1. Introduction
Alzheimer’s disease (AD) represents the predominant form of dementia and is
increasingly recognized as one of the costliest, deadliest, and most burdensome
diseases of the current century [1]. AD is a progressive and chronic
neurodegenerative disorder clinically characterized by cognitive dysfunction,
memory impairment, and behavioral changes, including motor symptoms such as gait
disturbances and impaired balance [2, 3]. These complex clinical manifestations
are attributed to the multifaceted pathology of AD, characterized primarily by
the excessive accumulation of amyloid (A) peptides, the
formation of neurofibrillary tangles (NFTs) composed of phosphorylated tau
(p-Tau), and persistent neuroinflammation across various brain regions [4].
Neuroplasticity encompasses a series of stimuli-induced biochemical alterations
at both pre- and postsynaptic sites [5, 6]. Further, neuroplasticity is
categorized into structural plasticity and functional plasticity [7]. Structural
plasticity is crucial in forming and developing neuronal dendrites and spines.
Cytoskeleton-regulating proteins modulate structural plasticity by regulating
actin cytoskeletal dynamics, including brain-derived neurotrophic factor,
glutamate receptors, and postsynaptic density protein-95 (PSD-95) [7]. Functional
plasticity, or synaptic plasticity, modulates synaptic transmission by altering
synaptic formation or transmission efficacy [5]. Functional plasticity is
demonstrated through long-term potentiation (LTP) and long-term depression (LTD),
contingent upon synaptic activity and efficacy [5]. The two forms of plasticity
are closely interconnected, as demonstrated by synaptic plasticity leading to
morphological changes in dendritic spines; specifically, LTP promotes spine head
growth, whereas LTD results in spine retraction [8]. Thus, structural and
functional plasticity interactions are fundamental to maintaining normal brain
microarchitecture and physiology.
In AD patients, the homeostasis of neuroplasticity is disrupted, representing a
significant pathological feature of disease progression [9]. This impairment in
neuroplasticity progressively impacts various aspects, including dendritic
ramifications, axonal sprouting, neurogenesis, synaptic remodeling, synaptic
efficacy, and synaptogenesis [10]. A and p-Tau, the main pathogenic
factors of AD, inhibit excitatory synapses and diminish neuronal activity,
thereby disrupting synaptic networks, ultimately resulting in spine or synaptic
loss and neuronal death [11, 12]. Furthermore, neuroinflammation significantly
impacts neuroplasticity, contributing to AD pathology [13]. Pathological lesions
are particularly pronounced in susceptible brain regions, notably the
hippocampus, and are among the first to exhibit changes in AD [14, 15].
Nevertheless, the mechanisms through which their relationship with
neuroplasticity is governed remain inadequately understood.
Hippocampal functional plasticity has been extensively studied in various
neurological disorders, including AD, highlighting its significant association
with learning and memory functions [16]. However, a deficiency exists in our
comprehension of hippocampal structural plasticity during AD progression.
Further, the molecular mechanisms underlying alterations in structural plasticity
remain largely unexplored. Thus, this study aimed to investigate the temporal
changes in neuroarchitecture within the hippocampal subregions and the associated
molecular alterations linked to brain dysfunction in AD using the 5FAD
mouse model, which is commonly utilized in AD research due to its rapid and
aggressive A deposition, gliosis, and progressive neuronal loss [17].
2. Methods
2.1 Animals
Transgenic 5FAD mice were obtained from Jackson Laboratories (strain:
034848JAX; Bar Harbor, ME, USA) and were maintained through regular breeding with
C57BL/6 mice (Damool Science, Daejeon, Korea). Female C57BL/6 wild-type (WT) and 5FAD mice at 3, 6, or
12 months of age were included in this study. Genotyping confirmed the presence
of five familial AD mutations: App KM670/671NL (Swedish), App
I716V (Florida), App V717I (London), Psen1 M146L (A C), and
Psen1 L286V. Mice were anesthetized with isoflurane (66794-013-10, Piramal Critical Care Inc, Bethlehem, PA, USA; 3% for induction, 1.5–2% for maintenance) and euthanized by cervical dislocation under deep anesthesia, in accordance with institutional guidelines. Experimental timelines are presented in Fig. 1A.
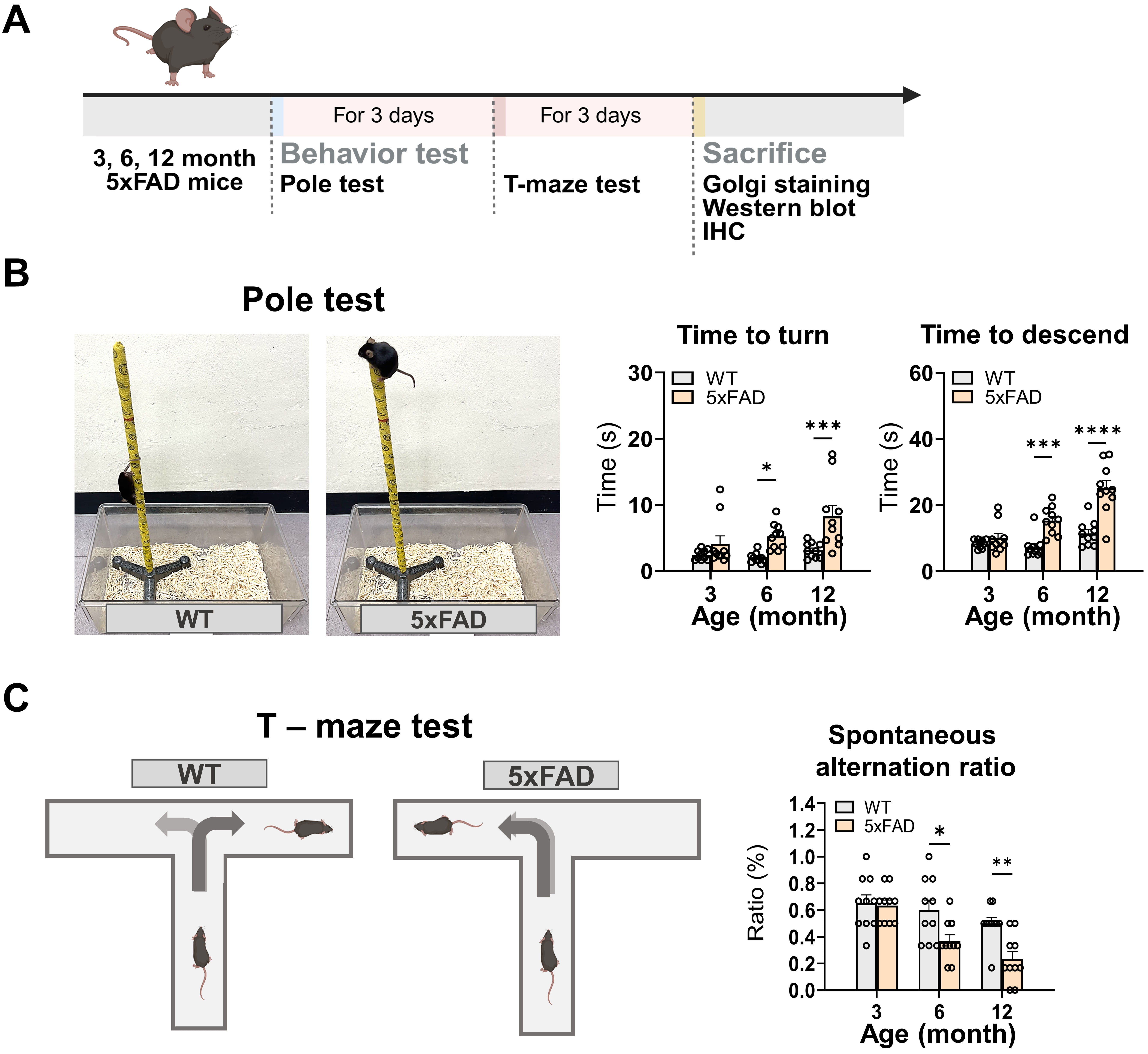 Fig. 1.
Fig. 1.
Schematic of the experimental design and behavioral
changes in 5FAD mice. (A) Pole and T-maze tests were performed on
5FAD and WT mice aged 3, 6, and 12 months. Subsequently, the animals
were sacrificed to collect samples for Golgi staining, Western blot, and IHC. (B)
Images representative of the pole tests are provided (left panel). The bar graphs
illustrate time to turn and time to descend in the pole test in 5FAD
mice compared with the WT (right panels). (C) Representative illustrations of the
T-maze test are provided (left panel). The bar graphs illustrate the spontaneous
alternation ratio observed in the T-maze test (right panel). Fig. 1A and Fig. 1C were generated
using BioRender.com (https://www.biorender.com/). The data represent the combined
mean standard error (SE) from two distinct experiments (n = 10/group). * p 0.05, ** p 0.01, *** p 0.001,
**** p 0.0001. IHC, immunohistochemistry; WT, wild-type;
5FAD, five familial Alzheimer’s disease.
2.2 Pole Test
The pole test was performed based on a previously established method,
incorporating minor modifications [18]. In brief, a metal pole (0.5 cm diameter,
50 cm height) was covered with gauze to prevent slippage. Each mouse was placed
facing upward at the top of the pole, and the time taken to descend to the base
was measured. Mice completed three trials, separated by 20-minute intervals, and
the average times (s) were assessed.
2.3 Spontaneous Alternation T-maze Test
The spontaneous alternation T-maze test was carried out using a previously
reported method with minor changes [19]. A T-maze was constructed from black
acrylic plastic, featured arms measuring 30 10 20 cm. Each
mouse was placed at the start arm for 10 min, after which the central divider was
removed to begin the trial. The animal was allowed to move freely toward either
the left or right goal arm. Once the tail fully entered the chosen arm, it was
closed off with a divider. After 30 s, the divider was lifted, and the mouse was
returned to the start arm for another trial. This procedure was performed twice
daily for three days, with 3-hour intervals between sessions. Scoring was
performed as follows: a score of 0 was assigned if the mouse chose the same arm
consecutively, and 1 if it alternated between arms within the trial.
2.4 Golgi Staining
To examine dendritic architecture, including spine morphology and density,
neurons from the cornu ammonis (CA) 1 subregion and dentate gyrus (DG) of the
hippocampus were subjected to Golgi staining. Following previously described
procedures [20], we used the FD Rapid Golgistain™ kit (#PK401, FD
Neurotechnologies, Columbia, MD, USA), following the instructions provided by the
manufacturer. Brain hemispheres (n = 5/group) were rinsed with 0.1 M phosphate
buffer saline (PBS, 1X; diluted from 10X stock, 502B828, Samchun Pure Chemical Co., Ltd., Gyeonggi-do, Korea) and immersed in Golgi–Cox solution for 2 weeks. Subsequently, the samples
were placed in a sucrose-based solution at room temperature (RT; 22 2
°C) for an additional 3 to 7 days. Coronal brain sections, cut at 200
µm thickness, were mounted on gelatin-coated slides with a small
amount of sucrose solution. After air-drying in the dark for 3 days, the slides
were processed according to the staining kit’s standard protocol.
2.5 Sholl Analysis
Quantification of dendritic complexity, length, and branching was performed
according to established methods [20]. Neuronal structures were imaged at
200 magnification using a Leica DM750 microscope (Leica Microsystems,
Wetzlar, Germany), and neuronal traces were obtained with Leica Application Suite
(v4.12) alongside Adobe Photoshop CS6 (Adobe Systems, San Jose, CA, USA). Sholl
analysis was carried out with ImageJ software (version 1.53f51, NIH, Bethesda,
MD, USA) using a concentric circle overlay technique [21]. For each animal (n =
5/group), 10 neurons were selected per hippocampal subregion following defined
criteria [22]: The cell body was situated in the designated subregion, exhibiting
thorough and effective branch staining along its entirety while remaining
isolated from adjacent cells. Concentric circles were drawn from the center of
the soma to the most distal dendritic end, spaced 10 µm apart. The
number of dendritic intersections at each radius was recorded to assess
arborization. Ten neurons per subregion were analyzed per mouse, and the mean
value from each mouse was treated as n = 1. Group results were calculated as the
mean standard error (SE), and sample details are provided in the
respective figure legends.
2.6 Analysis of Dendritic Spine Density and Morphology
The density and morphology of dendritic spines were measured according to an
established methodology [23]. For this analysis, we enumerated all identifiable
dendritic spines along a 30 µm segment of the distal dendrite of
each neuron using a magnification of 1000. Only segments that were
intact, clearly stained, and unbranched were selected. Spines were categorized
into three morphological types: thin spine, featuring a small head and elongated
neck; mushroom spine, distinguished by a large head and defined neck; stubby
spine, characterized by a compact appearance and absence of a discernible neck. A
total of 10 dendritic segments were analyzed per animal (n = 5/group), and spine
density was calculated as the number of spines per 10 µm of
dendritic length.
2.7 Western Blot
Western blotting was conducted in accordance with established protocols [24].
Each sample was sonicated in buffer H for 10 s, which contained 50 mM
-glycerophosphate (#G6376, Sigma-Aldrich, St. Louis, MO, USA), 1.5 mM ethylene glycol tetra-acetic acid (#E3889, Sigma-Aldrich), 1 mM
dithiothreitol (#09779, Sigma-Aldrich), 10 µg/mL aprotinin (#A1153, Sigma-Aldrich), 1 mM phenylmethanesulfonyl fluoride (#P7626, Sigma-Aldrich),
0.1 mM Na3VO4 (#S6508, Sigma-Aldrich), 10 µg/mL leupeptin (#L2884, Sigma-Aldrich), and 2 µg/mL pepstatin (#EI10, Sigma-Aldrich) at
pH 7.4. Subsequently, 6 sodium dodecyl sulfate (SDS) loading buffer was
added, and samples were boiled at 100 °C for 10 min. Protein samples
were separated on 10–15% SDS-polyacrylamide gel electrophoresis (#1658033, Bio-Rad;
Hercules, CA, USA), followed by transfer onto polyvinylidene difluoride membranes
(#10600023, Amersham Hybond-P; GE Healthcare Life Sciences, Pittsburgh, PA, USA). To block
nonspecific binding, membranes were incubated at RT for 1 h in PBS (1X; diluted from 10X stock, 502B828, Samchun Pure Chemical Co., Ltd.) with 0.1% (v/v) Tween-20 (#P2287, Sigma-Aldrich; PBS-T; pH 7.4) containing 1% (v/v)
normal goat serum (NGS; #S-1000, Vector Laboratories, Burlingame, CA, USA) and
0.5% (v/v) bovine serum albumin (BSA; #A9647, Sigma-Aldrich). Membranes were then incubated overnight at 4 °C with primary
antibodies diluted in blocking solution and then left at RT for 1 h. Primary
antibodies included mouse anti-A (1-16) (clone 6E10, 1:1000; #803001,
BioLegend, San Diego, CA, USA), rabbit anti-p-Tau (Thr181) (1:1000; #12885S,
Cell Signaling Technology, Danvers, MA, USA), mouse anti-glial fibrillary acidic
protein (GFAP, 1:1000; #3670, Cell Signaling Technology), rabbit anti-ionized
calcium-binding adapter molecule 1 (Iba-1, 1:1000; #016-20001, Fujifilm Wako,
Osaka, Japan), rabbit anti-PSD-95 (1:1000; #2507, Cell Signaling Technology),
and rabbit anti-activity-regulated cytoskeleton-associated protein (Arc; 1:1000;
#ab183183, Abcam, Cambridge, UK). Subsequent to the primary antibody incubation,
membranes were incubated for 2 h with either a horseradish peroxidase-conjugated
anti-rabbit secondary antibody (1:5000; #31460, Thermo Fisher Scientific,
Waltham, MA, USA) or mouse secondary antibody (1:5000; #31181, Thermo Fisher
Scientific). Signals were visualized using the EZ-Western Lumi Femto kit
(#DG-WF200, DoGenBio, Seoul, South Korea). Following stripping, membranes were re-probed
with mouse anti--actin antibody (1:5000; #A5441, Sigma-Aldrich) at RT
for 2 h. The optical density (OD) of each band was evaluated utilizing an iBright
CL750 Imaging System (Thermo Fisher Scientific). The mean band intensity of the
WT group was designated as ‘1’ for each blot, and relative expression levels were
calculated as fold changes. Group means were calculated and reported as mean
SE.
2.8 Immunohistochemistry
Immunohistochemistry (IHC) was performed in accordance with the protocol
previously established [20]. Brain hemispheres were fixed and sectioned
sagittally into 3 µm slices. The paraffin-embedded tissues were
processed using the Vectastain® Elite ABC kit (Rabbit IgG, #PK-6101; Mouse IgG, #PK-6102, Vector Laboratories) in accordance with standard immunohistochemical procedures. To
inhibit endogenous peroxidase activity, sections were incubated in 0.3% hydrogen
peroxide (#4104-4400, Daejung Chemicals & Metals Co., Ltd., Siheung, South Korea) for 20 min. Subsequently, tissues were blocked with either 5% NGS or
5% normal horse serum (Vectastain® Elite ABC kit) in PBS-T at RT
for 1 h to minimize non-specific binding. The sections were then incubated
overnight at 4 °C with the following primary antibodies: mouse
anti-A (1-16) (clone 6E10, 1:2000; #803001, BioLegend), rabbit
anti-p-Tau (Thr181) (1:100; #12885S, Cell Signaling Technology), mouse anti-GFAP
(1:4000; #3670, Cell Signaling Technology), and rabbit anti-Iba-1 (1:2000;
#019-19741, Fujifilm Wako). Following three washes with PBS, the slides were
incubated with the appropriate biotinylated secondary antibodies and subsequently
treated with ABC peroxidase in accordance with the kit protocol
(Vectastain® Elite ABC kit; Vector Laboratories). Finally,
peroxidase labeling was developed using the 3,3′-Diaminobenzidine (DAB) Substrate
kit (Vector Laboratories), followed by hematoxylin counterstaining.
2.9 Enzyme-linked Immunosorbent Assay
Cytokine levels, specifically tumor necrosis factor alpha (TNF, #DY410),
interferon-gamma (IFN, #DY485-05), interleukin (IL)-1 (#DY401), IL-6 (#DY406), and IL-10 (#DY417),
were quantified in hippocampal lysates from 5FAD mice using commercial
Enzyme-linked Immunosorbent Assay (ELISA) kits (R&D Systems, Minneapolis, MN,
USA) following the manufacturer’s instructions. The hippocampal tissues were
mechanically disrupted in lysis buffer, and the concentrations of total proteins
were subsequently determined. Lysates were applied to ELISA plates, and cytokine
concentrations were assessed by comparing sample absorbance to standard curves
created with recombinant cytokines. Absorbance measurements were conducted using
an Epoch Microplate Spectrophotometer (Bio-Tek Instruments, Winooski, VT, USA).
2.10 Statistical Analysis
Statistical analyses were performed using GraphPad software (version 9.3.1,
GraphPad Software, San Diego, CA, USA). To evaluate group differences in
behavioral performance, dendritic complexity, and spine density between WT and
5FAD mice, a two-way ANOVA followed by Sidak’s post hoc test was used.
Additional pairwise comparisons were conducted using an unpaired Student’s
t-test. A significance threshold of p 0.05 was considered
for all analyses. All numerical values are presented as mean SE. The
number of samples included in each experiment is indicated in the corresponding
Results sections and figure legends.
3. Results
3.1 Progressive Motor Deficits Observed in 5FAD Mice
The 5FAD mouse model exhibits age-related deficits in motor function
[25]; however, limited information exists regarding the temporal profile of motor
dysfunction in these mice. Therefore, this study conducted a pole test to
validate the fine motor dysfunctions in 5FAD mice (n = 10 mice/group;
the time to turn down: Finteraction (2,54) = 1.946, p = 0.1527, the
time to descend: Finteraction (2,54) = 8.825, p = 0.0005; Fig. 1B).
Notably, no significant differences were observed at 3 months in the time it took
the mice to turn down from the top of the pole (WT vs. 5FAD: p
= 0.4608) or to descend to the base of the pole (WT vs. 5FAD:
p = 0.7966). Comparatively, the 5FAD mice displayed a
significantly prolonged time to turn (p = 0.0357) and descend
(p = 0.0006) at 6 months compared to the WT mice. At 12 months, these
impairments were significantly more pronounced, with 5FAD mice showing
a significantly longer time to turn (p = 0.0004) and to descend
(p 0.0001). Collectively, these findings indicate that motor
dysfunction is evident in 5FAD mice and exacerbates with increased age.
3.2 Progressive Cognitive Impairments Observed in 5FAD
Mice
Next, the T-maze test was conducted in 5FAD and WT mice at 3, 6, and
12 months of age to assess the working memory through spontaneous alternation
behavior (n = 10/group; Finteraction (2,54) = 2.943, p = 0.0612;
Fig. 1C). This assessment evaluates the innate exploratory behavior of rodents in
novel environments, indicative of hippocampal-dependent cognitive function [26]
(Fig. 1C, left scheme). No significant differences were noted in the spontaneous
alternation ratio at 3 months between WT and 5FAD mice (p =
0.9955), indicating comparable cognitive performance during the early stage of AD
pathology. However, by 6 months, the 5FAD mice exhibited a significant
reduction in the spontaneous alternation ratio relative to WT mice (p =
0.0141), suggesting the emergence of hippocampus-dependent cognitive impairment
is linked to AD pathology. Meanwhile, cognitive deficits in 5FAD mice
were significantly more pronounced at 12 months—indicated by a further decrease
in the spontaneous alternation ratio (p = 0.0042). Statistical analysis
demonstrated an age-related cognitive decline in 5FAD mice, with
significant differences observed at 6 months, which were exacerbated by 12 months
(Fig. 1C, right bar graphs).
3.3 Increased Expression of A and p-Tau (Thr181) in the
Hippocampi of 5FAD Mice
This study investigated the age-dependent changes in A and p-Tau
(Thr181) expression in the hippocampi of 5FAD mice compared to WT
controls using Western blot and IHC (n = 3 mice/group). Western blot analysis
demonstrated a significant and progressive increase in A1-42 levels
in 5FAD mice with age compared to WT controls (t(4) = 3.311, p
= 0.0296 at 3 months; t(4) = 3.467, p = 0.0257 at 6 months; t(4) =
5.260, p = 0.0063 at 12 months) (Fig. 2A). Moreover, an age-dependent
increase in p-Tau expression levels was observed, achieving statistical
significance at 12 months (Fig. 2A). At 3 months, p-Tau levels did not
significantly differ from WT controls (t(4) = 0.8643, p = 0.4362). At 6
months, p-Tau levels were elevated in 5FAD mice, although this
difference did not reach statistical significance (t(4) = 2.166, p =
0.0963). At 12 months, hippocampal p-Tau levels in 5FAD mice
significantly increased (t(4) = 3.854, p = 0.0182).
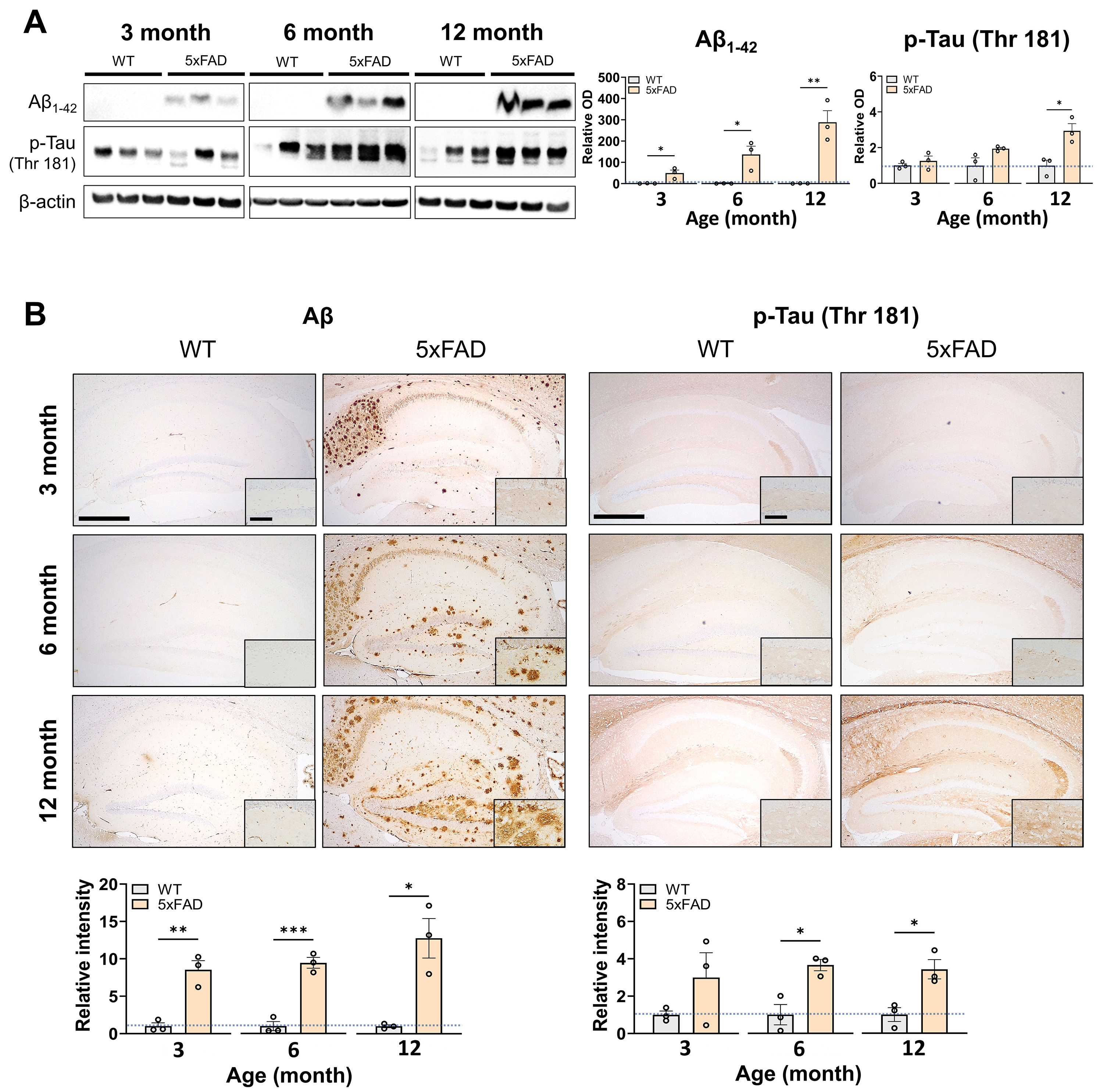 Fig. 2.
Fig. 2.
Expression levels of A1-42 and p-Tau
(Thr181) in the hippocampus. (A) Representative Western blot images and bar
graphs of the relative levels of A1-42 (~4 kDa) and
p-Tau (Thr181) in the hippocampi of WT and 5FAD mice (n = 3
mice/group). The full-length blot images are presented in Supplementary
Fig. 1. (B) Representative immunohistochemical images and bar graphs of the
relative immunoreactivities of A and p-Tau (Thr181) in the hippocampus
of WT and 5FAD mice (n = 3 mice/group). High-magnification versions of
the insets are presented in Supplementary Fig. 2. The data represent the
combined mean SE from two distinct experiments. * p 0.05, ** p 0.01, *** p 0.001. Scale bars represent 500 µm (low magnification) and 100 µm (inset) in panel B. A, amyloid
; OD, optical density; p-Tau, phosphorylated Tau.
Next, we performed IHC to characterize further the spatial distribution of
A and p-Tau (Thr181) expression across hippocampal subregions. IHC
analysis demonstrated a significantly progressive increase in A
immunoreactivity with age in 5FAD mice (t(4) = 5.879, p =
0.0042 at 3 months; t(4) = 8.966, p = 0.0009 at 6 months; t(4) = 4.443,
p = 0.0113 at 12 months) (Fig. 2B, left panels). At 3 months, A
exhibited extensive staining in the stratum pyramidale of CA1 and the distal CA1
subregion adjacent to the subiculum, with slight staining observed in the
molecular layer of the DG. Immunoreactivity was more pronounced at 6 months than
at 3 months of age, with A also detected in the stratum oriens of CA1,
stratum lucidum of CA3, and the subgranular zone as well as in the hilus of the
DG. At 12 months, the staining pattern was significantly enhanced across the
hippocampus of 5FAD mice.
IHC analysis of p-Tau demonstrated a delayed yet progressive accumulation in the
hippocampi of 5FAD mice (Fig. 2B, right panels). However, the p-Tau
levels in 5FAD mice did not significantly differ from those in WT
controls at 3 months (t(4) = 1.483, p = 0.2122). Conversely, the
difference in p-Tau expression reached statistical significance at 6 and 12
months (t(4) = 4.275, p = 0.0129 at 6 months; t(4) = 3.822, p =
0.0187 at 12 months). In the spatial pattern analysis, the immunoreactivity of
p-Tau was faintly detected at 3 months; however, p-Tau was observed in the
stratum lacunosum-moleculare of CA1, distal CA1 area, and hilus at 6 months of
age. Moreover, enhanced immunoreactivity was noted across the hippocampus of
5FAD mice at 12 months of age. Collectively, these findings indicate a
notable age-related escalation in early A and a subsequent increase in
p-Tau expression within the hippocampi of 5FAD mice.
3.4 Elevated Neuroinflammatory Responses Observed in the Hippocampi
of 5FAD Mice
We assessed the age-related alterations in astrocytic and microglial activation
and cytokine levels in the hippocampi of 5FAD mice. GFAP and Iba-1,
which serve as astrocytes and microglia markers, respectively, were assessed
through Western immunoblotting and IHC (n = 3 mice/group). Western blot analysis
revealed a progressive increase in GFAP and Iba-1 expression with age in
5FAD mice relative to WT controls (Fig. 3A). At 3 months, GFAP levels
were marginally elevated in 5FAD mice, though this difference did not
achieve statistical significance (t(4) = 1.986, p = 0.1180).
Comparatively, GFAP expression was significantly elevated in 5FAD mice
at 6 months and 12 months relative to WT controls (6 months: t(4) = 2.904,
p = 0.0439; 12 months: t(4) = 4.792, p = 0.0087). Iba-1 levels
were significantly elevated in 5FAD mice across all examined age
groups: 3 months: (t(4) = 6.161, p = 0.0035), 6 months: (t(4) = 4.351,
p = 0.0121), and 12 months: (t(4) = 5.931, p = 0.0041). In the
IHC analysis, the left panels in Fig. 3B demonstrate no significant differences
in the GFAP intensity in the hippocampus between groups at 3 months (t(4) =
1.054, p = 0.3515). However, a slight increase was observed in the
5FAD mice in the distal CA1 area. Elevated GFAP expression was
significantly noted at 6 months (t(4) = 4.722, p = 0.0092) and 12 months
(t(4) = 7.026, p = 0.0022). Notably, the intensities were markedly
pronounced throughout the hippocampus of 5FAD at 6 and 12 months of
age, particularly in the distal CA1, stratum oriens of CA1, and hilus. Iba-1
immunoreactivity in microglia (Fig. 3B, right panels) showed significant
differences with age-dependent increases (t(4) = 3.320, p = 0.0294 at 3
months; t(4) = 4.742, p = 0.0090 at 6 months; t(4) = 4.031, p =
0.0157 at 12 months). Furthermore, the Iba-1 expression was primarily noted in
the distal CA1 region at 3 months. In contrast, Iba-1 expression was present
throughout the hippocampus at 6 and 12 months of age, especially in the distal
CA1, stratum oriens of CA1, and molecular layer and hilus of the DG.
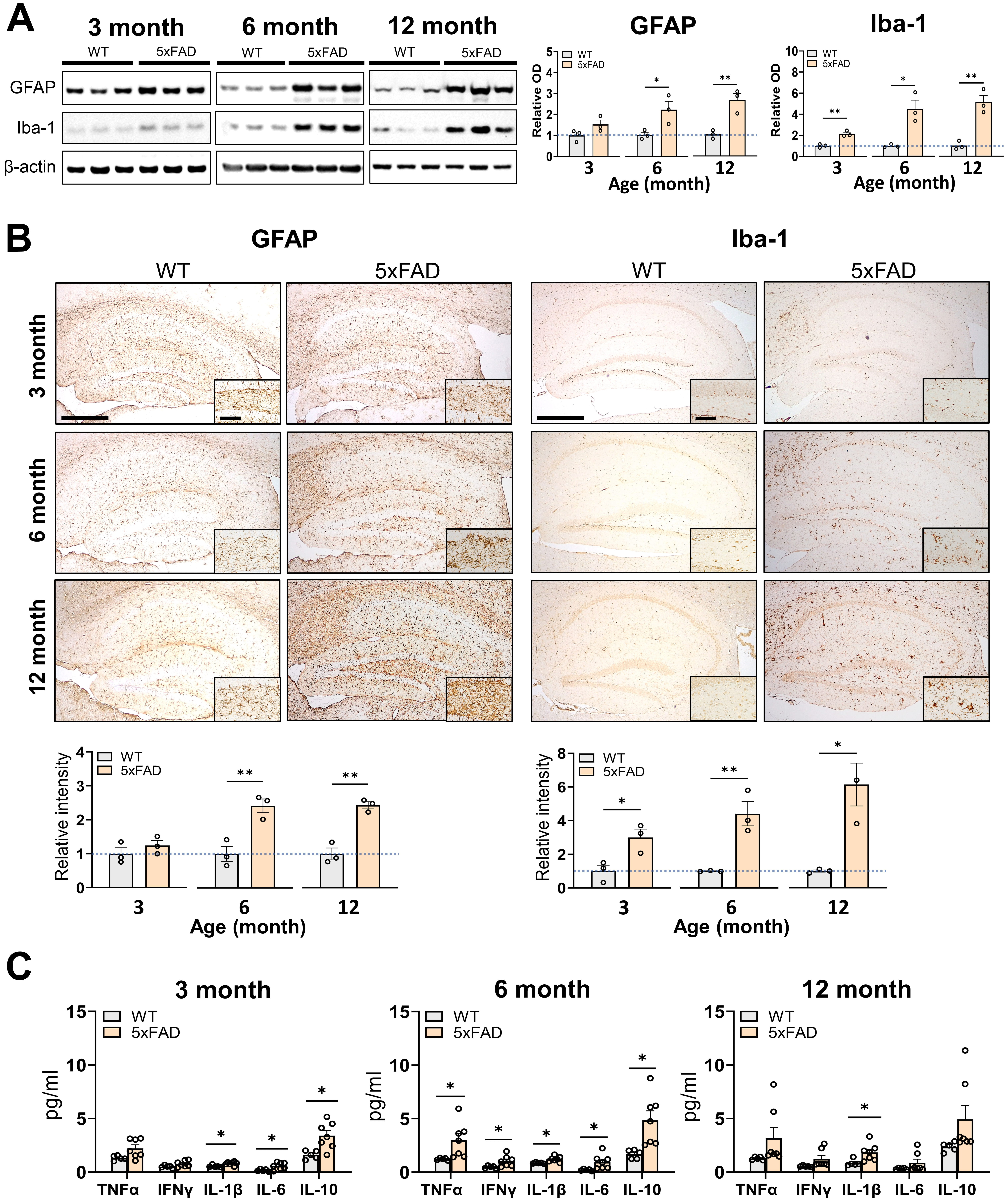 Fig. 3.
Fig. 3.
GFAP and Iba-1 expression and cytokine levels in the
hippocampus. (A) Representative Western blot images and bar graphs of the
relative levels of GFAP and Iba-1 in the hippocampi of WT and 5FAD mice
(n = 3 mice/group). The full-length blot images are presented in
Supplementary Fig. 3. (B) Representative immunohistochemical images and
bar graphs of the relative immunoreactivities of GFAP and Iba-1 in the hippocampi
of WT and 5FAD mice (n = 3 mice/group). High-magnification versions of
the insets are presented in Supplementary Fig. 4. (C) The bar graphs
illustrate the cytokine levels (TNF, IFN, IL-1,
IL-6, and IL-10) in the hippocampi of WT (n = 5) and 5FAD mice (n = 7)
at 3, 6, and 12 months of age, as determined by ELISA. The data represent the
combined mean SE from two distinct experiments. * p 0.05, ** p 0.01. Scale bars represent 500 µm (low magnification) and 100 µm (inset) in panel B. ELISA, enzyme-linked immunosorbent assay; GFAP, glial
fibrillary acidic protein; Iba-1, ionized calcium-binding adapter molecule 1;
IFN, interferon-gamma; IL, interleukin; TNF, tumor necrosis
factor alpha.
Additionally, ELISA was employed to assess the inflammatory cytokine profile in
the hippocampi of 5FAD mice for TNF, IFN,
IL-1, IL-6, and IL-10 at 3, 6, and 12 months of age (n = 5 for WT and n
= 7 for 5FAD; Fig. 3C). The inflammatory cytokines levels in the
hippocampi of 5FAD mice showed an overall upward trend across ages. At
3 months, the IL-1 (t(10) = 3.042, p = 0.0124), IL-6 (t(10) =
2.632, p = 0.0251), and IL-10 (t(10) = 3.084, p = 0.0116)
levels were significantly increased in 5FAD mice compared to WT
controls (Fig. 3C, left bar graphs). At 6 months, the TNF (t(10) =
2.375, p = 0.0390), IFN (t(10) = 2.329, p = 0.0422),
IL-1 (t(10) = 3.016, p = 0.0130), IL-6 (t(10) = 2.472,
p = 0.0330), and IL-10 (t(10) = 2.946, p = 0.0146) levels were
significantly elevated (Fig. 3C, middle bar graphs). At 12 months, the elevated
level of IL-1 (t(10) = 2.567, p = 0.0280) was statistically
significant (Fig. 3C, right bar graphs). Thus, these findings demonstrate a
progressive and age-dependent increase in astrocytic and microglial activation,
along with alterations in cytokine expression, in the hippocampi of
5FAD mice.
3.5 Dendritic Complexity Alterations in the Hippocampi of
5FAD Mice
We assessed hippocampal structural plasticity alterations in 5FAD mice
by analyzing the dendritic complexity of neurons in the CA1 and DG subregions,
focusing on the number of crossing dendrites, total dendritic length, and branch
points per neuron. Fig. 4 illustrates the counting of dendritic intersections at
various radial distances from the neuronal soma in the CA1 and DG subregions,
conducted through Sholl analysis (n = 5 mice/group). In the CA1 basal subregion
(Fig. 4A, upper panels), the 5FAD group showed fewer dendritic
intersections compared to the WT group at a Sholl radius of 50 µm from the
soma (Finteraction (20,1960) = 1.944, p = 0.0073; left-upper line
graphs) at 3 months. Dendritic intersections exhibited a significant reduction at
Sholl radii of 10–80 µm from the soma (Finteraction (20,1960) =
4.581, p 0.0001; middle-upper line graphs) at 6 months, with a more
pronounced effect observed at 12 months, particularly at Sholl radii of 10–90
µm (Finteraction (20,1960) = 5.652, p 0.0001; right-upper
line graphs). The total dendritic length in the CA1 basal subregion was reduced
in 5FAD mice relative to WT controls across all ages; however, these
differences did not reach statistical significance (Finteraction (2,24) =
0.2901, p = 0.7507; Fig. 4A, left-upper bar graphs). Analysis of
neuronal branch points (Finteraction (2,24) = 3.405, p = 0.0499;
Fig. 4A, right-upper bar graphs) revealed no significant differences at 3 months
(p = 0.8217). However, a significant reduction was observed in
5FAD mice at 6 months compared to WT controls (p = 0.0126),
with a more pronounced decrease at 12 months (p = 0.0005).
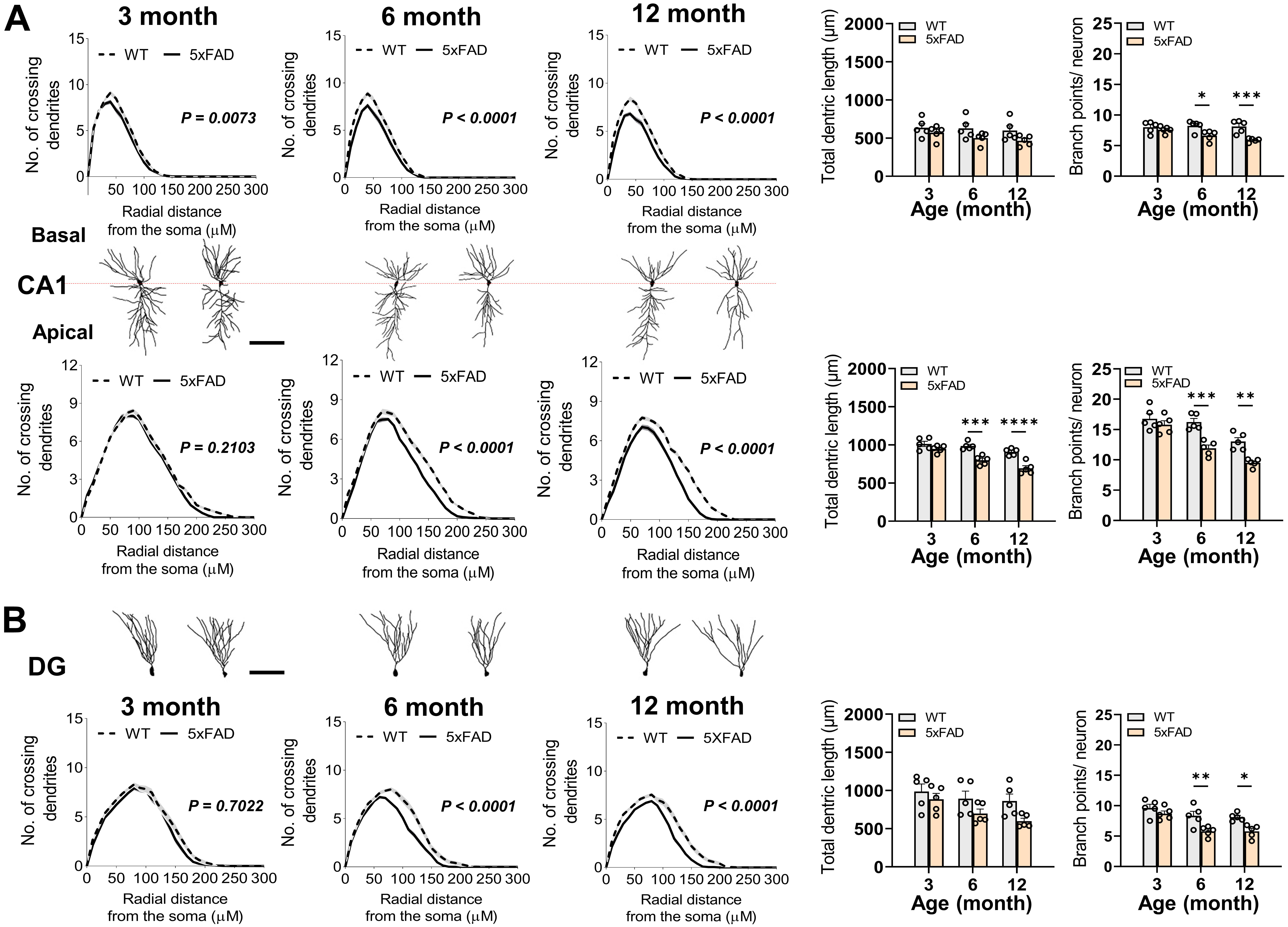 Fig. 4.
Fig. 4.
Dendritic complexity of hippocampal neurons in WT and
5FAD mice. Photographs illustrate representative neurons used for the
Sholl analysis in the CA1 (A) and DG (B) subregions. Line graphs illustrate the
mean number of intersections per 10 µm radial unit distance from the
soma (0 µm) in the neuronal dendrites of CA1 basal (A, upper panels)
and apical (A, lower panels), as well as in the DG (B) subregions. The bar graphs
illustrate the total dendritic length (left panels) and the number of dendritic
branch points (right panels) per neuron within each subregion. The data represent
the combined mean SE from two distinct experiments involving 10 neurons
per mouse and five mice per group. The actual images of the neurons are presented
in Supplementary Fig. 5. The gray areas illustrate the SE of the means
presented in the line graphs. * p 0.05, ** p 0.01, *** p 0.001, **** p 0.0001. Scale bars shown in the illustrated neuron images indicate 100 µm. CA1, cornu ammonis 1; DG, dentate gyrus.
No significant differences in dendritic intersections were noted in the CA1
apical subregion at 3 months between the 5FAD and WT groups
(Finteraction (30,2940) = 1.199, p = 0.2103; Fig. 4A, left-lower
line graphs). The dendritic intersections at 6 months showed a significant
reduction at Sholl radii of 90–200 µm from the soma
(Finteraction (30,2940) = 6.308, p 0.0001; Fig. 4A,
middle-lower line graphs). This gap became more pronounced at 12 months,
particularly at Sholl radii of 30, 50–70, and 90–190 µm
(Finteraction (30,2940) = 10.070, p 0.0001; Fig. 4A, right-lower
line graphs). The total dendritic length in the apical subregion
(Finteraction (2,24) = 4.403, p = 0.0235; Fig. 4A, left-lower bar
graphs) did not show a significant reduction in 5FAD mice compared to
WT controls at 3 months (p = 0.2714). However, the total dendritic
length in the apical subregion was statistically significant at 6 months
(p = 0.0002) and 12 months (p 0.0001). Analysis of neuronal
branch points (Finteraction (2,24) = 4.125, p = 0.0289; Fig. 4A,
left-lower bar graphs) revealed no significant differences at 3 months.
Meanwhile, a significant reduction was observed in 5FAD mice at 6
months compared to WT controls (p = 0.0001); this reduction persisted at
12 months (p = 0.0012).
No significant differences were observed in the dendritic intersections in the
DG subregion between 5FAD and WT mice at 3 months
(Finteraction (25,2450) = 0.8327, p = 0.7022; Fig. 4B, left line
graphs). The dendritic intersections in 5FAD mice at 6 months were
significantly diminished at Sholl radii of 80–140 µm from the soma
(Finteraction (25,2450) = 5.886, p 0.0001; Fig. 4B, middle line
graphs). This reduction became more pronounced at 12 months, particularly at
Sholl radii of 100–160 µm (Finteraction (25,2450) = 4.635, p 0.0001; Fig. 4B, right line graphs). The total dendritic length
(Finteraction (2,24) = 0.5119, p = 0.6057; Fig. 4B, left bar
graphs) did not show a significant reduction in 5FAD mice, although a
trend towards a decrease was observed across all ages in 5FAD group.
Neuronal branch points in the DG (Finteraction (2,24) = 1.460, p =
0.2520; Fig. 4B, right bar graphs) exhibited a slight decrease at 3 months, with
no significant difference observed. Alternatively, significant reductions were
observed at 6 and 12 months in 5FAD mice compared to WT controls
(p = 0.0049; p = 0.0103, respectively). Collectively, these
findings suggest that dendritic complexity decreases with age in the CA1 and DG
subregions of 5FAD mice.
3.6 Alteration in Dendritic Spine Density and Morphology in the
Hippocampi of 5FAD Mice
Dendritic spines serve as the main components of synapses in hippocampal neurons
and undergo rapid modifications in response to particular microenvironments,
including neurodegenerative conditions [27]. This study investigated the temporal
variations in dendritic spine density and morphology within the hippocampal CA1
and DG subregions of 5FAD and WT mice aged 3, 6, and 12 months (n = 5
mice/group; Fig. 5). Fig. 5A shows representative photomicrographs of neuronal
dendrites across the various hippocampal subregions. No significant differences
in spine density were observed in the CA1 basal subregion between 5FAD
and WT mice at 3 months (Finteraction (2,24) = 3.295, p = 0.0544;
Fig. 5B, left bar graphs). At 6 months, 5FAD mice exhibited a
significant reduction in spine density (p 0.0001), which persisted
at 12 months (p 0.0001). In the CA1 apical subregion of 3-month-old
5FAD mice (Finteraction (2,24) = 5.820, p = 0.0087; Fig. 5B, middle bar graphs), spine density exhibited a slight but significant decrease
(p = 0.0450); a notable reduction was observed at 6 months (p =
0.0114), with an additional decrease at 12 months (p 0.0001). The
spine density at 3 months in the DG subregion (Finteraction (2,24) =
9.058, p = 0.0012; Fig. 5B, right bar graphs) presented no significant
difference between groups (p = 0.9738). At 6 months, 5FAD mice
exhibited a significant reduction (p = 0.0018), which remained evident
at 12 months (p 0.0001).
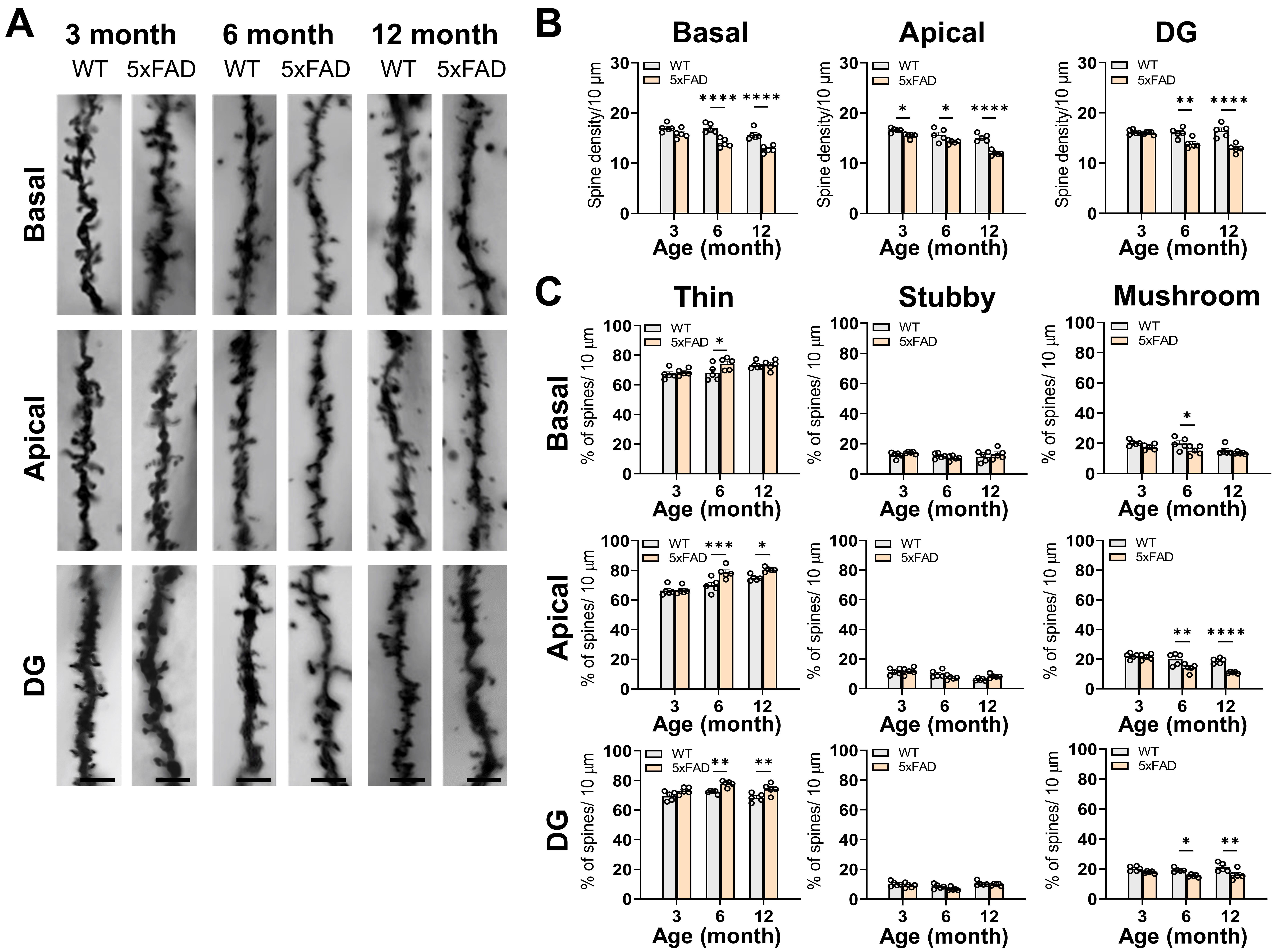 Fig. 5.
Fig. 5.
Dendritic spine density and morphology of hippocampal neurons in
WT and 5FAD mice. (A) Representative images of dendritic spines in the
CA1 basal, CA1 apical, and DG regions of 5FAD and WT mice at 3, 6, and
12 months of age. (B) The bar graphs illustrate spine density per 10
µm of dendrite length across hippocampal subregions in both WT and
5FAD mice. (C) The bar graphs exhibit proportional variations in spine
morphology categories (thin, stubby, and mushroom) per 10 µm of
dendrite. The data represent the combined mean SE from two distinct
experiments involving 10 neurons per mouse and five mice per group.
* p 0.05, ** p 0.01, *** p 0.001, **** p 0.0001. Scale bars in panel A represent 5
µm.
Additionally, differences in the proportion of dendritic spine morphology (% in
10 µm dendrite) were observed in the subregions of the hippocampi of
5FAD and WT mice (Fig. 5C). The proportion of thin spines (Fig. 5C,
left panels) was significantly elevated in the CA1 basal subregion
(Finteraction (2,24) = 1.933, p = 0.1667) of 6-month-old
5FAD mice (p = 0.0385), the CA1 apical subregion
(Finteraction (2,24) = 4.453, p = 0.0227) of both 6- and
12-month-old 5FAD mice (p = 0.009 and p = 0.0309,
respectively), and the DG subregion (Finteraction (2,24) =
0.5795, p = 0.5678) of 6- and 12-month-old 5FAD mice
(p = 0.0070 and p = 0.0063, respectively). In contrast, the
proportion of mushroom spines (Fig. 5C, right panels) was significantly decreased
in the CA1 basal subregion (Finteraction (2,24) = 0.7758, p =
0.4715) of 6-month-old 5FAD mice (p = 0.0306), the CA1 apical
subregion (Finteraction (2,24) = 6.364, p = 0.0061) of both 6- and
12-month-old 5FAD mice (p = 0.0017 and p 0.0001,
respectively), and the DG subregions (Finteraction (2,24) =
1.003, p = 0.3818) of 6- and 12-month-old 5FAD mice
(p = 0.0179 and p = 0.0026, respectively). No significant
difference was exhibited between WT and 5FAD mice in the proportion of
the stubby spines across all hippocampal subregions and age (Fig. 5C, middle
panels). Collectively, these findings indicate that changes in spine density and
morphology in the hippocampal neurons of 5FAD mice significantly worsen
with advancing age.
3.7 Decreased Expression of PSD-95 and Arc in the Hippocampi of
5FAD Mice
The subsequent investigation focused on the molecular alterations associated
with postsynaptic dysfunction in the hippocampi of 5FAD mice,
specifically analyzing the protein expression levels of PSD-95 and Arc, which
serve as markers of neuroplasticity [28, 29]. Western blot analysis was performed
on hippocampal lysates from 3-, 6-, and 12-month-old WT and 5FAD mice
(n = 3 mice/group; Fig. 6). At 3 months, no significant differences in PSD-95 or
Arc expression were noted between WT and 5FAD mice (t(4) = 1.005,
p = 0.3719; t(4) = 0.2712, p = 0.7997, respectively).
Conversely, both PSD-95 and Arc expression were significantly reduced in
5FAD mice at 6 months relative to WT controls (PSD-95: t(4) = 3.169,
p = 0.0339; Arc: t(4) = 3.746, p = 0.0200). While Arc levels
remained consistently reduced at 12 months (t(4) = 2.875, p = 0.0452),
PSD-95 expression showed a more pronounced decline (t(4) = 4.639, p =
0.0097). The results indicate an age-related decrease in the expression of PSD-95
and Arc in the hippocampi of 5FAD mice.
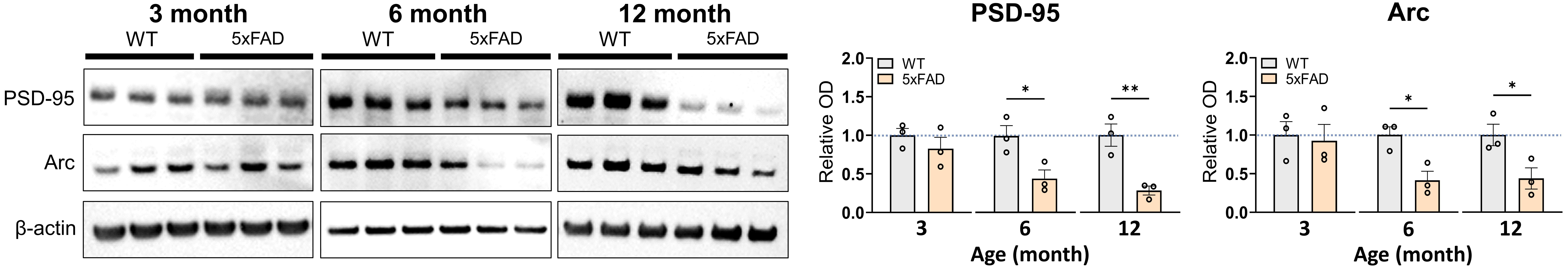 Fig. 6.
Fig. 6.
Expression levels of PSD-95 and Arc in the hippocampi of
5FAD and WT mice. Representative Western blot images and bar graphs of
the relative levels of PSD-95 and Arc in the hippocampi of WT and 5FAD
mice (n = 3 mice/group). The full-length blot images are presented in
Supplementary Fig. 6. The data represent the combined mean SE
from two distinct experiments. * p 0.05, ** p 0.01. Arc, activity-regulated cytoskeleton-associated protein; PSD-95,
postsynaptic density protein-95.
4. Discussion
This study examined the temporal changes in behavior and histopathological
lesions in 5FAD mice to understand the potential interactions between
these factors over time. The aging 5FAD mice demonstrated progressive
deficits in motor and cognitive functions, along with aggregation of A
and p-Tau, activation of astrocytes and microglia, production of proinflammatory
cytokines, alterations in neuronal dendritic complexity and spine morphology, and
reduced expression of postsynaptic proteins associated with neuroplasticity.
These data indicate a progressive decline in both neuronal structure and function
within the hippocampus of the AD mouse model, which may contribute to the
manifestation of behavioral symptoms associated with AD. To our knowledge, no
previous studies have investigated the temporal changes in the architecture of
hippocampal neurons in 5FAD mice. Therefore, this study offers
significant evidence concerning neuronal structural plasticity in AD progression.
In contrast to other prevalent AD models such as 3Tg and APP/PS1, the
5FAD model exhibits an earlier and more severe onset of A
pathology, due to the presence of five familial AD mutations [30, 31, 32]. The
features offer a clear advantage in detecting early structural and functional
changes in the hippocampus, thus reinforcing the temporal emphasis of the current
study.
Cognitive impairment is recognized as the primary pathological symptom in AD
patients [33]. Numerous studies have investigated learning and memory deficits in
various AD mouse models, including APP/PS1, Tg2576, 3Tg, and
5FAD, employing assessments such as passive avoidance, fear
conditioning, novel object recognition, and maze tests [34, 35, 36, 37]. Furthermore,
there is a growing body of evidence regarding non-cognitive motor symptoms in AD,
encompassing gait slowing, deficits in functional mobility, and impaired movement
balance [38, 39]. This study examined cognitive and motor functions in
5FAD mice at various ages, utilizing the T-maze test to assess
hippocampus-dependent working memory and the pole test for motor evaluation.
Although no significant differences were noted in the earlier age group (3
months), a marked decline in working memory and fine motor coordination became
evident at 6 months, with a more pronounced deterioration observed as the mice
aged. Multiple prior studies have indicated that an impairment in synaptic
plasticity, including diminished basal synaptic transmission and LTP, in the
hippocampus of 5FAD mice is directly linked to deficits in learning,
memory [40, 41, 42], and motor function [3]. The current findings indicate that
hippocampus-related behavioral dysfunction in 5FAD mice deteriorates
with age, mirroring the pathological progression of AD in humans.
The current study aimed to investigate the changes in the neuronal
microarchitecture of hippocampal subregions, including CA1 and DG, in
5FAD mice across various ages. Among indexes for dendritic complexity,
the significant decrease in crossing dendrite number was first observed in the
CA1 basal dendrites of 3-month-old 5FAD mice, with subsequent
reductions noted in all examined regions. Total dendritic length and the number
of dendritic branches decreased across all subregions in 5FAD mice
after 6 months of age, with significant reductions in dendritic length observed
exclusively in the CA1 apical dendrites. As indicated by the synaptic drive of
afferent inputs such as the perforant pathway [43], the accumulation of
pathological proteins in the entorhinal cortex (EC) and subiculum throughout AD
progression subsequently impacts the CA1 subregion, particularly the stratum
radiatum and stratum lacunosum-moleculare (SRLM) [44, 45, 46]. The DG, a gate
in the hippocampal trisynaptic circuit, regulates the transmission of information
from the EC to the CA3 region, subsequently influencing CA1 pyramidal neurons.
Consequently, the AD pathology originating in the EC also impacts the DG and CA3
subregions [43]. Furthermore, the impairment of adult hippocampal neurogenesis in
early AD may lead to neuronal degeneration in the EC due to a reduction in newly
formed axonal targets in the DG [47]. Consequently, significant neuronal death in
the EC results in axonal denervation from the perforant pathway and synaptic loss
in the DG dendrites, thereby fundamentally disrupting dendritic and axonal
integrity and, in turn, hippocampal circuitry [48, 49]. Postmortem studies in AD
patients have indicated a progressive synaptic loss in the SRLM from normal
individuals to those with mild cognitive impairment and further to AD [50].
Additionally, notable atrophy of the SRLM was observed in mild AD cases [51]. DG
granule cells exhibit early morphological changes that worsen as AD progresses,
suggesting a correlation with cognitive deficits [49]. Meanwhile, highly plastic
synapses onto hippocampal dendrites are hypothesized to be critical for learning
and memory [52]. Therefore, dysfunction in neuroplasticity at these synapses may
directly induce cognitive impairment in AD.
In addition, this study observed a decline in spine density across all
identified hippocampal areas in 3 or 6-month-old 5FAD mice,
characterized by an increase in thin spines and a decrease in mushroom spines.
Thin spines can transform into mushroom spines while encoding information and
reacting to neuronal activity [53, 54]. In contrast to thin spines that
predominantly feature N-methyl-D-aspartate glutamate receptors (NMDARs), mushroom
spines, characterized by a broad postsynaptic density in their large heads,
contain a higher concentration of
-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs),
thereby enhancing synaptic functionality [55]. Therefore, a deficiency in
mushroom spines enriched with AMPARs may contribute to learning and memory
deficits exhibited in the AD pathophysiology, as reduced AMPAR levels are linked
to the spine and synaptic immaturity and LTD [56]. Additionally, activation of
the extrasynaptic NMDAR GluN2B subunit following excessive glutamate release from
activated glia results in LTD, spine shrinkage, and retraction, which can
ultimately promote synaptic loss [57]; the dysregulation of NMDAR subunits
contributes to glutamate excitotoxicity in AD pathology [58]. Consequently, the
functional imbalance of glutamatergic receptors in spines, associated with their
morphology, will result in synaptopathy in AD. Furthermore, clinical studies
indicate a correlation between cognitive impairment and reduced spine density,
decreased spine head diameter, and increased spine length in AD patients [59, 60]. Local synaptic dysfunction in AD patients initiates several years before the
onset of clinical symptoms, with cumulative deficits in neuronal plasticity
leading to behavioral changes [61]. Based on prior evidence, we propose that
early morphological changes in dendrites and spines within the hippocampal CA1
and DG subregions may contribute to behavioral dysfunction in 5FAD
mice.
To enhance our understanding of the mechanisms contributing to structural
impairments of hippocampal neurons in AD, we investigated the temporal patterns
of accumulation of key pathological proteins, including A and p-Tau, and
neuroinflammation. The present study showed that A deposits were
prominently identified initially in the distal CA1 subregion connecting with the
subiculum and the stratum pyramidale of CA1 in 3-month-old 5FAD.
Additionally, p-Tau exhibited faint staining in the distal CA1 and the stratum
lacunosum-moleculare of CA1 in 6-month-old 5FAD mice. Subsequently,
both immunoreactivities disseminated across all hippocampal subregions. These
results align with our neuromorphological data and support the prior assertion
that A is primarily involved in the early stages of AD progression;
meanwhile, p-Tau pathology is likely significant in the later stages [62, 63].
Numerous in vitro and in vivo studies suggest that A
oligomers contribute to postsynaptic dysfunction and dendritic pathology in AD
through the dysregulation of glutamatergic receptors, including NMDARs, AMPARs,
and metabotropic glutamate receptors. This dysregulation may result in an
imbalance of LTP and LTD and loss of dendrites and spines [64, 65, 66, 67]. Furthermore,
A oligomers induce neurotoxicity via several mechanisms, such as
blood–brain barrier disruption, oxidative stress, glial activation, and
dysfunction of kinases and phosphatases [68, 69]. A oligomers induce the
overproduction of p-Tau, with both proteins interacting synergistically through a
feedback loop, independent of their aggregation into plaques and tangles, thereby
impairing neuroplasticity [70]. Therefore, our findings suggest that the
accumulation of A and p-Tau adversely affects neuronal structure and
synaptic integrity, contributing to cognitive decline in AD.
Additionally, we investigated the inflammatory responses, identified as a third
core feature of AD in 5FAD mice, alongside A plaques and NFTs
[71]. At 3 months, microgliosis was primarily observed in the distal CA1
subregion, aligning spatially and temporally with A staining.
Subsequently, as AD progressed, the activation of astrocytes and microglia was
heightened across the hippocampus, particularly in the distal CA1, stratum oriens
of CA1, and molecular layer and hilus of the DG, where immunoreactivity mirrored
the accumulation of A and p-Tau. Inflammatory cytokine production was
observed in the hippocampus of 5FAD mice from an early age. Meanwhile,
substantial evidence exists in AD patients and animal models regarding the
inflammatory response contribution to the disease pathology [72]. In the early
stages of AD, A deposition triggers microglial activation and the
recruitment of astrocytes. Moreover, glia-secreting inflammatory cytokines,
including TNF, IL-1, and IL-6, can increase A
accumulation and promote hyperphosphorylation of tau proteins. This process
establishes a detrimental cycle that leads to additional glial activation and
cytokine production [73, 74, 75]. Furthermore, A can stimulate the release of
complement protein C3 in astrocytes, and the binding of C3 to microglial or
neuronal receptors may influence A phagocytosis or dendritic morphology,
respectively, contributing to synaptic deficits in AD [76]. This study identified
a close temporal relationship among pathological protein accumulation,
neuroinflammation, neuroarchitecture, and behavioral impairment, suggesting that
these interconnected factors may collectively contribute to synaptic and
cognitive dysfunction in AD.
Arc, an immediate early gene, and PSD-95, a scaffolding protein, both situated
post-synapse, regulate synaptic homeostasis through distinct mechanisms. Arc
promotes the endocytosis of AMPARs by interacting with endocytic machinery and
alters dendritic spine morphology by regulating cytoskeletal proteins [77, 78, 79].
PSD-95 influences the trafficking and localization of postsynaptic components,
including ion channels, signaling molecules, cytoskeletal elements, and glutamate
receptors [80, 81]. Consequently, Arc or PSD-95 dysfunction induces abnormal
synaptic plasticity and neuronal morphology. In AD pathology, Arc and PSD-95
contribute directly to disease progression through their respective roles in
A production by interacting with presenilin 1 [82] and the essential
involvement in the neurotoxic pathway of A and p-Tau [83]. Conflicting
observations have been noted regarding the up- or downregulation of Arc and
PSD-95 expressions in the brains of AD patients and animal models; however, both
homeostatic dysfunctions may contribute to synaptopathy [84, 85]. These
contrasting expression patterns may be linked to varying disease severities or
susceptibility among brain regions in the studied subjects [85]. This study
confirmed a decrease in the expression of both proteins in the hippocampus of
5FAD mice from 6 months of age. This decline appears to be attributable
to the hippocampus being one of the most vulnerable brain regions in AD, with
pathological progression accelerating from that age. Furthermore, clinical
reports indicate that decreased expressions of Arc [86, 87] and PSD-95 [88]
significantly correlate with cognitive status and postmortem levels of A
and p-Tau. Therefore, as AD progresses with age, postsynaptic proteins associated
with synaptic homeostasis, including Arc and PSD-95, may become compromised in
the hippocampus, leading to synaptic degeneration, loss, and subsequent
behavioral dysfunction.
While this study provides valuable insights into the age-dependent structural
and behavioral alterations in the 5FAD model, several limitations
should be acknowledged. First, all experiments were conducted using only female
5FAD mice to reduce variability related to sex-dependent transgene
expression and behavioral phenotypes [89, 90], which may limit the
generalizability across sexes. Second, although the T-maze test efficiently
assesses hippocampal-dependent working memory, incorporating additional cognitive
paradigms would provide a more thorough behavioral profile. Third, a consistent
within-subject design was not implemented at all time points to mitigate stress
and maintain tissue quality, especially in aged mice. This may restrict direct
associations between molecular markers and behavior. Fourth, the relatively small
sample size necessitates careful interpretation of the results. Lastly, the
analysis focused solely on pTau181 and excluded other phospho-tau species, such
as p-Tau217, which may offer further insights into tau pathology [91, 92]. Taken
together, these limitations underscore the necessity for future research to
include both sexes, utilize broader cognitive assessments, employ longitudinal
within-subject designs, involve larger cohorts, and integrate distinct p-Tau
markers to validate and expand upon our findings.








 , Sueun Lee 2,†
, Sueun Lee 2,†
 Fig. 7.
Fig. 7.