted diagnostic strategy, including the use of immunohistochemistry, mass spectrometry, and advanced imaging to confirm amyloid type. Early recognition of CA, combined with an accurate diagnosis, is essential for guiding appropriate treatment and improving patient outcomes, especially as novel therapies emerge. Continued research into more precise diagnostic methods and biomarkers is crucial to enhancing the accuracy and efficiency of CA detection, ultimately facilitating timely interventions and better prognosis for affected patients.
SB: Conception, project supervision, literature review, drafting the manuscript, figure preparation, final review. SB has contributed to the conception and editorial changes in the manuscript. SB has read and approved the final manuscript. SB has participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The author declares no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.