1. Introduction
Neuropathic pain (NP) is a prevalent condition, affecting approximately 10.3%
of individuals in the general population. Its incidence is notably higher in
specific populations such as elderly patients with herpes zoster (HZ), patients
with diabetes, and patients with cancer [1]. Studies indicated that 5–30% of HZ
patients develop postherpetic neuralgia (PHN) [2], with more than 30% of these
patients experiencing persistent neuralgia for more than 1 year. This chronic NP
significantly reduces the quality of life, leading to sleep disorders, anxiety,
depression, decreased physical function, and even debilitation [3]. Furthermore,
it increases the economic burden on patients [4]. Consequently, PHN has become
one of the most common chronic diseases affecting the quality of life and mental
health of the elderly.
Indeed, a widely recognized contributor to NP is nerve injury. Neurons, the
functional units of the nervous system, are capable of sensing stimuli,
transmitting excitation, and activating corresponding brain regions to produce
sensations. PHN, a typical form of NP associated with nerve injury, is also
related to aging, immune deficiency, and genetic factors [5, 6, 7]. Nerve injury
promotes the upregulation of inflammatory cytokines [8], activation of glial
cells [9], and reduction of peripheral sensory nerve fibers [10, 11].
Subsequently, the abnormal expression of various ion channels related to pain
signaling pathways in neurons leads to central sensitization, which is the
primary cause of increased neuronal excitability and NP [12, 13, 14, 15]. However,
although most adults infected with the varicella-zoster virus (VZV) experience
some degree of nerve injury, not all HZ is accompanied by PHN, which might be a
hint of the decline in immunity [16]. Therefore, events secondary to nerve
injury, such as the release of chemokines by peripheral neuron cell bodies and
Schwann cells, are likely to induce immune cell infiltration, further
contributing to the development of NP [17, 18, 19]. From this perspective, nerve
injury serves as the backdrop for the occurrence of PHN.
Although rodent models for exploring the pathogenesis of PHN, such as rat models
induced by herpes simplex virus type 1 (HSV-1) and VZV, have been successfully
constructed to the current time, there are differences in the pathways of PHN
progression from viral latency to activated infection between these models and
humans. Notably, so far, there have been no reports of constructed animal models
that mimic the VZV reactivation process. Therefore, the development of a more
reliable viral reactivation animal model that exhibits PHN-like herpetic pain is
undoubtedly a major challenge we currently face. The pathogenesis of PHN is
associated with nerve injury, and the generally accepted academic mechanism for
the pathogenesis of PHN is central and peripheral sensitization, including
abnormal activation of glial cells and dysfunction of ion channels. Meanwhile,
although the pathogenesis of other NP may vary depending on their etiology,
pathological processes such as nerve injury and abnormal ion channel function are
always present. Given this, the study of other NP can undoubtedly provide a
valuable reference for PHN. PHN, as a chronic neuralgia triggered by VZV invasion
of nerves, is essentially, like all neuralgia, closely related to nerve injury,
whether this injury is caused by viral infection, immune response, or
mechanochemical factors. However, our study of the role of cytokines in PHN
remains limited and clinical evidence seems insufficient due to the many
challenges still facing the construction of current animal models of PHN. To fill
this gap, we drew on other NP research findings to provide further insights into
the pathogenesis of PHN when exploring the role of cytokines in PHN.
The pathogenesis of PHN remains under investigation. A growing body of evidence
has enhanced our understanding of the role of inflammatory mediators, such as
interleukins (ILs), tumor necrosis factor alpha (TNF-), ATP, and
chemokines, in the mechanisms of NP [17, 20, 21, 22]. However, systematic and
comprehensive studies on the changes and mechanisms of cytokines in the
pathogenesis of PHN are still lacking. This paper reviews the peripheral and
central mechanisms of PHN by which cytokines mediate pain, and also discusses the
signaling pathways involved in its pathogenesis.In this paper, the identification
of cytokines and neuroglial cells as potential therapeutic targets for PHN is
emphasized, along with suggestions for future research avenues focusing on PHN
treatment.
2. Nerve Injury
PHN is a prevalent type of NP in clinical practice and remains one of the most
challenging chronic pain to manage. It is triggered by infection with VZV, a
neurotropic herpes virus that lies dormant in human peripheral sensory ganglia.
Reactivation of VZV is related to cellular immunity, which is typically effective
in preventing such reactivation. However, compromised immune responses,
especially in the elderly, facilitates VZV reactivation from dorsal root ganglia
(DRG) latency, spreading along peripheral nerves to the skin [23, 24]. During
viral replication, the immune system releases inflammatory mediators, including
cytokines and chemokines [25]. This leads to inflammation, hemorrhagic necrosis,
and neuronal loss in the affected DRG [26, 27], resulting in nerve damage. The
inflammatory changes following nerve damage lead to the infiltration of immune
cells, which are consistent with observations in other animal models of
peripheral nerve injury [28, 29, 30, 31]. This causes spontaneous firing of peripheral
neurons, lowered activation thresholds, and amplified incoming neural signals,
manifesting as neuronal dysfunction and ectopic discharges [12, 13, 14, 25].
Interestingly, in the chronic constriction injury (CCI) model [32], thermal
hyperalgesia can still occur when the ligature is loosely applied without causing
actual mechanical damage. This suggests that the inflammatory response and the
release of inflammatory mediators, rather than nerve injury itself, are key to
maintaining NP. Inhibiting the inflammatory response can reduce hyperalgesia
[33], where the injection of exogenous inflammatory mediators can induce pain
[34], supporting the idea that inflammatory mediators play a role in mediating
NP. These mediators encompass cytokines, interferons, tumor necrosis factors,
chemokines, and colony-stimulating factors. Their production is from immune cells
[35] and from glial cells [18, 36, 37, 38], providing a structural basis for the
mechanism by which inflammatory mediators mediate NP. Thus, VZV reactivation in
PHN serves as the etiological factor for nerve damage, with cytokines playing a
crucial role in mediating NP.
3. Cytokines
3.1 Overview of Cytokines
Associated with PHN
Cytokines are proteins with low molecular weight that possess diverse biological
functions. These proteins are predominantly secreted by immune cells.
Additionally, other cell types, including keratinocytes, dendritic cells in the
skin, and neuroglia within the central nervous system (CNS), also contribute to
cytokine secretion [39, 40, 41]. The release of these cytokines is typically
triggered by injury and inflammation [42]. Cytokines exert their biological
functions by binding to specific receptors, and regulating cell growth,
differentiation, and tissue repair. They are particularly important in stress
reactions such as injury, pain, and infection. The properties of cytokines are
related to the microenvironment and most have dual effects in different
situations [43]. IL-1 promotes neuronal sensitization [44, 45].
Pro-inflammatory cytokines include IL-1, IL-6, IL-18 and TNF-. ILs
mainly participate in the proliferation, differentiation, and activation of
immune cells, whereas TNF- can activate cytotoxic T cells and promote
the production of other cytokines, collectively enhancing the inflammatory
response. Anti-inflammatory cytokines, such as IL-4, IL-10, soluble IL-2 receptor
(sIL-2R) antagonists, and TNF-binding proteins, primarily inhibit inflammation to
prevent excessive inflammatory responses that could damage the body. However,
IL-10 additionally facilitates the activation and expansion of B cells [46],
thereby sustaining autoimmune responses (Fig. 1 shows the inflammatory response
of immune cells). Under pathological conditions, an imbalance in cytokine levels
can contribute to disease progression. For instance, following nerve injury,
cytokines secreted by immune cells exert direct effects on neural signaling by
binding to homologous receptors on neurons, microglia, and astrocytes within the
spinal cord, DRG, and brain. Neuronal activation is not solely dependent on
receptor-mediated interactions and cellular contacts but is also regulated
through a broader network influenced by cytokine activity [47]. Consequently,
these cytokines establish a communication network between immune cells and
neurons, involved in the modulation of neural responses [48].
 Fig. 1.
Fig. 1.
The roles of immune cells and glial cells in amplifying the
inflammatory response during nerve injury. Glial cells in the DRG and immune
cells secrete inflammatory mediators. T cells, macrophages, and monocytes have
been shown to infiltrate tissues infected by viruses. These cytokines promote the
activation of glial cells and increase neuronal excitability. The interactions of
neuroimmune and glial cells accelerate nerve injury, and promote pain generation. The figure was drawn using WPS office (12.1.0.20305, Kingsoft Office Software Co., LTD., Kowloon, Hong Kong, China). CXCL, C-X-C motifchemokine lingand; DC, dendritic cell; DRG, dorsal root ganglia;
IL, Interleukin; TNF-, tumor necrosis factor alpha.
Recent research indicates that PHN is often accompanied by an inflammatory
response [10], evidenced by the presence of various cytokines at the injury site,
adjacent areas, and even in the plasma [49, 50]. These include TNF-,
ILs, interferon gamma (IFN-), various chemokines, and oxidative
stress-related factors. Collectively, these factors are pivotal in the onset and
advancement of NP. Among them, TNF-, IL-1, IL-6, IL-18, and
IL-10 have been more extensively studied in relation to PHN. Other cytokines,
such as IFN-, IL-2, IL-17 and IL-23, have been less well studied in
relation to PHN. In untreated animal models [34, 51, 52, 53, 54], intrathecal injections
of cytokines such as IL-1, IL-18, TNF, and IL-6 have been observed to
directly induce pro-nociceptive effects, leading to hyperalgesia. Cytokines are
both important members of the immune system and mediators of pain-related
signals. Specifically, TNF-, IL-1, and IL-6 have been
implicated in the development of NP across animal models, including PHN. These
cytokines, produced by both neuroglia and immune cells, share common functions
such as mediating pain-related cation channel expression, amplifying inflammatory
responses, and activating glial cells. In the context of nerve injury,
inflammatory cytokines act on nociceptor terminals to initiate pain pathways
[55]. Additionally, neuronal activation triggers reciprocal cytokine release,
further promoting inflammation. Prolonged inflammation leads to altered sensory
nerve fiber signaling, which persists even after inflammation and injury have
healed. In summary, it can be hypothesized that cytokines are closely related to
the development PHN. This paper explores the signaling pathways of
TNF-, IL-1, IL-6, IL-18, and IL-10, elucidating their roles in the
pathogenesis of PHN.
3.2 Signaling of Cytokines
3.2.1 Overview of TNF- Signaling
TNF- is intricately involved in the pathogenesis of NP. This cytokine
is primarily synthesized by macrophages and acts as a versatile systemic
inflammatory mediator. Structurally, TNF- comprises a signal peptide
and two domains, existing in monomeric, dimeric, or trimeric forms, with dimers
exhibiting the highest biological activity. Two different receptors, TNF receptor
1 (TNFR1) and TNFR2, are responsible for mediating TNF- signaling [56].
TNFR1 is broadly expressed and can induce apoptosis, while TNFR2 is predominantly
found on specific cell types like immune and endothelial cells, involved mainly
in anti-apoptotic signaling. TNF- influences a variety of biological
processes through its interaction with receptors TNFR1 and TNFR2. Through its
interaction with TNFR1, TNF- triggers the activation of the
mitogen-activated protein kinase (MAPK) and nuclear factor kappa B
(NF-B) pathways, initiating downstream signaling processes [57, 58, 59]. On
the other hand, when TNF- binds to TNFR2, it recruits TNF
receptor-associated factor 2 (TRAF2) along with cellular inhibitors of apoptosis
proteins 1 and 2 (cIAP1/2), thus activating the non-canonical NF-B
pathway (Table 1, Ref. [60, 61, 62, 63, 64, 65, 66]).
Table 1.
Cytokines, receptors and signaling pathways.
| Cytokines |
Receptors |
Signaling Pathways |
References |
| TNF- |
TNFR1 |
MAPK |
[65] |
| NF-B |
[66] |
| TNFR2 |
Non-classical NF-B |
[60] |
| IL-1 |
IL-1R |
NF-B, MAPK |
[61] |
| IL-6 |
IL-6R |
JAK-STAT3 |
[62] |
| IL-18 |
IL-18R |
NF-B |
[63] |
| IL-10 |
IL-10R |
JAK-STAT |
[64] |
IL-1R, IL-1 receptor; JAK, Janus kinase; MAPK, mitogen-activated protein kinase; NF-B, nuclear factor-kappa B; STAT, signal transducers and activators of transcription.
3.2.2 Overview of IL-1 Signaling
IL-1, another cytokine, contributes to the initiation and advancement
of NP. IL-1 is an inactive precursor peptide that undergoes cleavage and
activation by IL-1-converting enzyme caspase-1 during inflammation,
leading to its extracellular secretion [67]. IL-1 initiates signal
transduction primarily through the IL-1 R-associated kinase
(IRAK) pathway. Upon binding, IL-1 forms a heterotrimeric complex with
IL-1R type I (IL-1R1) and IL-1R accessory protein (IL-1RAcP). This interaction
triggers the activation of IRAK4, resulting in IRAK4 autophosphorylation and
phosphorylation of IRAK1 and IRAK2. Subsequently, TRAF6 is recruited and
activated, which then activates members of the MAPK kinase kinase family. This
cascade leads to NF-B-inducing kinase phosphorylation, facilitating
nuclear translocation of NF-B and subsequent regulation of gene
expression [68]. IL-1 signaling, similar to TNF-, activates
both the NF-B and p38-MAPK pathways, promoting the increased expression
of genes such as IL-6, monocyte chemotactic protein-1, cyclooxygenase-2,
IL-1, and IL-1 [61].
3.2.3 Overview of IL-6 Signaling
IL-6 is a small molecular weight protein secreted by various immune cells. It
transmits signals via the ligand-binding IL-6R alpha (IL-6R) and the
signaling component gp130 (CD130) [69]. IL-6 signaling is primarily mediated
through three main pathways: first, IL-6 binds to its receptor (IL-6R) to form a
complex. This complex subsequently interacts with gp130, which activates
intracellular signal transduction, ultimately leading to downstream signaling
cascades and gene expression. sIL-6R binds to IL-6 and forms a complex with
gp130, initiating signal transduction. Additionally, the interaction between IL-6
and the IL-6R-gp130 complex initiates signal transduction via Janus
kinases (JAKs) and signal transducers and activators of transcription (STATs),
which results in STAT3 phosphorylation, subsequent transcriptional activation in
T cells, and the triggering of diverse biological effects [62].
3.2.4 Overview of IL-18 Signaling
IL-18 is a protein that mediates its action through a receptor belonging to the
IL-1R family. The IL-18R on neurons and glial cells activates important signaling
pathways. IL-18 binds to IL-18R, which then binds to IL-18R to
form a trimer [70, 71, 72, 73, 74]. The intracellular domain of IL-18R includes a Toll/IL-1R
homology (TIR) domain, which is the same as that in Toll-like receptors. This TIR
domain enables MyD88 to attach and relay signals into the cell. IL-18 activates
the transcription factor NF-B [63] and activator protein-1 (AP-1)
through signaling molecules such as MyD88, IRAK, and TRAF6 (Table 1).
3.2.5 Overview of IL-10 Signaling
IL-10 exhibits analgesic effects and is secreted and recognized by a range of
immune cells. Its main function in vivo is to suppress inflammation by
downregulating the production of various pro-inflammatory factors. The functional
IL-10R complex comprises a tetramer with two ligand-binding subunits and two
auxiliary signaling subunits [75]. The canonical IL-10 signaling pathway involves
the JAK/STAT pathway (Table 1) [64]. IL-10 binding to the extracellular domain of
IL-10R1 triggers the phosphorylation of JAK1 and tyrosine kinase 2 (TYK2), which in turn activates
the transcription factor STAT3. Furthermore, IL-10 reduces NF-B
activation by diminishing its DNA binding ability and inhibiting the activity of
IB kinase. Simultaneously, IL-10 activates AP-1 and NF-B,
promoting the differentiation of CD8+ T cells. In monocytes, IL-10 activates p85
triiodophosphate and p70 S6-kinases. However, blocking these pathways affects the
proliferation-regulating activity of IL-10 but not its anti-inflammatory effects.
While these cytokines exhibit specificity in their biological functions
depending on the microenvironment, they share common downstream signaling
pathways. Notably, the MAPK and NF-B signaling pathways play a
significant role in pain sensitization, the expression of cation channels, and
the processes of neuroinflammation. The activation of JAK enhances the
sensitivity of the transient receptor potential vanilloid 1 (TRPV1), leading to
peripheral sensitization. TNF- is critical for Na+ channel expression
via the p38-MAPK pathway, which simultaneously stimulates the production of
TNF- by microglia. IL-1 influences pain transmission by
modulating ion channels, including TRPV1 and the voltage-gated sodium channel
alpha subunit 9 (Nav1.7), through activation of the NF-B and p38-MAPK
systems. IL-6 induces JAK and protein kinase C (PKC) activation, contributing to
chronic pain. IL-18 promotes inflammation and increases pain sensitivity through
NF-B transcription. IL-10 may exert its effects by modulating these
signaling pathways (Fig. 2).
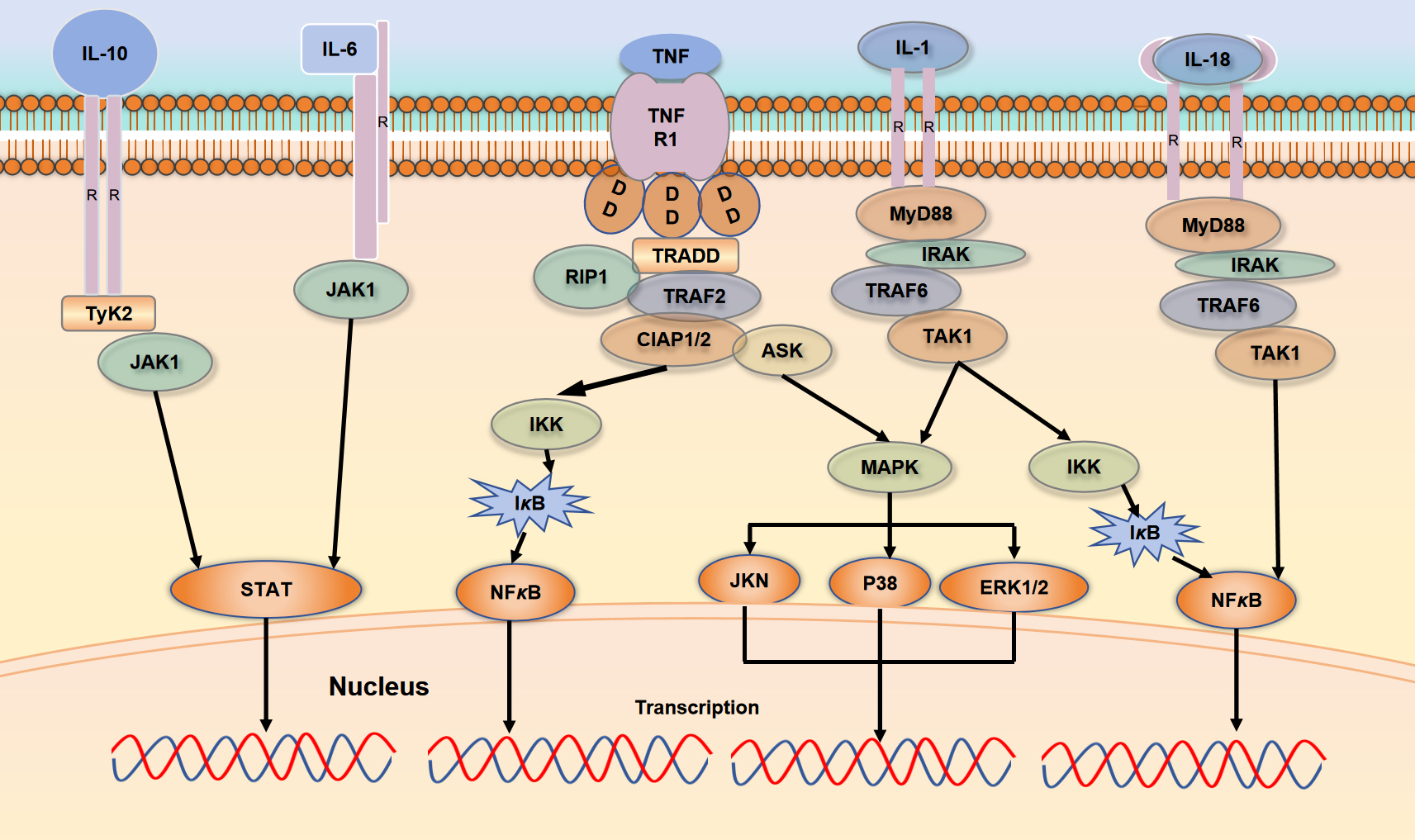 Fig. 2.
Fig. 2.
TNF-, IL-1, IL-6, IL-18, and IL-10
exerts various biological effects through their interactions with specific
receptors. Both TNF- and IL-1 engage with their respective
receptors, activating the NF-B and MAPK signaling pathways; IL-6 and
IL-10 activate the JAK/STAT pathway upon receptor binding, while IL-18 binds to
its receptor to activate the NF-B pathway. The figure was drawn using WPS office (12.1.0.20305, Kingsoft Office Software Co., LTD., Kowloon, Hong Kong, China). TNFR1, tumor necrosis factor
receptor 1; TRADD, TNFR1-associated DD proteins; TRAF2, TNFR associated factor 2;
TyK2, tyrosine kinase 2; IKK, IκB kinase; RIP1, receptor interacting serine/threonine protein kinase 1; CLAP, caspase recruitment domain-containing protein; ASK, apoptosis signal-regulating kinase; MAPK, Mitogen-Activated Protein Kinase; ERK, extracellular signal regulated kinase; TAK1, transforming growth factor β activated kinase 1; IRAK, interleukin receptor associated kinase.
4. The Role of Cytokines in PHN
Immune cells and glial cells produce a range of cytokines that contribute to
both peripheral and central sensitization. Cytokines sustain pain signaling by
influencing injury receptors and/or central spinal cord neurons. Cytokines
upregulate the expression of pain-related cation channels, including Nav1.3,
Nav1.7, Nav1.8, and Ca2+, and to activate neuroglial cells. Increased levels of
TNF- , IL-6, and IL-1, as well as other inflammatory
mediators, have been observed at the site of nerve injury and in adjacent areas.
Low concentrations of cytokines promote neuronal survival and growth and favor
post-injury nerve repair, whereas high concentrations of cytokines induce
neuronal apoptosis. The peripheral ends of injury receptors form tree-like
structures in tissues and organs [76]. These sites are in close proximity to
keratinocytes and immune cells and contribute to immunomodulation of injury
receptor function. In the context of high-frequency afferent input resulting from
tissue damage, a cascade of cytokines targets the ends of sensory nerve fibers
known as injury receptors. This interaction initiates the activation of pain
pathways, leading to neuronal activation and subsequent reciprocal stimulation of
various cytokine-producing cells [55]. Notably, prolonged inflammation alters
injury sensory processing, which persists even after inflammation and wound
healing, creating a “neuropathic” like phenotype. In the CNS, the types of
glial cells include astrocytes, oligodendrocytes, and microglia. Among them,
astrocytes and microglia have been the focus of extensive research in the context
of NP. It has been proposed that the p38-MAPK system can be activated by spinal
astrocytes to promote pain sensitization [77]. Moreover, astrocytes regulate
neuroplasticity [78]. Following inflammation, activated microglia undergo
transformation into macrophage-like cells [79] that release cytokines such as
TNF-, IL-1, IL-6, and IL-18, affecting synaptic signaling and
pain transmission via the p38-MAPK system [80, 81, 82]. Cytokines released from glial
cells at the spinal cord level can induce pain secondary to neuronal
sensitization (Fig. 3).
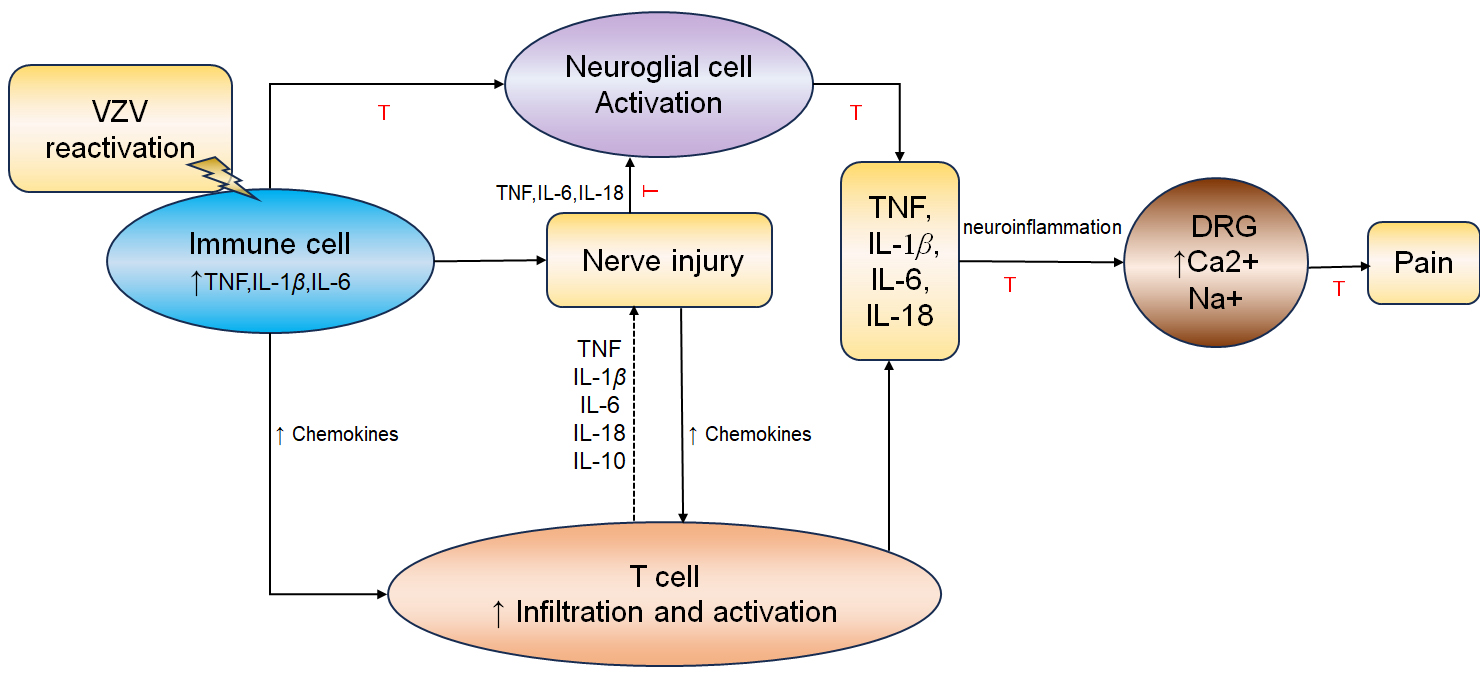 Fig. 3.
Fig. 3.
Schematic representation of major interrelationships leading to
nerve injury and pain. Upon VZV reactivation, immune cells secrete inflammatory
mediators, including TNF, IL-1 and IL6, as well as chemokines. This
inflammatory environment results in activation of glial cells and infiltration of
immune cells, further amplifing the inflammatory response. These inflammatory
cytokines, particularly TNF and IL-1, induce the expression of
pain-related cation channels in the DRG. This may lead to peripheral and central
sensitization, promoting NP. Red inhibitory lines indicate possible options for
targeted drug interventions. The figure was drawn using WPS office (12.1.0.20305, Kingsoft Office Software Co., LTD., Kowloon, Hong Kong, China). NP, neuropathic pain; VZV, varicella-zoster virus.
4.1 TNF-
In a meta-analysis study [83], TNF- levels were evaluated in the body
fluids of 113 participants, revealing higher TNF- levels in patients
with PHN compared to those who did not develop PHN after HZ. In a mouse model of
PHN induced by HSV-1, elevated TNF- levels were linked to PHN [12, 25].
These findings indicate that increased TNF- levels are linked to the
occurrence of PHN. Strangfeld et al. [84] observed an increased risk of
HZ with the use of TNF- inhibitors [85]. Interestingly, among patients
who developed HZ while using TNF- inhibitors, there was a lower
subsequent incidence of PHN, suggesting a potential role for TNF- in
PHN development. An experimental study has demonstrated that intrathecal
injection of exogenous TNF- can promote pain-induced hyperalgesia and
mechanical allodynia [34]. Conversely, TNF- inhibitors administered via
the same route can alleviate chronic pain [86]. This further confirms the crucial
role of TNF- in the pathogenesis of PHN. Consequently, TNF-
inhibitors emerge as promising candidates for the management of chronic pain
associated with PHN.
At low concentrations, TNF- promotes neuronal survival and growth,
whereas at high concentrations, TNF- induces neuronal apoptosis. In the
CCI model of rat sciatic nerve-induced NP, TNF- expression is detected
at the injury site [87]. Similar results have been observed in biopsies of human
NP lesions [88]. Exogenous TNF- injected into the DRG of CCI roots
causes ectopic pain, suggesting that the cause of NP is not the nerve injury
itself, but rather the action of inflammatory mediators following nerve injury
[28, 32]. In NP models following nerve injury, TNF- plays a pivotal role
in activating other cytokines [89]. TNF- binds to its receptors and
promotes the activation of inflammatory cells via NF-B, triggering an
inflammatory cascade [90]. TNF- binds to its receptor and activates the
NF-B signaling pathway to mediate aberrant expression of voltage-gated
Na+ channels (VGSCs) or Na+ currents [91, 92]. A study has shown that exogenous
TNF- administered around nerve leads to persistent mechanical allodynia
[90]. Zang et al. [92] demonstrated that TNF- induces the
upregulation of Nav1.3 expression in the DRG by activating NF-B, and
intrathecal injection of an NF-B inhibitor significantly alleviates
mechanical allodynia induced by the perisciatic injection of recombinant rat
TNF-. This indicates that TNF- mediates the expression of
VGSCs in the DRG via the NF-B system, contributing to the development
of NP [93, 94, 95]. Furthermore, TNF- enhances membrane cation conductance
in a non-voltage-gated manner, resulting in overall neuronal hyperexcitability,
further exacerbates NP [95].
TNF- is involved as a pro-inflammatory cytokine in the interaction
between neuroimmune cells and glial cells in sensory ganglia and is essential for
the promotion of PHN [96, 97]. Following ischemic, inflammatory, or traumatic
nerve tissue damage, TNF- rapidly increases in regions such as the
spinal dorsal horn, locus coeruleus, and hippocampus [98, 99, 100, 101]. TNF-
mediates the central mechanisms of NP through neuroglial cells.
Immunofluorescence staining has demonstrated the presence of TNF- on
the surface of astrocytes. TNF-, through G protein-coupled receptor
C-X-C chemokine receptor type 4, triggers the production of IL-1, IL-6, and ATP.
These substances increase neuronal activity, and contribute to NP [80, 102]. The
TNF/TNFR1 signaling pathway contributes to NP by downregulating inward rectifying
K+ channels (Kir4.1) in astrocytes, disrupting K+ homeostasis. Microglial cells
have been shown to participate in the pathogenesis of NP [103, 104]. Following
nerve injury and inflammation [80, 105, 106, 107, 108], activated microglia secrete
pro-inflammatory cytokines mediated by the p38-MAPK system including
TNF-, IL-1, IL-6, C-C motif chemokine ligand 2, and C-X3-C motif
chemokine ligand 1, affecting synaptic signal transmission and pain transmission.
TNF- promotes Na+ ion influx and lowers excitability thresholds through
activation of the p38-MAPK pathway, thereby contributing to NP [91]. For
instance, spinal nerve injury in rats induces allodynia, accompanied by elevated
levels of TNF- and phosphorylated p38. Inhibitors of TNF- or
p38 can alleviate this allodynia. TNF- binds to its receptor and
activates the NF-B signaling pathway, whereas activation of the
N-methy1-D-aspartate (NMDA) receptor is associated with the expression of
NF-B [109]. NMDA receptors are involved in peripheral, spinal cord, and
cerebral pain pathways by potentiating excitatory postsynaptic currents [110].
Thus, NMDA may be a potential therapeutic target for PHN.
TNF- is an important mediator in the pathogenesis of NP in the CNS and
the peripheral nervous system. It acts in concert with various mediators,
including ILs, nerve growth factors, chemokines, and IFNs, coordinating pain
signal transmission through the NMDA, ATP, and MAPK signaling pathways.
4.2 IL-1
IL-1, belonging to the IL-1 family, is primarily produced by activated
macrophages and exhibits different biological effects. Zhao et al. [111]
reported increased levels of IL-1 in the cerebrospinal fluid of patients with
PHN. In a meta-analysis involving 1373 participants, researchers confirmed that
in patients with HZ, the expression of IL-6 and IL-1 was higher in those
who developed PHN than in those with HZ without PHN [83]. However, Cao et
al. [112] used a protein array to examine 40 common inflammatory factors in the
skin lesion tissues of patients with PHN and found that only IL-1 was
significantly elevated, albeit with low specificity, whereas there was no
difference in expression of the other 39 inflammatory factors, including
IL-1. The contradictory results might stem from differences in detection
sites. The epidermis in skin affected by PHN exhibits increased thickness
compared to normal skin, suggesting possible structural or molecular changes.
Immunohistochemical data have consistently shown reduced nerve ending density in
the epidermis of PHN skin [113, 114, 115, 116, 117]. While various cell types can express IL-1,
barrier cells such as epithelial cells normally express high amounts of IL-1. The
decreased IL-1 expression in PHN may be linked to cellular changes such as the
loss of specific cell types in the affected skin. Moreover, the molecular
pathological alterations observed in the skin affected by PHN could lead to
decreased IL-1 expression. This indicates that the persistent and chronic pain
experienced by PHN patients may not be directly linked to skin inflammation.
Under normal conditions, IL-1 expression is low. After peripheral nerve
injury, IL-1 expression is elevated in glial cells, and certain immune
cells within the spinal cord, leading to diverse effects on the nervous system.
Similar to TNF-, IL-1 signaling participates in pain through
activation of the NF-B and p38-MAPK pathways. In the peripheral DRG,
IL-1 acts on TRPV1
and IL-1R to modulate pain sensitivity [118]. TRPV1, part of the TRP family, is
closely associated with the perception of harmful stimuli and pain generation.
Activation of TRPV1 can regulate Ca2+ influx through voltage-dependent Ca2+
channels (VDCCs) [119]. This process results in the release of neuropeptides and
excitatory amino acids from nerve terminals, which ultimately leads to the
perception of pain in the cortex [120]. JAK and PKC inhibitors alleviate
sensitization of TRPV1. TRP inhibitors help prevent the release of injurious
substances, paving the way for the discovery of novel analgesics. Upon binding to
IL-1Rs, IL-1 also promotes prostaglandin synthesis, indirectly
sensitizing pain receptors to induce pain. Biologically active IL-1 is
formed from the precursor IL-1 by cleavage through certain enzymes, with
matrix metalloproteinases (MMPs) playing a significant role in this process.
Among these, MMPs affect the release of IL-1. Increased activity of MMP9
and MMP2 due to nerve injury promotes cleavage of the IL-1 precursor,
resulting in the formation of biologically active IL-1. Inhibitors of
MMP9 or MMP2 can reduce the biological activity of IL-1, significantly
alleviating NP behaviors in animals. In addition, at central sites, IL-1
induces nociceptive sensitization [109] by promoting the release of substance P
from -amino-3-hydroxy-5-methyl4-isoxazolepropionic acid or NMDA
receptors [121], an ionotropic glutamate receptor mediating excitatory
neurotransmitter transmission that plays a crucial role in the pain pathway.
Activation of the NMDA receptor leads to Ca2+ inward flow, promoting central
sensitization [122]. In conclusion, IL-1 promotes PHN by directly or
indirectly increasing neuronal excitability. Modulation of IL-1, TRPV1
[123], and NMDA receptors may serve as new analgesic options to alleviate NP in
patients with PHN.
Therefore, therapeutic blockade of these proteases holds promise for limiting
the release of biologically active IL-1 and potentially alleviating
neuropathic inflammation to achieve relief from NP.
4.3 IL-6
IL-6 is considered an early marker of injury, playing a regulatory role in both
the immune and nervous systems. It suppresses immune function by inducing
macrophages to release transforming growth factor beta (TGF-),
which modulates the acute phase inflammatory response [69]. Arruda et
al. [124] observed elevated IL-6 mRNA levels in the spinal dorsal and ventral
horns of rats following peripheral nerve injury. Similarly, studies involving
human brain vascular adventitial fibroblasts (HBVAFs) and human peripheral nerve
cells infected with VZV revealed significant increases in IL-6 transcription and
expression levels, which lead to the loss of integrity of vascular and neural
barrier and continued viral replication [125, 126, 127].
Research has shown that patients with HZ who later develop PHN have
significantly higher IL-6 levels than those who do not develop PHN [128]. This
suggests that elevated IL-6 expression is associated with inflammatory responses
leading to nerve damage and promoting the onset and progression of NP.
Downregulation of IL-6 expression can significantly alleviate pain. Saxena
et al. [129] observed that during the treatment of neuralgia in patients
with HZ, IL-6 mRNA expression was significantly decreased following substantial
pain relief. Lin et al. [130] analyzed IL-6 levels in patients with PHN
based on pain severity and found significant differences among different severity
groups, with particularly elevated IL-6 levels in patients with severe pain.
Furthermore, IL-6 levels are correlated with short-term prognosis in patients
with PHN. However, a study by Zak-Prelich et al. [131] reported
conflicting results, possibly due to a smaller sample size and a broader age
range.
IL-6 is associated with peripheral mechanisms of pain onset. Under physiological
conditions, low levels of IL-6 are beneficial for normal development and repair
of the nervous system; however, excessive production of IL-6 can lead to
neurological damage. A study has shown that direct injection of IL-6 into rodent
joints results in sustained sensitization of injury-sensing C fibers [120].
IL-6-mediated inflammatory responses may be linked to pain hypersensitivity.
Following peripheral nerve injury, IL-6 and IL-6R are highly expressed in
neurons. Similar to IL-1 and TNF-, IL-6 enhances Na+ [132] and
Ca2+ [133] currents in peripheral nerve terminals, triggering action potential
firing. This increases membrane excitability and reducs the pain threshold,
leading to peripheral sensitization. IL-6 also induces activation of JAK and PKC,
increasing TRPV1 sensitivity and inducing pain [134, 135].
IL-6 is a neurogenic signaling mediator that transmits injury signals to the CNS
[117]. Microglia activation promotes the PHN. After nerve injury, IL-6 mediates
NP through the JAK2/STAT3 and ciliary neurotrophic factor (CNTF)/STAT3 signaling
pathway [136, 137]. Activation of the JAK2/STAT3 pathway promotes microglia and
astrocyte activation, leading to cytokine release and amplifying
neuroinflammation. The initiation and progression of inflammatory cascades from
the peripheral regions to CNS are mediated by the CNTF-STAT3-IL-6 signaling axis.
Blocking the CNTF-STAT3-IL-6 pathway can alleviate nerve inflammation in the DRG
and spinal cord, thereby reducing pain following injury [137]. Administering
anti-IL-6 antibodies significantly reduces JAK2/STAT3 signaling and pain behavior
in rats. Overall, these studies suggest that targeting IL-6 could be advantageous
in alleviating inflammatory responses and modulating the impact on nociceptors.
4.4 IL-18
IL-18 is also a cytokine involved in the regulation of the immune response [71].
It has been shown to be associated with chronic pain, such as NP, osteoarthritis
pain, and cancer pain [72, 73, 74]. Khazan et al. [138] confirmed that IL-18
levels are linked to the risk of PHN. At the peripheral site, IL-18 ,which
produced by immune cells, stimulates the transcription of Toll-like receptors and
NF-B, promoting the release of inflammatory mediators and mediated pain
signaling [139, 140]. IL-18 modulates pain by influencing ion channels and
receptors mediating pain in the nervous system [72, 141]. Increased IL-18
expression has been observed in various models of NP resulting from nerve injury.
IL-18 plays a pivotal role in NP regulation through injurious sensory
transmission [141, 142, 143, 144]. In addition, IL-18 is crucial in regulating the activity
of glial cells [145, 146, 147]. Blocking the IL-18 signaling pathway inhibits glial
cell hyperactivity and subsequent activation of Ca2+ dependent signaling pathways
[148]. IL-18-mediated interactions between microglia and astrocytes are pivotal
for NP [72]. Studies [53, 144] have shown that microglia contribute to
inflammation and NP by producing IL-18. Additionally, oligodendrocytes have been
implicated in neuroinflammation via IL-18 production [149]. Immune-mediated
inflammation significantly influences NP development, with IL-18 mediating
microglial and astrocytic interactions that release proinflammatory cytokines,
chemokines, and other signaling molecules [72], thereby amplifying pain signals
and recruiting immune cells to injury sites [150]. Moreover, IL-18 triggers
cytokine production through inflammatory vesicle complexes, further enhancing the
immune response [151]. These findings collectively emphasize the critical role of
IL-18 in the pathogenesis of PHN.
4.5 IL-10
Uçeyler et al. [152] showed that IL-10 gene expression increases
immediately after nerve injury, with a secondary peak observed within 45 days.
Khan et al. [153] found that minimal inflammation of peripheral nerves
following injury had no significant effect on IL-10 levels in sciatic nerve of
rats across four types of sciatic nerve injuries. Partial nerve injury decreased
IL-10 levels in the affected nerves, whereas complete nerve transection increased
IL-10 expression. This implies that IL-10’s involvement in NP etiology may vary
depending on the specific nature of the nerve injury. After nerve injury, IL-10
activates microglia in the CNS to mediate NP [154]. IL-10 stimulates G
protein-coupled receptor 40, along with intracellular signaling pathways such as
STAT3, leading to increased -endorphin expression and secretion
[155]. Sharma et al. [156] identified the expression of IL-10 receptors
in cortical neurons, where they mediate neuroprotection through the activation of
the PI3K/AKT and STAT3 pathways. Additionally, Huang et al. [157]
demonstrated that IL-10 attenuates NP by downregulating Nav1.7 in the DRG of
rats. Following sciatic nerve crush injury in mice [158] , IL-10 secretion by
macrophages is increased, which subsequently lowers the levels of chemokines and
cytokines. Additionally, IL-10 deficiency impairs axonal regeneration and hinders
the recovery of nerve functions. Atkins et al. [159] studied the effects
of IL-10 on nerve regeneration in anesthetized C57 Black-6 mice following left
sciatic nerve sectioning and resealing with four extraneous nerve sutures. The
authors injected IL-10 (125 or 500 ng) at the repair site before and after nerve
sectioning. Their findings revealed that a low dose of IL-10 at the sciatic nerve
repair site facilitated better axonal regeneration, whereas a high dose did not.
This suggests that optimal levels of IL-10 promote nerve regeneration, while
prolonged high levels are detrimental to nerve repair. A study has shown that
the induction of VZV-specific T lymphocytes is accompanied by increased IL-10
expression [152].
Jenkins et al. [160] showed that elevated levels of IL-10 are involved
in the immune response to certain viruses. The intensity of the inflammatory
response triggered by VZV directly influences the production of IL-10, which
increases proportionally to mitigate the inflammation. However, high IL-10 levels
can inhibit T helper1-specific cell-mediated immunity, allowing the virus to
spread more effectively. While IL-10 enhances anti-inflammatory effects, it can
also prolong inflammation, explaining why chronically high IL-10 levels impede
nerve damage repair. In a study seeking predictors of neuralgia duration in HZ
patients [161], a marked elevation in IL-10 levels was observed in the serum of
patients experiencing severe HZ. IL-10 were positively correlated with neuralgia
duration and pain severity, leading to the conclusion that IL-10 levels are an
independent risk factor for PHN. Although most studies suggest that IL-10 has a
positive effect on relieving NP, NP in these studies was short-lived and mostly
inflammatory. PHN is a chronic NP lasting more than 3 months, caused by nerve
injury. Persistent elevation of IL-10 not only prolongs the inflammatory response
but also impedes nerve repair. From this perspective, IL-10 may mediate the
development of PHN. Future studies on the mechanism of IL-10 involvement in PHN
are needed (Table 2, Ref. [72, 80, 90, 91, 92, 95, 102, 110, 119, 120, 132, 133, 141, 152, 162]).
Table 2.
Possible mechanisms of action of cytokines involved in PHN.
| Cytokines |
Mechanisms of action |
Effect |
Refrences |
| TNF- |
Activation of voltage-gated Na+ and Ca2+ channels |
Central sensitization |
[91, 92, 95] |
|
Increased excitatory postsynaptic membrane currents |
Increases neuronal excitability |
[80, 102] |
|
Increase in membrane cation conductance in non-voltage gated mode |
Increases neuronal excitability |
[95] |
|
Promotes the release of inflammatory factors |
Amplifying the inflammatory response |
[90] |
|
NR1 phosphorylation of the NMDA receptor |
Hyperalgesia |
[110] |
|
Increased cyclooxygenase 2 and PGI2 synthase in endothelial cells |
Peripheral sensitization |
[162] |
| IL-1 |
Capsaicin receptor activation regulates Ca2+ inward flow via VDCC |
Release of neuropeptides and excitatory amino acids from nerve endings |
[119, 120] |
| IL-6 |
Increased Na+ and Ca2+ currents in injury receptor terminals |
Peripheral sensitization |
[132, 133] |
| IL-18 |
Regulation of ion channels and receptors |
Peripheral and central sensitization |
[141] |
|
Activation of glial cells |
Promotes inflammation and increases pain sensitivity |
[72] |
| IL-10 |
Suppression of the immune response |
Prolonged inflammatory response |
[152] |
IL, interleukin; NMDA, N-methyl-D-aspartic acid; PGI2, prostaglandin I2; VDCC,
Voltage-dependent calcium channel; PHN, postherpetic neuralgia.
5. Future Research Perspectives
Cytokines are essential for sustaining NP. PHN, as a typical NP, is closely
associated with inflammatory cytokines. Cytokines are generally secreted by
multiple cells and can act on various target cells. Moreover, a single cytokine
can trigger its target cells to produce additional cytokines, initiating a
cascade of effects. In fact, neuroinflammation, driven by the interplay between
pro-inflammatory and anti-inflammatory cytokines, serves as a fundamental
mechanism underlying the onset of PHN. The above-mentioned role of cytokines in
PHN is unquestionable, but there are few studies on the effects of cytokine
antibodies on PHN. Cell and organ culture models, as well as animal studies, have
demonstrated promising outcomes with anti-cytokine medications and pathway
inhibitors for alleviating NP. However, clinical evidence remains sparse
regarding the role of anti-cytokine drugs in NP. Moreover, clinical trials
[163, 164, 165, 166] targeting TNF- antagonists in sciatic neuropathy treatment
have notably failed to eliminate NP. Given the complex interplay in
neuroinflammatory balance, targeting specific cytokines poses a challenge. An
alternative therapeutic approach may involve regulating the cytokine cascade more
comprehensively through growth factors. In a clinical trial using ibudilast to
treat diabetic NP, patients experienced effective pain relief [167] due to the
fact that ibudilast inhibits the activation of glial cells and reduces the
release of cytokines. Valerian theophylline reduces pain by inhibiting microglia
activity [168]. Thus, future research targeting entire systems [169, 170, 171, 172, 173, 174] such as
inflammatory cytokines, chemokines, MAPK, and neuroglial cells is expected to
emerge as promising avenues for comprehensive PHN treatment. Due to the strict
human specificity of VZV, how to use VZV strains in rodent models to overcome
this specific infection remains to be determined by researchers, which will also
provide reliable models for PHN treatment research.
Abbreviations
PHN, postherpetic neuralgia; HZ, herpes zoster; VZV, varicella-zoster virus; HSV-1, herpes simplex virus type 1; TNF-, tumor necrosis factor alpha; CCI, chronic constriction injury; ILs, interleukins; IFN-, interferon gamma; TNFR1, TNF receptor 1; MAPK, mitogen-activated protein kinase; NF-B, nuclear factor kappa B; cIAP1/2, cellular inhibitor of apoptosis proteins 1 and 2; IRAK, IL-1 receptor-associated kinase; IL-1R1, IL-1 receptor type I; IL-1RAcP, IL-1 receptor accessory protein; TRAF6, tumor necrosis factor receptor-associated factor 6; JAK, Janus kinases; STAT, signal transducers and activators of transcription; TIR, toll-IL-1 receptor; AP-1, activator protein-1; DRG, dorsal root ganglion; NMDA, N-menthy1-D-aspartic acid; VGSCs, voltage-gated sodium channels; TRPV1, transient receptor potential vanilloid subtype 1; VDCC, Voltage-dependent calcium channels; MMPs, matrix metalloproteinases; TGF-, transforming growth factor beta.




 , Ping Lin 2,*
, Ping Lin 2,*