
 , Li Zeng 1, Wu Li 1,*
, Li Zeng 1, Wu Li 1,* , Jiang-Shan Li 1
, Jiang-Shan Li 11 Department of Tuina, School of Acupuncture-Moxibustion and Tuina, Hunan University of Chinese Medicine, 410208 Changsha, Hunan, China
Abstract
Chronic pain frequently coexists with adverse emotions, including anxiety and depression, significantly affecting patients’ physical and psychological health as well as their quality of life. Changes in hippocampal synaptic architecture, neuronal injury, and diminished neurogenesis significantly contribute to pain-related emotions. Microglia in the hippocampus are implicated in these pathologies. Stimulation or injury leads to microglial activation, which causes pain; prolonged pain causes microglia to continuously release pro-inflammatory factors that induce astrocyte activation, which mediates the apoptosis of hippocampal neurons and abnormal neurogenesis. Concurrently, microglia exhibit aberrant phagocytosis and augmented pruning of hippocampal dendritic spines, which disrupts synaptic plasticity and influences hippocampal long-term potentiation, hence contributing to the emergence of negative emotions. Inflammatory responses in the brain are a prevalent pathological foundation for mood disorders and pain, and the activation or inhibition of microglia M1 polarization can influence pain-related emotions. This review elucidates the significance of hippocampal microglia activation, and their interactions with neurons in the hippocampus and astrocytes, in pain-related emotions.
Keywords
- hippocampus
- microglia
- pain
- pain-related emotion
Chronic pain not only engenders a sense of desperation for treatment among patients, but it can also heighten their pain perception, leading to exaggerated responses. Patients seek alleviation from pain and its associated unpleasant feelings, but achieving this respite is sometimes challenging, which can intensify their symptoms of anxiety and depression. Consequently, these feelings lead patients to concentrate more on the pain [1], establishing a detrimental cycle between pain and negative emotions, which is the primary reason why chronic pain is challenging to manage. The hippocampus is engaged in both emotional processing and pain perception, serving a major function in the neuronal network associated with pain-related emotions [2, 3]. Microglia are spread throughout the central nervous system and the inflammation they initiate is a significant factor contributing to the adverse emotions associated with pain. This research review presents an overview of microglia polarization in the hippocampus and its mechanisms involved in pain-related emotions.
Microglia, originating from the yolk sac of the embryonic hematopoietic system,
are pivotal resident immune cells that have a shared lineage with peripheral
macrophages [4] and are often the initial responders to nociceptive stimuli.
Microglia exhibit extensive branching in their resting state under typical
physiological settings, performing functions that include phagocytosing apoptotic
cell debris, and pruning and stabilizing neuronal dendritic spines. Upon external
stimulation, microglia are activated, multiply fast, grow and expand the cytosol,
retract and eliminate protrusions, have an “amoeba-like” shape, and secrete
cytokines. According to macrophage-polarization terms, activated microglia can be
categorized into classically activated (M1) cells, which exhibit pro-inflammatory
functions, and alternatively activated (M2) cells, which demonstrate
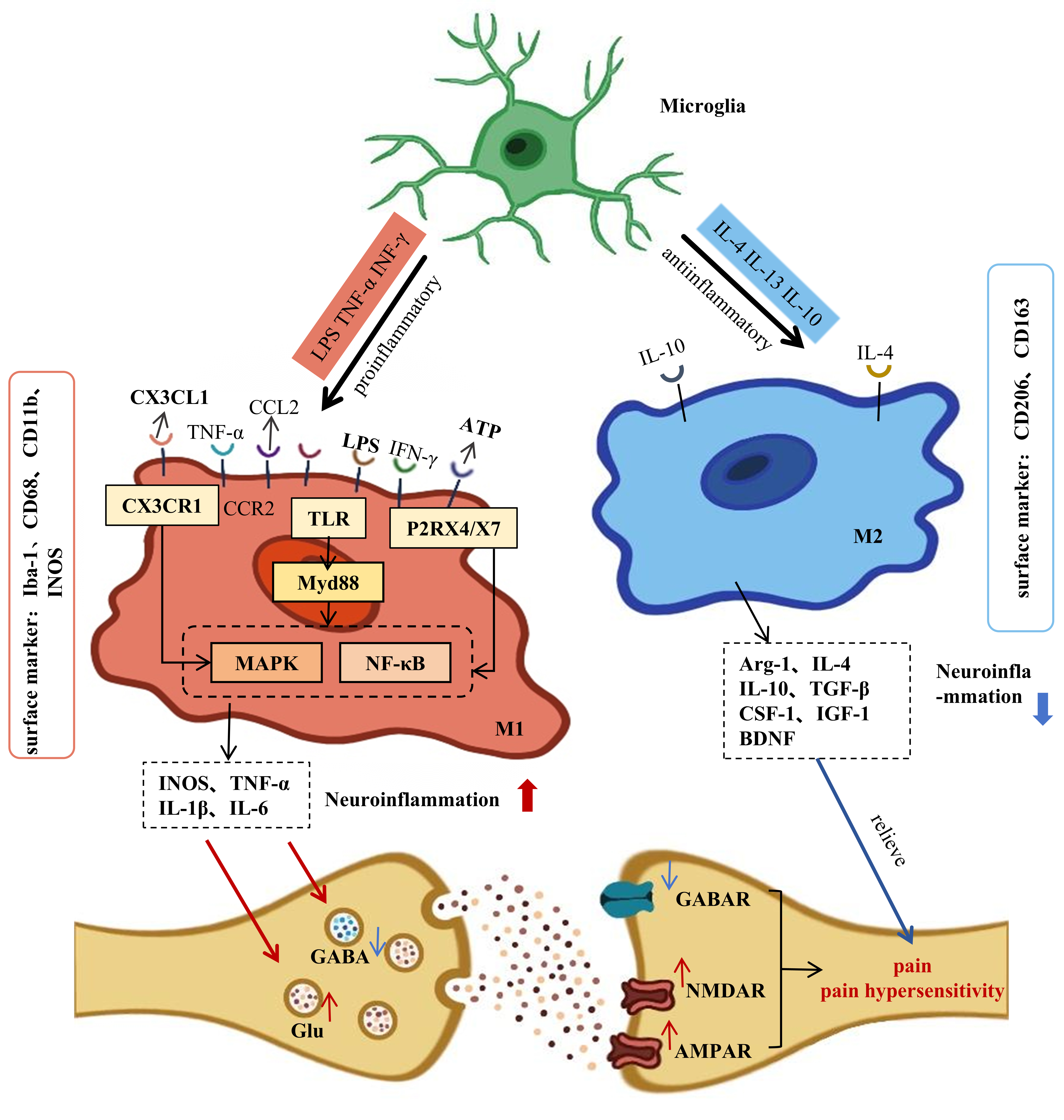
anti-inflammatory functions—a phenomenon referred to as polarization (Fig. 1).
The polarization of microglia exerts a bidirectional influence on neurons. M1
microglia detect interferon-gamma (IFN-
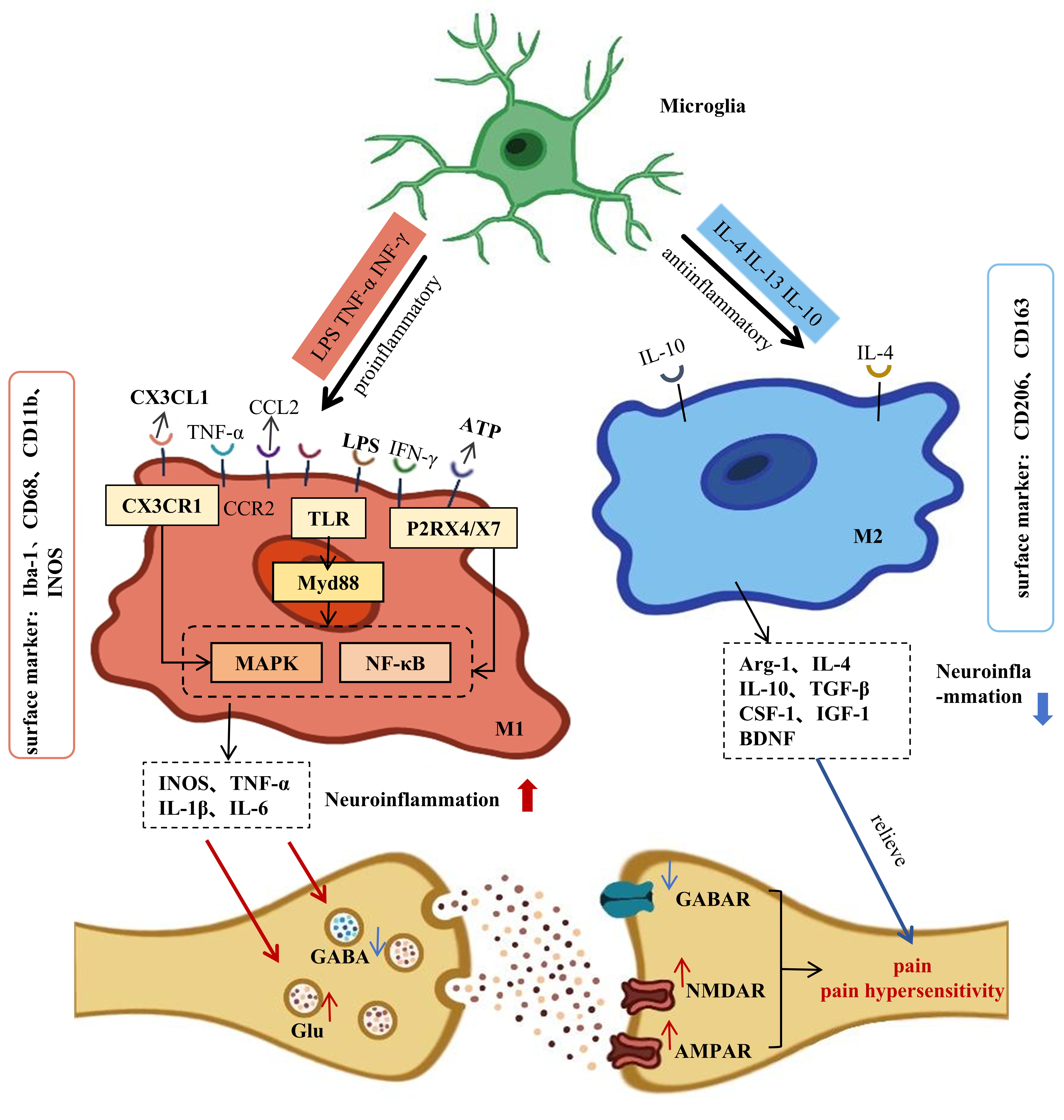 Fig. 1.
Fig. 1.
The polarization process of microglia. Resting microglia detect
IFN-
In recent years, with the development and application of technologies such as spatial and single-cell histology, multiple subtypes of microglia have been found, and the formation of these subtypes is related to signaling pathways at different levels within the microenvironment, such as epigenetic, transcriptional, and translational aspects [6]. The morphologic structures of the different subtypes are very different and can be categorized as follows: (a) CD11c+ microglia, which are associated with the phagocytosis of microglia, shifting from CD11c- to CD11c+ upon clearance of apoptotic neurons [7]; (b) dark microglia (DM), associated with aging and degenerative diseases such as Parkinson’s disease [8], which can only be observed by electron microscopy, showing cytoplasmic shrinkage and expansion of the endoplasmic reticulum and Golgi apparatus accompanied by mitochondrial alterations [9]; (c) repair-associated microglia, which are often seen during the repair process after cerebrovascular injury, promote angiogenesis by forming a “rosette” of tissue that encapsulates the injured blood vessels to inhibit leakage of the blood-brain barrier [10]; (d) disease-associated microglia, named after their discovery in Alzheimer’s Disease (AD) transgenic mice [11], which are also present in other diseases such as multiple sclerosis and stroke [12]; and (e) white matter-associated microglia that are associated with age and clear white-matter myelin debris, with marker gene expression that increases with white-matter aging. Microglia subtypes are not limited to those described above; they can have different morphologies and functions in different species, diseases, and brain regions. For example, microglia located in the cerebellum highly express F4/80, whereas hippocampal microglia express more Tnf and Fc gamma receptor II (Fcgr2) [13]. DM is present in rats and humans [14], but the main source of CD11c+ and DM cell subtypes is the mouse. In summary, microglia can change into subtypes according to the external environment, and thus perform different functions.
Pain can be classified as acute or chronic according to its duration. Pain not only stimulates reward-related neuronal pathways in the brain’s limbic system, including the dopamine system, but also profoundly influences an individual’s psychological condition. Acute pain typically provokes an immediate reaction from the individual, including stress and the emergence of avoidance strategies. Chronic pain is linked to gradual adverse mood alterations, first with anxiety and progressively advancing to depression, ultimately resulting in a co-morbid condition of pain, anxiety, and depression. The morphology of microglia in the brain under chronic stress states is remodeled and is accompanied by a depressive and anxious state [15]. The imaging results of patients with episodes of major depressive disorder indicate that microglia are excessively activated in the brain [16] and are strongly associated with the severity of these episodes. Subsequent research has established that M1-type microglia play a crucial role in the emergence of negative emotions. When the M1 type remains prevalent and M2-type expression diminishes, behavioral symptoms such as depression and anxiety emerge. These adverse emotions are further intensified when M1-type expression significantly surpasses the level of the M2 type [17]. Both chronic pain and the elicitation of negative emotions are associated with the activation and polarization of microglia, with M1-type microglia being pivotal in the manifestation of pain-related emotions. There are sex differences in microglia in response to chronic pain and its accompanying emotions, which are related to sex hormones in vivo [18] and to stressful events experienced during fetal life [15]. This phenomenon is also present in rodents, in which there are sex as well as age differences [19] in the phenotype [20], morphology, and number of microglia and the cytokines and chemokines they release in the hippocampus; males show activation of significantly more microglia than do females; females show a greater tendency for hippocampus-located microglia to migrate to the amygdala with age, accompanied by a decrease in microglial cell marker cluster of differentiation 68 (CD68). There are also significant differences between males and females in their attitudes toward pain [21] and in their immune and stress responses [22, 23] when in a state of chronic pain. In neuropathic pain, female rats have more intense pain sensations [22]. Emotional responses to neuropathic pain and the mechanisms involved also differ by sex, i.e., microglia in the hippocampus show higher levels of CD11b expression in males, whereas treatment with ginger only alleviates the emotional response in males but reduces microglia activity in rats of both sexes [23]. In humans, chronic pain and its accompanying negative emotions are related to an individual’s own life experiences and environment, in addition to sex and age. Adverse events that occur during early formative experiences are associated with a greater risk of developing chronic pain and psychiatric disorders in adulthood [24]. External stress induces a pro-inflammatory transformation of microglia through an increase in systemic glucocorticoids [25], and microglia are more active in women than in men when experiencing stress [26].
Chronic pain can elevate pro-inflammatory molecules such as IL-1
In conclusion, hyperactivated microglia and M1-type microglia, which contribute to central inflammation, are pivotal in the emergence of unpleasant emotions, and the interactions between microglia and astrocytes also participate in the occurrence of this process. Consequently, examining the central inflammatory process of emotional impairments related to pain via the lens of microglia is crucial for elucidating the pathophysiology of such emotions.
The hippocampus, a part of the limbic system, contributes to the manifestation of chronic pain and negative affect [37], and its malfunction and lesions can lead to co-morbidities of pain and emotion [38]. Bingel et al. [39] showed that painful stimuli can be transmitted to the hippocampus, resulting in activation and hemodynamic alterations. Imaging results from certain patients with chronic pain indicated that bilateral hippocampus volume was reduced below normal levels [40]. In the neuropathic-pain paradigm, impairment of hippocampus function in mice has also been demonstrated [41]. The aforementioned findings demonstrate that chronic pain induces functional and anatomical alterations in the hippocampal region. The cornu ammonis 1 (CA1) region of the ventral hippocampus participates in pain perception and emotional feedback, and studies have indicated that pain activation can suppress neuronal activity in this area [41, 42]. Under stress, the synaptic architecture of the rat hippocampal CA3 region was modified, and the atrophy of pyramidal cell apical dendrites coincided with a significant decrease in the volume of the hippocampal CA1 area, leading to deficits in memory and emotion [43]. Consequently, changes in hippocampal neurons and their synaptic structure are intricately linked to the emergence of pain-related emotions. Simultaneously, neurogenesis occurs in the hippocampus, indicating that the process of new neural growth and development can persist from the embryonic stage into adulthood. Neurogenesis in the hippocampus significantly influences the modulation of pain perception and emotional conditions. Hippocampal neurogenesis involves multiple steps and regulatory mechanisms. The source of newborn neurons in the adult hippocampus is neural stem cells (NSCs). In rodents, NSCs in the hippocampal dentate gyrus (DG), also known as radial glial-like cells, have radial projections that extend into the DG granule-cell layer and express the astrocyte markers sex-determining region Y-box 2 (SOX2) and glial fibrillary acidic protein (GFAP) [44]. When NSCs are activated, they give rise to amplifying neural progenitors (ANPs) [45]. After passing through the early survival stage [46], progenitor cells further differentiate into adult neuronal cells [45]. Adult neuronal cells differentiate into functional granular neurons through a maturation process of weeks or months [45]. After 2–3 weeks, newborn DG granular neurons (GCs) receive excitatory inputs from DGs and conduct action potentials to pyramidal neurons in the hippocampal CA2 and CA3 regions. After 4–8 weeks, the newborn GCs are fully integrated into the hippocampal circuits and participate in information storage (memory) [45]. Terreros-Roncal et al. [46] demonstrated by that NSCs and immature neurons are also present in the brain of healthy adults. Fang et al. [47] noted a decrease in immature nerve cells inside the hippocampal dentate gyrus in mice with chronic inflammatory pain, and the administration of a neurogenesis inhibitor in the hippocampus resulted in heightened nociception and exacerbated depressive symptoms in the mice. Conversely, the administration of neurogenesis-enhancing therapies showed efficacy in mitigating pain and depressive-like behaviors. In addition, BDNF is crucial for hippocampal regeneration and synaptic plasticity, and its diminished expression and levels predispose individuals to negative emotions. Chronic pain stimuli may precipitate mood disorders, including anxiety, by reducing BDNF levels and neurogenesis in the hippocampus [48]. In conclusion, chronic pain not only diminishes hippocampal volume but also disrupts its normal functioning.
Pain affects more than the hippocampal region and involves the interaction of several regions in the brain. For example, the anterior cingulate cortex (ACC) receives afferent pain signals and projects these signals to the hippocampus and the anterior thalamic nucleus (Papez’s loop) to co-construct the basis of pain cognition [49]. The connecting loops of the hippocampus and the prelimbic neocortex are then involved in the generation of emotional and cognitive deficits associated with neuropathic pain [50]. In addition, Ma’s team [51] found that chronic inflammatory pain disrupts CA1-infralimbic cortex (vCA1-IL) connectivity in the ventral hippocampus of rats, and that activation of the vCA1-IL loop using optogenetics results in pain relief. In rodents, the basolateral nucleus of the amygdala (BLA) has dense monosynaptic glutamatergic projections to the ventral hippocampus (vHPC), and functional connectivity between the two brain regions is strongly correlated with anxiety production [52]. In the brains of chronic social-defeat-stress-model mice, gamma-aminobutyric acid (GABA)-receptor-mediated reduction of tensile inhibitory currents from BLA neurons projecting to ventral hippocampal neurons led to a rise in neural excitability of the BLA-vHPC loop, and the mice developed anxiety [53]. Chronic-restraint-stress mice show increased dendritic branching and density of BLA-vHPC neurons and stronger excitatory synaptic transmission between them, an alteration that is associated with anxiety [53]. This shows that the crosstalk between the amygdala and the hippocampus is closely related to anxiety in pathological states.
The above studies reveal that chronic-pain-induced emotional responses are not the result of lesions in a single brain region, but rather a joint result of the interaction of neural circuits between the hippocampus and brain regions such as the ACC, prelimbic neocortex, IL, and amygdala. A complex network of these brain regions works in concert to regulate pain-related emotional responses and cognitive processes. In a state of chronic pain, neuroplastic alterations in the pain pathways of the spinal cord and brain are commonly termed “central sensitization” [54]. One of the main manifestations is long-term potentiation (LTP) of synaptic transmission [55]. Hippocampal neuronal damage, along with altered neurogenesis and synaptic plasticity, constitutes a significant pathological foundation for the emergence of unpleasant feelings.
Microglia are located throughout the adult brain, especially in the hippocampus and cerebral cortex, regions characterized by high neuroplasticity and intricate functions, where their activity and density are notably pronounced. Simultaneously, the hippocampus is abundant in pattern-recognition receptors (PRRs) and cytokine receptors [56]. These receptors are predominantly located in microglia, astrocytes, and neurons, rendering glial cells and neurons in the hippocampus very vulnerable to inflammation. Microglia influence hippocampal neuronal activity by releasing inflammatory substances and give rise to the emergence of negative emotions by disrupting synaptic plasticity, namely LTP. Gaspar et al. [15] showed that hippocampal microglia are activated in rats experiencing depression and anxiety. Subsequent research revealed that the inhibition of microglial activation averted harm to LTP and mood disorders, but their activation replicated anxiety-depression-like behaviors associated with nerve injury. Chen’s team [57] discovered that asymmetric activation of hippocampal microglia could induce anxiety-depressive behaviors after neuralgia. The aforementioned findings indicate that microglia can affect the functionality and plasticity of hippocampal neurons by controlling the inflammatory response within the hippocampus [58]. Consequently, microglia activation in the hippocampal area is essential for anxiety and sadness resulting from chronic pain.
Interactions occur between microglia and neurons in the hippocampus. In mice exhibiting chronic inflammatory pain and mood disorders, an increase in microglial count in the hippocampus was observed, alongside a reduction in structural complexity and a diminished capacity for metabolite clearance, which correlated with impaired hippocampal neuronal function [59]. M1-type microglia not only induce aberrant neuronal function but also decrease the density of dendritic spines in hippocampal neurons. Liang et al. [60] modeled chronic neuropathic pain in mice by spinal nerve ligation, leading to the activation of microglia in the hippocampus, which enhanced their pruning of neuronal dendritic spines, resulting in a diminution in both the aggregate length of dendritic spines and the number of branches. Bassett et al. [61] showed that the hippocampus microglia in depressed mice were activated and demonstrated, in vitro, and that a substantial quantity of M1 microglia may stimulate hippocampal neuronal activity. M1-type microglia were capable of inducing apoptosis in hippocampal neurons, a behavior also observed in chronic pain conditions. The activation of M1 microglia disrupts neuronal synaptic plasticity by influencing synaptic structure and apoptosis in the hippocampus, alterations that are also critical characteristics of psychiatric illnesses [62]. The aforementioned study illustrates that pain-induced polarization of M1 microglia primarily affects hippocampal neuronal function and synaptic architecture. Increased M1 type resulting from persistent painful stimuli leads to neuronal death and disrupts synaptic plasticity, subsequently eliciting unpleasant feelings.
The synapse is the basic structure through which neurons transmit information.
When external signals are incoming, synaptic connections between neurons are
dynamically altered and persistent stimuli cause changes in synaptic function and
structure, a process known as synaptic plasticity. Synaptic plasticity within the
hippocampus is associated with memory storage and chronic pain, and its
triggering of negative emotions [63]. Synaptic plasticity is categorized into two
main forms, LTP and long-term depression (LTD). There is bidirectional
communication between microglia and synaptic structures, which respond to
synaptic release of neurotransmitters such as adenosine triphosphate (ATP),
glutamate (Glu), GABA [64], and excitatory neurons, through the release of ATP.
The excitatory neurons, by releasing ATP, enhance the immunosurveillance function
of microglia [65]. Microglia also regulate synaptic plasticity and maturation,
and prune synapses by releasing BDNF, inflammatory factors, and immune complement
[66]. BDNF secreted by microglia has been shown to aid learning and memory by
affecting the formation of synaptic structure [67], and its involvement in
regulating synaptic plasticity in the hippocampus may be related to the
BDNF/tyrosine receptor kinase B
(TrkB)/extracellular signal-regulated kinases (ERK) signaling pathway [68].
Physiologically, microglia in the hippocampus release ATP that binds to receptors
on astrocytes, causing excitatory postsynaptic potentials [69]. The inflammatory
factors TNF-
Microglia can influence neurogenesis in the hippocampus via phagocytosis and
cytokine release in neonatal mice [76]. NSCs at different developmental stages
can establish contact with microglia. Microglia eliminate apoptotic ANPs and
neuronal cells through phagocytosis to maintain the homeostasis of neurogenic
nests [77]. Apoptotic neonatal GCs have been shown to be wrapped around the ends
of microglia protrusions to form “ball-and-chain” structures that are
phagocytosed and removed, a process that is accompanied by increased expression
of inflammation-associated markers [77]. Microglia can also influence the
proliferation, differentiation, and survival of newborn neurons by secreting
different factors [76] and Stratoulias et al. [78] showed that microglia
expressing the phagocytosis gene C-type lectin domain family 7 member a
(Clec7a) were only found in neurogenic areas. This suggests that phagocytosis by
microglia is important for the maintenance of hippocampal neurogenesis.
Researchers created arthritic mice using complete Freund’s adjuvant and
discovered that chronic pain resulted in a significant buildup of M1-type
microglia in the hippocampus after 21 days, accompanied by a commensurate
reduction in neurogenesis within the hippocampus [79]. M1 microglia can
simultaneously decrease hippocampus neurogenesis by secreting IL-1
The emergence of mood disorders can be mitigated at the cellular level. These studies have indicated that there is a negative correlation between pain-induced M1 microglial activation in the hippocampus and hippocampal neurogenesis, suggesting that M1 microglia may instigate mood disorders by impairing neurogenesis and neuroplasticity, specifically through the secretion of LTP and BDNF, which facilitates the onset and intensification of various negative emotions.
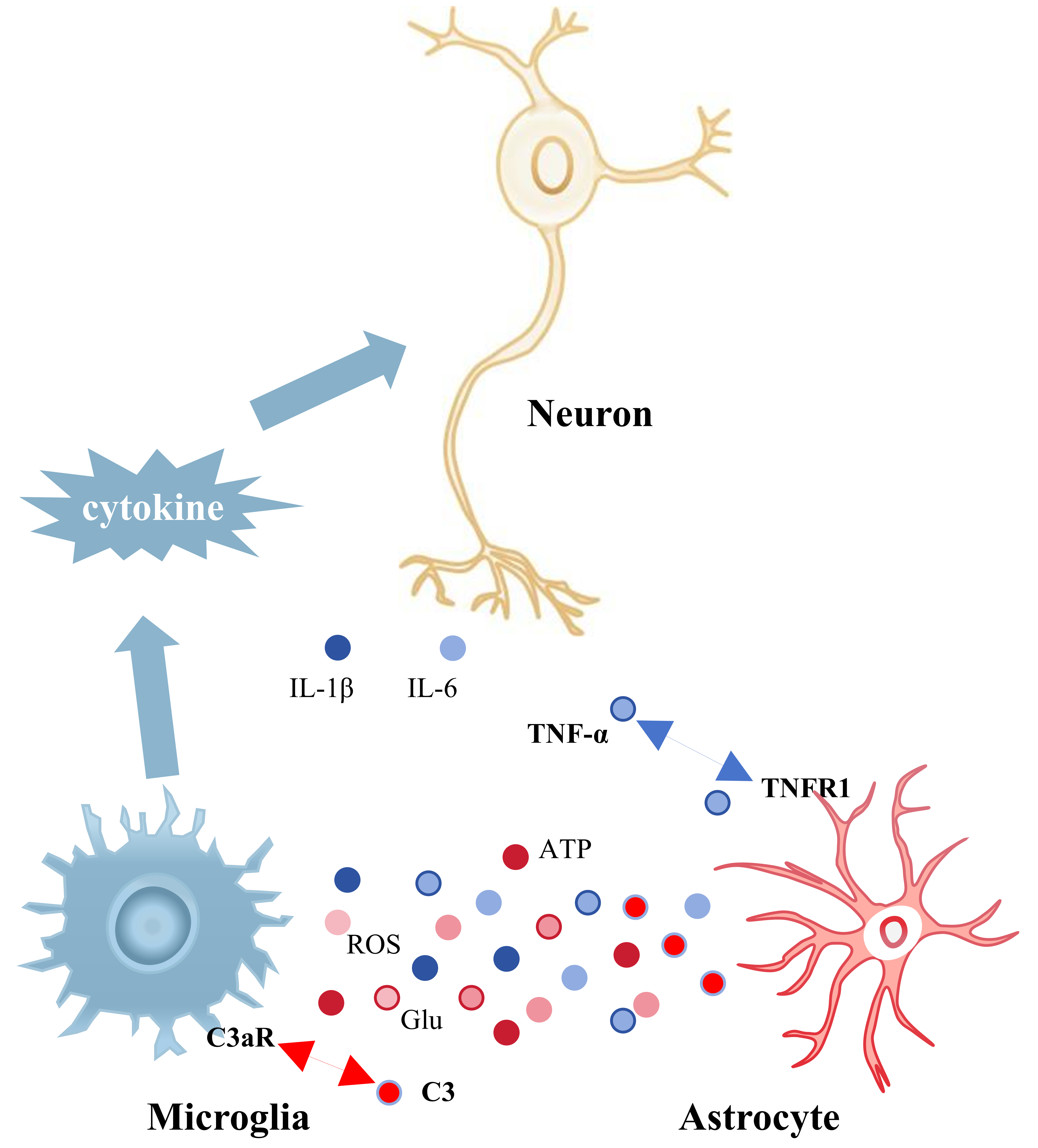
The interaction of microglia and astrocytes in the hippocampus is essential for
the progression of neuroinflammation (Fig. 2). Chronic pain results in heightened
activation of hippocampal microglia and astrocytes, which interact to sustain
central inflammation and facilitate unpleasant emotions [58]. Wu et al.
[84] found that rats experiencing visceral discomfort, associated with the
development of negative mood, had a notable rise in phenotypic markers of both
hippocampal astrocytes and microglia. Astrocytes release the complement molecule
C3, whereas microglial membranes are equipped with the receptor C3aR for the
active fragment C3a. Consequently, the interaction between astrocytes and
microglia affects microglial polarization and the emission of inflammatory
cytokines via the C3/C3a-C3aR signaling pathway. In chronically stressed mice,
the amplification of the C3/C3a-C3aR signaling pathway resulted in the
polarization of microglia to the M1 phenotype and the secretion of the
pro-inflammatory cytokines TNF-
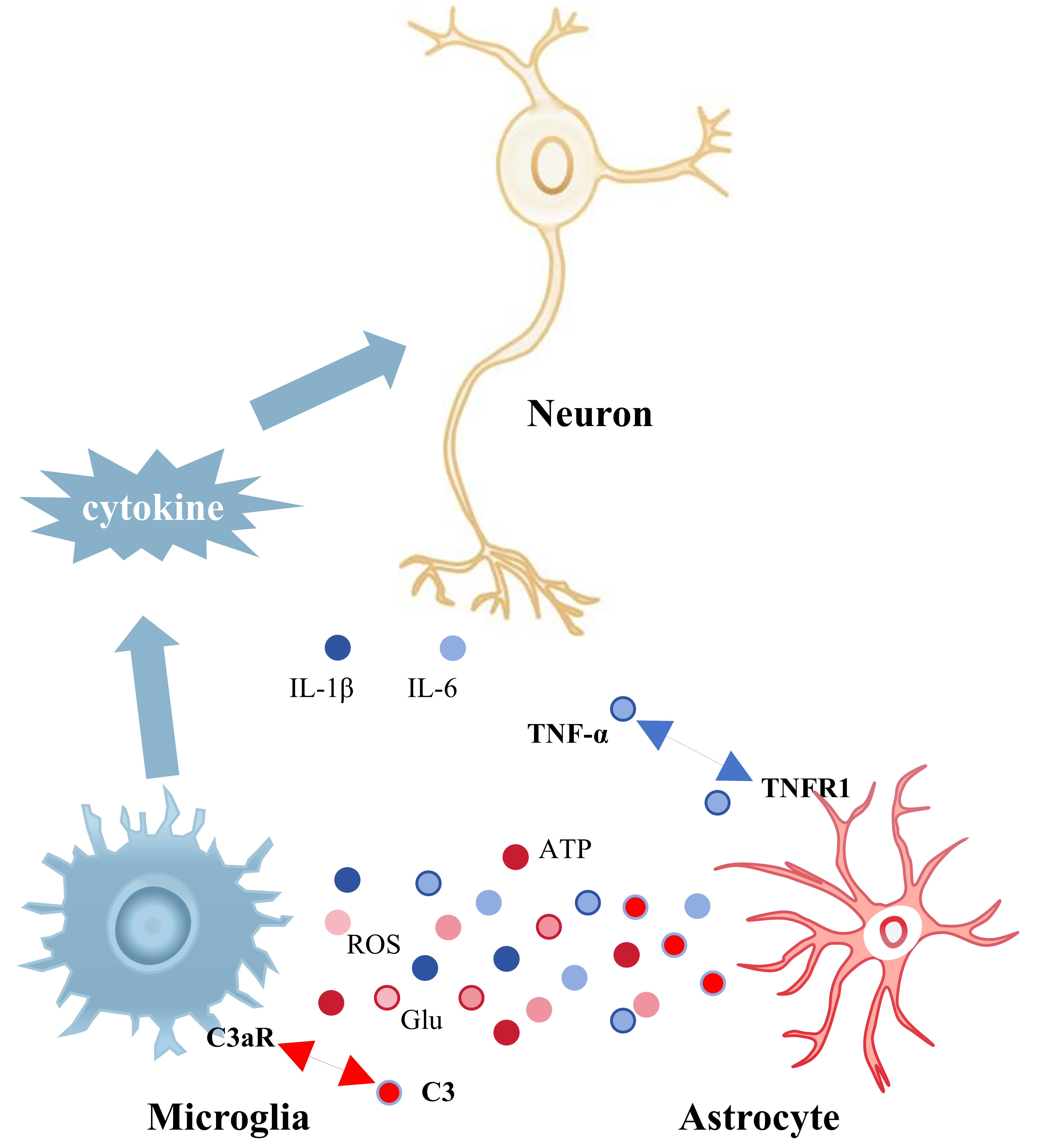 Fig. 2.
Fig. 2.
Interactions between microglia and astrocytes in the hippocampus
and their effects on hippocampal neurons. Information is transmitted between
astrocytes and microglia through the release of cytokines and the
pro-inflammatory factors they produce can damage hippocampal neurons. Blue
globules represent inflammatory mediators released by microglia (IL-1
In summary, chronic pain-induced interactions between microglia and astrocytes not only provoke an inflammatory response but also result in damage to the blood-brain barrier. Microglia and astrocytes in the hippocampus were demonstrated to interactively participate in the adverse emotional response elicited by persistent pain.
Microglia significantly contribute to the formation of painful feelings and are
capable of producing proinflammatory factors and chemokines, which are generated
by pathways such as the nuclear transcription factor-
Purinergic receptors on microglia, including P2X4R, P2X7R, and purinergic P2Y G protein-coupled 12 receptor (P2Y12R), participate in the exchange of intracellular and extracellular sodium and calcium ions. In circumstances such as cellular injury or apoptosis, purinergic receptors stimulate microglia through the binding of the extracellular signaling molecule ATP, which facilitates the phosphorylation of kinases within the intracellular MAPK signaling pathway, as well as nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) and STAT3, thereby affecting the expression of inflammation-related genes and cytokines, resulting in neuroinflammation and subsequent organismal nociceptive sensitization. The activation of the P2X4R receptor can stimulate microglia to release BDNF, augment excitatory postsynaptic currents and glutamate release, influence neuronal depolarization and synaptic plasticity, and mediate mechanical hypersensitivity. The activation of P2X7R is linked to lysosomal activity and the autophagy process, playing a role in inflammatory responses and nociception by influencing the clearing of cellular waste by microglia [87] (Table 1, Ref. [32, 57, 88, 89]). Most P2X7 receptors in the hippocampus are localized in microglia, where they are essential for microglial activation. Overexpression of P2X7 receptors enhances the permeability of microglial membranes, leading to microglial hyperplasia, the secretion of inflammatory factors, and subsequent neuronal injury. Suppression of hippocampal P2X7R expression attenuates microglial activation and proinflammatory factor release generated by neuropathic pain, while also modifying hippocampal plasticity anomalies and the emergence of pain-related emotions. The phagocytosis of microglia is positively linked with hippocampus neurogenesis. Experimental findings [90] indicated that microglial phagocytosis is impaired in animals lacking purinergic receptor P2Y12 and tumor-associated macrophage tyrosine kinase receptor, resulting in diminished hippocampal neurogenesis. Chronic pain affects microglia and activates astrocytes; persistent stimulation of microglia leads to their over-activation and the output of substantial pro-inflammatory cytokines. This process further induces the release of ATP from astrocytes, which is then converted to adenosine by CD39, activating A2AR and eliciting anxiety-like behavior [32].
| Signaling molecule (ATP receptor) | Animal species | Animal model | Behavioral tests | Brain regions | Empirical conclusion | Ref. |
| P2X7R | C57BL/6 mouse | Establishment of neuropathic pain (NP) model in chronic constriction injury (CCI) | Von Frey test, open field test (OFT), tail suspension test (TST), forced swimming test (FST) | Medial prefrontal cortex, amygdala, and hippocampus | P2X7R expression significantly increased in the amygdala and hippocampus, chronic pain, depression | [88] |
| P2X7R | Sprague Dawley (SD) rats | The NP model of type 2 diabetes was established by administering a diet rich in sugars and fats, coupled with the injection of streptozotocin | Mechanical withdrawal threshold (MWT), FST, thermal withdrawal latency (TWL), sucrose preference test (SPT), OFT | Hippocampus | P2X7R expression was greatly amplified, hyperalgesia, allodynia, depression | [89] |
| P2X7R | Adult male and female Wistar rats and male C57BL/6 mice | Unilateral constriction of the infraorbital nerve (CION) was performed to establish trigeminal neuralgia in rat and mouse models | Von Frey test, assessment of cold allodynia, OFT, elevated plus maze (EPM), FST | Hippocampal CA1 | P2X7R expression increased, impairment of LTP, anxiodepressive-like behaviors | [57] |
| A2AR | Female and male C57BL/6 mouse | The resembling trigeminal neuralgia mouse model was constructed by chronic CION via an intraoral approach | Von Frey test, EPM, OFT | Ventral hippocampus (vCA1) | A2AR activation, chronic pain, anxiety | [32] |
P2X7R, purinergic P2X ligand-gated ion channel 7 receptor; CA1, cornu ammonis 1; A2AR, adenosine A2a receptor.
Toll-like receptors, particularly TLR4, are highly expressed as
pattern-recognition receptors in microglia in the hippocampus, where they
participate in pain signaling and are intricately associated with the emergence
of pain-related emotions (Table 2, Ref. [91, 92, 93]). The pro-inflammatory effects
of M1-type microglia initiate with the activation of TLR4. The activation of TLR4
stimulates a cascade of downstream signaling, including the activation of myeloid
differentiation factor 88, which then activates two principal pathways associated
with inflammation: nuclear NF-
| Signaling molecule (toll-like receptor) | Animal species | Animal model | Behavioral tests | Brain regions | Empirical conclusion | Ref. |
| TLR4 | Specific pathogen free grade male BALB/c mouse | Establishment of NP model by unilateral sciatic nerve cuffing | MWT, TWL, SPT, FST, TST | Hippocampus | Activation of TLR4/NF- |
[91] |
| TLR4 | Adult female SD rats | Chronic migraine model | Von Frey test, OFT, y-maze test, light/dark test (DLB) | Cortex and hippocampus | TLR4/NF- |
[92] |
| TLR2 | Female BALB/c mice inoculated with breast cancer cells | Cancer-induced pain model | OFT, the semi-automatic version tested by von Frey test | Hippocampus | TLR2 expression increased, chronic pain, depression | [93] |
NF-
The chemokine C-X3-C motif chemokine ligand 1 (CX3CL1), and its receptor CX3CR1, have dual neuroprotective and neurotoxic effects on the central nervous system. CX3CR1 serves as the principal chemokine receptor on microglia, whereas CX3CL1 is mostly found in hippocampal neurons. Their interaction modulates the migration and activation of microglia by stimulating the phosphorylation of p38 MAPK, hence disrupting microglial migration and activation [97], neuroinflammation, and affective pain. Inhibiting CX3CR1 expression mitigates these effects, whereas elevating CX3CL1 expression stimulates the release of pro-inflammatory molecules from microglia, modifies synaptic transmission in neurons, boosts LTP, and reduces the progression of pathological pain. In peripheral nerve damage, the chemokine C-C motif chemokine ligand 2 (CCL2) binds to CCR2 on microglia, subsequently activating microglia and facilitating neuropathic pain (Table 3, Ref. [98, 99, 100, 101]). It has been shown [2, 102] that increased expression of the chemokine monocyte chemoattractant protein-1 (MCP-1, also known as CCL2) has been found in the hippocampus of rodents in the chronic constriction injury (CCI) model and that this change is accompanied by alterations in emotional behaviors, such as pleasure or drive to explore. Microglia regulate astrocytes through the secretion of cytokines and chemokines, including CCL2 and C-X-C Motif Chemokine Ligand 12 (CXCL1), resulting in their hyperreactivity, which then induces injury or apoptosis of neurons and oligodendrocytes.
| Signaling molecule (chemokine receptor) | Animal species | Animal model | Behavioral tests | Brain regions | Empirical conclusion | Ref. |
| CCR2 | ICR male mice | Establishment of NP model by spinal nerve ligation | Von Frey test, Hargreaves test, FST, SPT | Nucleus accumbens shell | Activation of CCL2/CCR2 signaling pathway, chronic pain, depression | [98] |
| CCR2 | Green fluorescent protein transgenic mice prepared by bone-marrow transplantation | Partial ligation of sciatic nerve to establish NP model | EPM, von Frey test | Amygdala | MCP-1 expression increases, CCR2 activation in bone marrow-derived microglia, chronic pain, anxiety | [99] |
| CXCR4 | C57BL/6 mice | Partial ligation of sciatic nerve to establish NP model | Novel object recognition test | Brain, particularly in the hippocampus | CXCL12-CXCR4 pathway activation, chronic pain, cognitive impairment | [100] |
| Prokineticin receptors (PK-R) | Male C57BL/6J mice | Peripheral neuropathy induced by vincristine | Von Frey test, acetone drop test, plantar test, OFT, marble burying test, novelty suppressed feeding, DLB, FST, SPT | Dorsal root ganglia, spinal cord, prefrontal cortex, and hippocampus | PK-R, TLR4, Cluster of Differentiation 68 (CD68), etc., expression was enhanced in spinal cord, abnormal pain, and nociceptive sensitization | [101] |
ICR, institute of cancer research; MCP-1, monocyte chemoattractant protein-1; CXCR4, C-X-C motif chemokine receptor 4; CXCL12, C-X-C Motif Chemokine Ligand 12.
In summary, microglia and their receptors in the hippocampus are activated in
different types of pain, which seems to be a common pathway for pain-triggered
emotional disorders. However, different receptors are influenced by the unique
pathomechanisms of different pains, and their specific effects will again vary.
ATP receptor activation on hippocampal microglia is involved in negative emotions
triggered by various types of chronic neuropathic pain, such as in diabetes
mellitus or sciatic nerve injuries, in which P2X7 receptor expression was found
to be significantly elevated and accompanied by the emergence of depressive
moods, and also in which the A2AR receptor is associated with the anxiety-like
moods induced by trigeminal-nerve pain. In addition, the TLR4-mediated
NF-
Glucose metabolism is the main source of energy production in brain cells.
Microglia metabolize glucose mainly through oxidative phosphorylation (OXPHOS)
and glycolysis pathways [106]. However, with the development of cellular
transcriptomics, microglia have been found to utilize amino acids and fatty acids
in addition to glucose [107], and thus amino acids and fatty acids have become
alternate energy sources for microglia. Metabolic reprogramming is considered to
be a feature of activated microglia [107], and the pro-inflammatory properties of
microglia are accompanied by a metabolic switch from OXPHOS to glycolysis [108].
Stimulation of BV-2 microglia with LPS results in an increase in lactate content
and glycolysis, while inhibiting ATP production by mitochondria [109]. Further
treatment with LPS+IFN-
In animal models of pain that is associated with mood disorders, microglial activation in the hippocampus results in an imbalance in the aberrant M1/M2 ratio and the levels of pro-inflammatory and anti-inflammatory factors. The elevated expression of inflammatory mediators promotes astrocyte activation, leading to hippocampal neuronal apoptosis, as well as disruptions in neurogenesis and synaptic plasticity.
Toll receptors play a significant role in pain-emotion research; they are
engaged in nociceptive signaling and are linked to the development of anxiety
symptoms related to chronic pain. Inhibiting toll-like receptor signaling can
diminish microglia-induced neuroinflammation, hence decreasing anxiety associated
with chronic pain. He et al. [114] administered minocycline to the
hippocampus of rats with sciatic-nerve-constriction injury and observed that it
inhibited toll receptors, decreased the concentrations of pro-inflammatory
factors IL-1
Selective targeting of microglia M1 polarization appears to be a viable option,
as in the case of minocycline administration, a commonly used inhibitor of
microglia M1-type polarization [116, 117], which exerts a palliative effect on
heat-sensitive symptoms [114] and anxiety behaviors [115] in rats with
neuropathic pain. In addition, minocycline, when used on young rats in a model of
neuropathic pain [118], does not alleviate pain but improves anxiety and
depression-like behaviors and imparts anti-inflammatory properties to the
hippocampus. The authors of that article suggested that the reason minocycline
did not work on pain is because the dose used was too low. These studies suggest
that controlling the inflammatory response has a role to play in pain and its
accompanying emotions, but that a balance between anti-inflammatory and
pro-inflammatory effects in the brain, and a reversal of central inflammation,
are key. Selectively targeting microglia without compromising their essential
functions is a key challenge. If appropriate drugs are to be used to inhibit
microglia M1 polarization for the treatment of clinical pain and associated mood
disorders, the dose, toxicity, and mechanism of action of the drugs used to
produce the effect need to be considered. The equilibrium between the M1-type and
M2-type polarization of microglia is essential for hippocampal neuronal
homeostasis. Han et al. [119] used a liver X receptor (LXR) agonist on
pain-induced mice to mobilize the Phosphatidylinositol 3-Kinase (PI3K)/Protein
Kinase B (AKT) signaling pathway in the hippocampus, resulting in microglia
phenotype transition from M1 to M2. This intervention elevated the hippocampal
expression of M2 phenotypic markers, including macrophage mannose receptor
(CD206), transforming growth factor-
The polarization of M1-type microglia resulting from pain also leads to the
polarization of astrocytes, resulting in an inflammatory cascade in the
hippocampus; the suppression of polarization appears to alleviate both pain and
mood disturbances. In one study [121] the activation of microglia and astrocytes
was blocked by intranasal administration of oxytocin in mice experiencing
neuropathic pain and depression, resulting in increased BDNF levels in the
hippocampus and preservation of synaptic plasticity, thus ameliorating depressive
mood. Liang et al. [122] found that betaine administration in mice
exhibiting chronic inflammatory pain suppressed the activation of microglia and
astrocytes, and produced a marked shift of both cell types from pro-inflammatory
to anti-inflammatory profiles. The transition from a pro-inflammatory phenotype
to an anti-inflammatory phenotype occurred in both cases, accompanied by a
reduction in IL-1
Chronic pain inflicts not just physical suffering on patients but is frequently accompanied by distressing feelings such as worry and depression, significantly affecting their psychological and physical well-being and quality of life. The hippocampus plays a crucial role in this process, being engaged in both emotion and pain processing. Chronic pain stimulation induces functional and structural alterations in the hippocampus, particularly within the CA1 area, which is crucial to pain-emotion studies. Moreover, study have investigated the correlation between the hippocampal dentate gyrus [123] and pain-related emotions, which may serve as a pertinent avenue for future research. Microglia modulate neuronal and synaptic growth in the hippocampus by interacting with neurons or releasing cytokines under normal settings, hence affecting synapse formation and death. Consequently, investigating the mechanisms of neuroinflammation induced by microglia is essential for the examination of emotional impairments related to pain. The activation of microglia results in bidirectional effects on the functional and structural alterations of hippocampal neurons. Receptors like P2X7R, TLR4, and CCR2 are pivotal in microglial activation and the elicitation of pain responses. Recently, the LXR has garnered interest from researchers [124] and a comprehensive examination of LXR function in pain-related emotions may facilitate the innovation of novel therapeutic options for the more effective treatment of pain and associated mood disorders. Recent research has indicated that microglia are an especially significant target for the examination of pain-emotion models. Therapeutic agents like minocycline or other interventions can reestablish homeostasis within the hippocampal neuronal environment by rectifying the abnormal activation of microglia and equilibrating the M1 and M2 subtype ratio, hence mitigating pain and emotional distress. However, microglia are sexually differentiated in pain and negative emotions, and this difference can also have an impact on the effects of drugs, such as the use of microglia inhibitors, which preferentially work in male mice, to block microglial activation to reduce neuropathic pain-induced injurious behaviors [125]. In addition, the effects of anxiolytic drugs in the treatment of depression and anxiety are more pronounced in males [126]. This indicates that modulating microglial activation to manage neuroinflammation in the hippocampus presents a superior therapeutic target for pain and mood disorders in males. However, further comprehensive research is required on the mechanisms of microglial genesis related to pain emotions, along with the interactions among microglia, astrocytes, and neurons. Personalized treatments tailored to the individual’s needs for the precise treatment of chronic pain and the emotions it produces might be possible.
Microglia, the primary innate immune cells of the central nervous system, are the initial responders to nociceptive stimuli. Chronic pain influences all facets of pain-related emotional development by triggering cytokine release from microglia, which subsequently promotes abnormal activation of astrocytes, perpetuating neuroinflammation in the hippocampus. Activated microglia results in modifications to hippocampal synaptic structures, including impaired LTP, neuronal death, and reduced neurogenesis, which are significant in the emergence of painful feelings. The hyperactivation of hippocampal microglia is fundamental to these pathological events, indicating their significant role in the mechanism of persistent pain associated with unpleasant emotions.
A2AR, Adenosine A2A Receptor; ATP, Adenosine Triphosphate; BDNF, Brain-Derived Neurotrophic Factor; DG, Dentate Gyrus; FST, Forced Swimming Test; IL-1
LC conducted the literature search and screening, wrote the main part of the paper, and prepared tables and figures. LZ provided significant input in designing the article’s framework and carefully reviewed the written sections after the initial draft was completed, offering feedback and suggestions for revisions. WL made substantial contributions to the conception and design of the work and played a vital role in drafting and subsequent manuscript revisions, making detailed modifications to the overall structure and content. Additionally, this manuscript was primarily funded by a grant program that he chaired. JSL made substantial contributions to the conception and design of the work and proposed suggestions and modifications to the initial draft framework and key sections, and made the final revisions to the written parts. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This study was supported by the funders of General Program of National Natural Science Foundation of China under Grant No. 82374613.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.