



 , Myoung Cheol Shin 2, Dae Won Kim 3,4, Ki-Yeon Yoo 5,6, Moo-Ho Won 7,*
, Myoung Cheol Shin 2, Dae Won Kim 3,4, Ki-Yeon Yoo 5,6, Moo-Ho Won 7,*
1 Department of Physical Therapy, College of Health Science, Youngsan University, 50510 Yangsan, Gyeongnam, Republic of Korea
2 Department of Emergency Medicine, Kangwon National University Hospital, Kangwon National University, 24289 Chuncheon, Gangwon, Republic of Korea
3 Department of Biochemistry and Molecular Biology, College of Dentistry, Gangneung-Wonju National University, 25457 Gangneung, Gangwon, Republic of Korea
4 Research Institute of Oral Sciences, Gangneung-Wonju National University, 25457 Gangneung, Gangwon, Republic of Korea
5 Department of Anatomy, College of Dentistry, Gangneung-Wonju National University, 25457 Gangneung, Gangwon, Republic of Korea
6 Research Institute for Dental Engineering, Gangneung-Wonju National University, 25457 Gangneung, Gangwon, Republic of Korea
7 Department of Emergency Medicine, School of Medicine, Kangwon National University, 24341 Chuncheon, Gangwon, Republic of Korea
Abstract
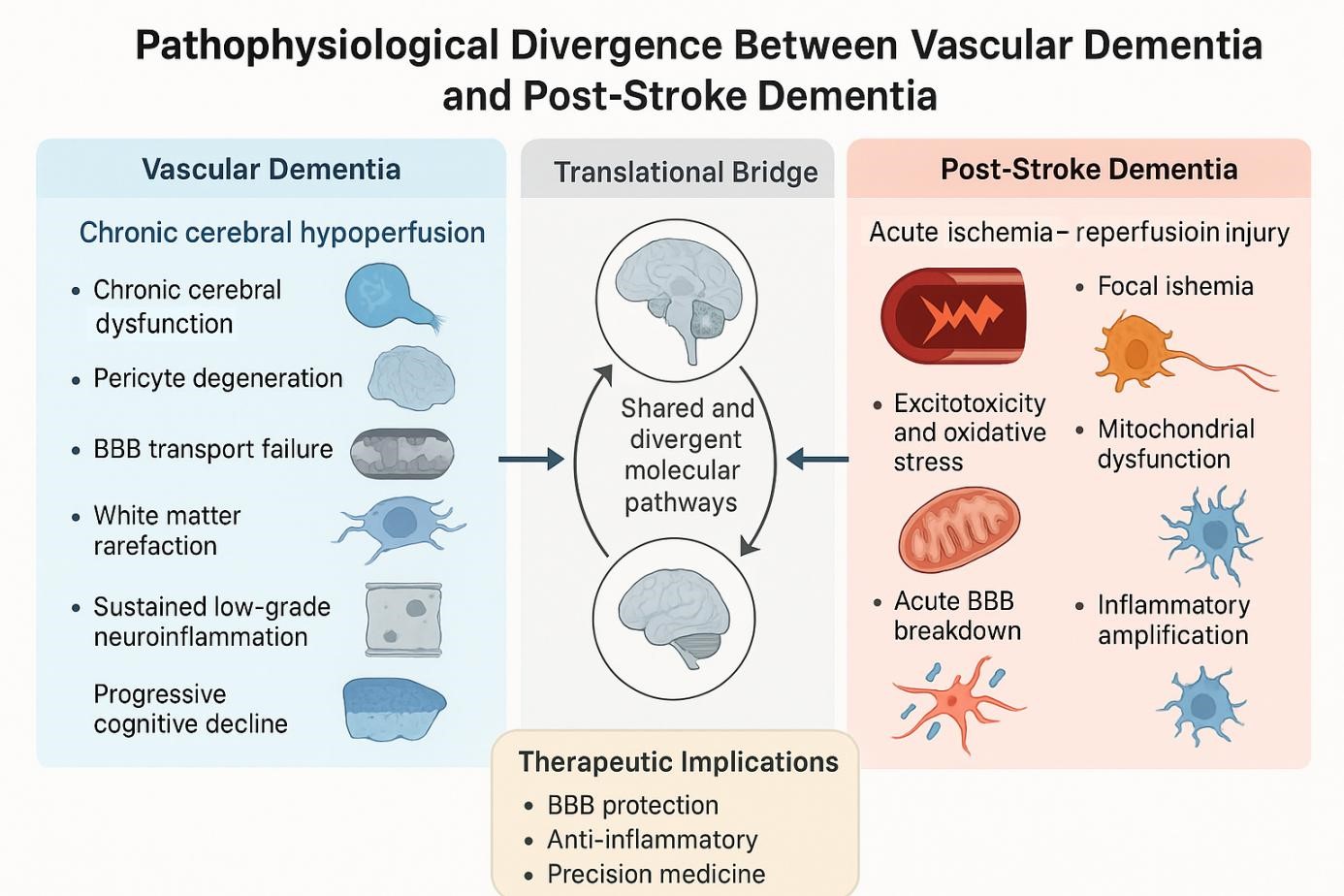
Vascular dementia (VaD) and post-stroke dementia (PSD) are two leading subtypes of vascular cognitive impairment (VCI), each arising from distinct cerebrovascular pathologies. VaD typically results from chronic cerebral hypoperfusion and small vessel disease, leading to progressive executive dysfunction and white matter degradation. In contrast, PSD occurs following acute ischemic events and is frequently associated with hippocampal damage and episodic memory deficits. This review delineates the pathophysiological divergence between VaD and PSD by integrating findings from human clinical studies and preclinical animal models. While rodent models of chronic hypoperfusion replicate key features of VaD, such as oligodendrocyte injury and myelin loss, transient ischemia models—particularly middle cerebral artery occlusion—capture hallmark PSD features, including excitotoxic neuronal death, blood–brain barrier disruption, and glial activation. Emerging research also highlights the involvement of neurovascular unit dysfunction, inflammation-driven neurodegeneration, and region-specific synaptic alterations. Recognizing these mechanistic differences is critical for advancing diagnostic precision, identifying therapeutic windows, and improving translational relevance. Furthermore, the review underscores the need for aged and comorbid animal models, integration of human biomarker studies, and implementation of novel therapies targeting endothelial function, glial reactivity, and cognitive plasticity. Through this comparative approach, we propose a unified framework to guide future investigations and interventions across the spectrum of VCI.
Graphical Abstract
Keywords
- vascular dementia
- post-stroke dementia
- cognitive impairment
- chronic hypoperfusion
- ischemia-reperfusion
- neurovascular unit
- neuroinflammation
- translational models
Vascular cognitive impairment (VCI) encompasses a spectrum of cognitive disorders arising from cerebrovascular pathology, with vascular dementia (VaD) and post-stroke dementia (PSD) constituting two principal clinical endpoints. Although both share a common vascular origin, they diverge in clinical presentation, temporal onset, and underlying pathophysiological mechanisms. VaD typically results from chronic cerebral hypoperfusion and small vessel disease, leading to progressive or stepwise cognitive decline, especially in executive function [1, 2]. Conversely, PSD is defined as a decline in cognitive performance following a clinically apparent ischemic stroke, with onset commonly occurring within three to six months post-event [3, 4].
The global burden of both VaD and PSD is considerable, particularly in aging populations where cerebrovascular diseases are prevalent. Epidemiological studies estimate that approximately 25–30% of ischemic stroke survivors experience cognitive decline, with around 10% progressing to dementia within the first year post-stroke [5], a burden further confirmed by recent surveys reporting post-stroke dementia rates of 20–30% within 1 year [6]. More recent data show that up to 70% of stroke survivors exhibit some degree of post-stroke cognitive impairment [7], and that the prevalence of vascular dementia continues to rise globally in parallel with aging populations and accumulated vascular risk factors [8]. Despite overlapping clinical symptoms and shared risk factors, the divergent pathophysiological substrates of VaD and PSD warrant direct comparative evaluation.
From a mechanistic standpoint, VaD is often associated with chronic ischemia, white matter rarefaction, arteriolosclerosis, and microinfarcts. In contrast, PSD more commonly reflects acute neuronal loss, glutamate-mediated excitotoxicity, and ischemia-reperfusion (I/R)–induced neuroinflammation [9, 10]. Recent neuroimaging and neuropathological studies have demonstrated that these conditions affect distinct neural substrates—VaD predominantly involves subcortical white matter tracts, while PSD more frequently impacts the hippocampus and associated cortical networks [11, 12]. Furthermore, advances in animal modeling have enabled simulation of both chronic hypoperfusion (e.g., bilateral carotid artery stenosis in rodents) and acute ischemic events (e.g., middle cerebral artery occlusion), offering mechanistic insights under controlled experimental conditions [13, 14, 15].
This review provides a side-by-side comparison of VaD and PSD, emphasizing their distinct pathophysiological features as revealed by human studies and experimental models. Previous reviews have tended to discuss VCI in general terms—highlighting clinical criteria [2], small vessel disease mechanisms [16], or post-stroke cognitive decline [3]—without clearly distinguishing VaD and PSD as separate mechanistic entities. In contrast, our approach is to integrate findings across disciplines, including neuroimaging, neuropathology, and molecular biology, to elucidate parallel and divergent disease trajectories. By exploring these differences at cellular, molecular, and systems levels, we aim to refine diagnostic frameworks and support the development of targeted therapeutic strategies for each subtype of vascular cognitive disorder.
To construct this comparative review, we conducted a comprehensive literature search using PubMed (https://pubmed.ncbi.nlm.nih.gov/), Scopus (https://www.scopus.com/), and Web of Science (https://webofscience.com/wos/alldb/basic-search/) for studies published from January 2000 to May 2025. Search terms included combinations of “vascular dementia”, “post-stroke dementia”, “vascular cognitive impairment”, “chronic cerebral hypoperfusion”, “middle cerebral artery occlusion”, and “animal models of dementia”. We prioritized peer-reviewed original articles and systematic reviews offering mechanistic insights, neuroimaging correlations, histopathological findings, or translational relevance. Studies focused exclusively on Alzheimer’s disease or non-vascular dementias were excluded unless they provided comparative perspectives. Over 120 references were screened, and the most representative studies were selected to support the thematic structure of this review.
VaD and PSD are subtypes of VCI, yet they differ significantly in their diagnostic frameworks, temporal association with cerebrovascular events, and cognitive domains predominantly affected. Establishing clear operational definitions is essential for consistent clinical diagnosis, research standardization, and interpretation of translational studies.
VaD is characterized as an acquired cognitive disorder attributed to cerebrovascular pathology, particularly small vessel disease and chronic cerebral hypoperfusion [9]. Over the past two decades, multiple diagnostic frameworks have emerged. Among the most widely adopted is the National Institute of Neurological Disorders and Stroke-the Association Internationale pour la Recherche et l’Enseignement en Neurosciences (NINDS-AIREN) criteria, formulated through consensus by the NINDS and AIREN. This research-oriented framework mandates the presence of cognitive decline in at least two domains, functional impairment, and a clear temporal relationship to cerebrovascular events, supported by neuroimaging evidence of infarction or lesions [17]. NINDS-AIREN offers diagnostic rigor and is more relevant to VaD research, but it is less practical for PSD where rapid post-stroke diagnosis is required. In clinical contexts, the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) classifies VaD under “major neurocognitive disorder due to vascular disease”, emphasizing a stepwise or fluctuating course, focal neurological signs, and a vascular etiology [18]. DSM-5 provides broader clinical applicability and captures both conditions, though it lacks specificity in distinguishing acute versus chronic vascular impairment. The international classification of diseases, 11th revision (ICD-11), developed by the World Health Organization, similarly defines VaD as a cognitive disorder secondary to cerebrovascular disease, incorporating both ischemic and hemorrhagic mechanisms [19]. ICD-11 ensures international standardization and inclusivity, but its criteria do not clearly separate progressive small vessel disease in VaD from acute ischemic injury in PSD. A side-by-side comparison of these diagnostic frameworks is presented in Table 1, outlining their scope, imaging requirements, common usage contexts, and comparative differences between VaD and PSD. In contrast, PSD specifically refers to cognitive impairment that arises within a defined interval following a clinically diagnosed stroke, typically within 3 to 6 months [3]. It is conceptualized as a consequence of acute ischemic injury and is commonly characterized by deficits in memory, executive function, language, or visuospatial skills. While VaD may progress insidiously through cumulative microvascular damage, PSD is typically preceded by an overt cerebrovascular event. The Vascular Impairment of Cognition Classification Consensus Study (VASCOG) has proposed that PSD be diagnosed when dementia emerges within 6 months of a stroke, regardless of prior cognitive status, provided that the stroke is the dominant causative factor [20]. This temporal definition has facilitated greater consistency in clinical trial design and patient stratification.
| Criteria | Origin/Purpose | Core requirements | Imaging support | Common usage | Comparative differences between VaD and PSD |
| NINDS-AIREN | Research-oriented; consensus between NINDS and AIREN | Cognitive decline in |
Required (CT/MRI evidence of infarcts or lesions) | Primarily in research settings | More applicable to VaD due to emphasis on chronic vascular lesions and multiple domains; less suited for PSD, where acute onset after stroke and rapid diagnosis are critical. |
| DSM-5 | Clinical classification by American Psychiatric Association | Evidence of vascular etiology; stepwise/fluctuating course; focal neurological signs | Recommended but not mandatory | Widely used in clinical practice | Captures both VaD (progressive/subcortical decline) and PSD (acute deficits after stroke), but lacks specificity in distinguishing temporal onset. |
| ICD-11 | Global classification system by WHO for clinical use | Cognitive decline due to cerebrovascular disease (ischemic or hemorrhagic); functional impact | Recommended | Used internationally across healthcare systems | Broadly covers both VaD and PSD; recognizes the vascular origin but lacks detail in distinguishing chronic small vessel pathology seen in VaD from the acute post-stroke onset typical of PSD. |
Abbreviations: VaD, Vascular Dementia; NINDS, National Institute of Neurological Disorders and Stroke; AIREN, Association Internationale pour la Recherche et l’Enseignement en Neurosciences; DSM-5, Diagnostic and Statistical Manual of Mental Disorders, 5th Edition; ICD-11, International Classification of Diseases, 11th Revision; CT, Computed Tomography; MRI, Magnetic Resonance Imaging; WHO, World Health Organization; PSD, Post-Stroke Dementia.
Despite these formal criteria, clinical differentiation between VaD and PSD can be difficult due to overlapping symptoms and frequent coexistence of mixed pathologies, especially among elderly patients. VaD and Alzheimer’s Disease (AD) frequently coexist, particularly in older adults, and the overlap of cerebrovascular disease and Alzheimer-type pathologies often accelerate cognitive decline and complicate diagnosis and treatment [21]. These mixed mechanisms also present challenges for VaD and PSD research, underscoring the need to integrate vascular and neurodegenerative biomarkers in future studies [22]. While this review focuses on VaD and PSD, mixed dementia remains an essential context for interpreting clinical and translational findings. From a diagnostic standpoint, neuroimaging serves as a crucial adjunct, with PSD often associated with focal cortical infarcts or atrophy, whereas VaD more commonly presents with white matter hyperintensities, lacunar infarcts, and other small vessel disease markers [2]. In summary, VaD and PSD are both vascular in origin but differ in temporal onset, lesion characteristics, and diagnostic categorization. Standardized and reliable criteria are essential not only for clinical accuracy but also for the translational relevance of animal models used to study these conditions.
Although VaD and PSD both arise from cerebrovascular pathology, they differ substantially in terms of etiology, lesion distribution, inflammatory response, hemodynamic profile, and neural network disruption. VaD is primarily linked to chronic cerebral hypoperfusion, most commonly due to small vessel disease (SVD). Hemodynamically, VaD emerges in the context of prolonged global hypoperfusion without a discrete vascular event [23], leading to gradual and widespread damage to subcortical structures and white matter tracts. Neuropathological hallmarks include periventricular lacunes, white matter rarefaction, and microinfarcts—especially within fronto-subcortical circuits that mediate executive function, motor planning, and attention regulation [2, 16]. By contrast, PSD arises from acute ischemic events—such as large vessel infarctions or multiple embolic occlusions—that frequently involve the hippocampus and adjacent cortical structures and typically followed by reperfusion. This sequence provokes oxidative stress, excitotoxicity, and blood–brain barrier (BBB) disruption [23], leading to focal neuronal loss, hippocampal atrophy, and disconnection of memory-associated networks like the default mode network (DMN) [11, 12, 24]. At the network level, VaD is associated with fronto-subcortical disconnection, contributing to deficits in psychomotor speed, attention, and executive function. In PSD, hippocampal–prefrontal pathways and DMN integrity are acutely disrupted, correlating with profound memory impairment and impaired consolidation processes [25, 26, 27]. Disruption of these networks compromises episodic memory encoding and retrieval, as well as the integration of memory with executive control—manifesting clinically as amnesia and disorientation.
Taken together, these comparisons highlight that VaD and PSD, while both arising from vascular insults, diverge in their temporal dynamics, inflammatory profiles, and network vulnerabilities. VaD exemplifies a chronic disconnection syndrome linked to diffuse white matter injury, whereas PSD reflects an acute circuit breakdown driven by reperfusion-induced neurotoxicity. Such distinctions provide critical insight into differential diagnostic markers and potential therapeutic targets. A comparative overview of these human pathophysiological features is presented in Table 2 (Ref. [11, 12, 16, 23, 25, 27, 28, 29]).
| Feature | VaD | PSD | Key pathophysiology | Representative references |
| Primary cause | Chronic cerebral hypoperfusion, small vessel disease (SVD) | Acute ischemic stroke, large artery infarction | Prolonged ischemia due to small vessel pathology vs. acute infarction | [16, 28] |
| Pathology | Microinfarcts, white matter rarefaction, periventricular lacunes | Hippocampal atrophy, cortical infarcts, focal neuronal loss | Diffuse subcortical damage vs. focal cortical necrosis | [11, 12] |
| Inflammation | Chronic microglial activation, elevated CRP | Acute cytokine surge (↑ TNF- |
Persistent low-grade vs. acute inflammatory burst | [28, 29] |
| Hemodynamics | Persistent low perfusion | Sudden occlusion and reperfusion injury | Gradual vs. abrupt cerebral blood flow disturbance | [23] |
| Circuitry disruption | Fronto-subcortical circuit dysfunction | Hippocampal–prefrontal and DMN disruption | Disruption of executive vs. memory-related networks | [25, 27] |
Animal models have been pivotal in dissecting the divergent pathophysiological pathways of VaD and PSD. By simulating chronic hypoperfusion and acute I/R injury in a controlled environment, these models provide mechanistic insight and translational relevance, though none fully replicate the human disease spectrum.
Models of VaD typically involve bilateral common carotid artery stenosis (BCAS)
in mice or two-vessel occlusion (2VO) in rats. These paradigms induce sustained
cerebral hypoperfusion, resulting in progressive white matter degeneration,
demyelination, and axonal injury—especially in the corpus callosum, internal
capsule, and hippocampus. These structures support interhemispheric
communication, thalamocortical relay, and memory consolidation, respectively
[30, 31, 32]. The ensuing cognitive impairments—affecting executive function,
working memory, and spatial navigation—mirror fronto-subcortical disconnection
[33, 34, 35, 36]. Histopathological features include gliosis, oligodendrocyte loss, and
perivascular fibrosis—paralleling small vessel disease in humans. In contrast,
PSD models typically employ transient middle cerebral artery occlusion (tMCAO) in
rodents to replicate acute focal ischemia followed by reperfusion. This results
in rapid infarction of cortical and hippocampal regions, widespread neuronal
loss, and disruption of the BBB. The ensuing inflammatory cascade involves
microglial and astrocytic activation, oxidative stress, and robust upregulation
of proinflammatory mediators such as tumor necrosis factor alpha (TNF-
Importantly, regional vulnerability and network dysfunction vary across models. VaD paradigms predominantly target deep white matter tracts and subcortical relay nuclei, leading to diffuse disconnection syndromes. In contrast, tMCAO results in focal damage to the hippocampus and neocortex, disrupting circuits critical for memory and cognitive flexibility. Functional imaging studies have shown that BCAS diminishes prefrontal–thalamic and subcortical coherence, whereas tMCAO interrupts hippocampal–prefrontal coupling and DMN-like activity [40, 41]. These connectivity patterns align with the cognitive phenotypes observed: attentional and executive deficits in VaD, and amnesia with impaired consolidation in PSD. These animal models recapitulate essential aspects of human VCI. Chronic hypoperfusion paradigms (e.g., BCAS, 2VO) parallel white matter injury and small vessel pathology in VaD, whereas transient MCAO reproduces hippocampal and neocortical vulnerability and network disconnection observed in PSD. While no single model fully captures the complexity of human condition, they provide validated platforms for mechanistic exploration. Future research should integrate longitudinal imaging, advanced behavioral assays, and multimodal biomarkers to enhance translational relevance. A comparative summary of these pathophysiological features in experimental models is provided in Table 3 (Ref. [14, 30, 31, 34, 35, 36, 37, 38, 40, 41]).
| Feature | VaD models (e.g., BCAS, 2VO) | PSD models (e.g., tMCAO) | Key pathophysiology | Representative references |
| Vascular insult type | Chronic hypoperfusion | Acute ischemia–reperfusion | Gradual reduction in cerebral blood flow | [35, 36] |
| Affected regions | White matter (corpus callosum), hippocampus, internal capsule | Cortex, hippocampus | Axonal degeneration vs. focal infarction | [14, 34] |
| Inflammatory response | Low-grade, chronic microgliosis | Acute, robust microgliosis and cytokine storm | TNF- |
[37, 38] |
| Functional impairment | Working memory, executive function, spatial navigation | Episodic memory, learning consolidation | Disrupted fronto-subcortical vs. hippocampal–prefrontal pathways | [40, 41] |
| Network connectivity | ↓ Prefrontal–thalamic coherence | ↓ Hippocampal–prefrontal, DMN-like networks | Distinct regional vulnerability and reorganization | [30, 31] |
The divergent clinical and neuropathological trajectories of VaD and PSD are underpinned by distinct molecular mechanisms, despite several shared pathogenic elements. This section reviews key overlapping and disease-specific pathways observed in both human studies and animal models, focusing on oxidative stress, neuroinflammation, BBB disruption, and unique molecular signatures—such as Wnt/Notch dysregulation in VaD and excitotoxicity and aquaporin/matrix remodeling in PSD (see Fig. 1).
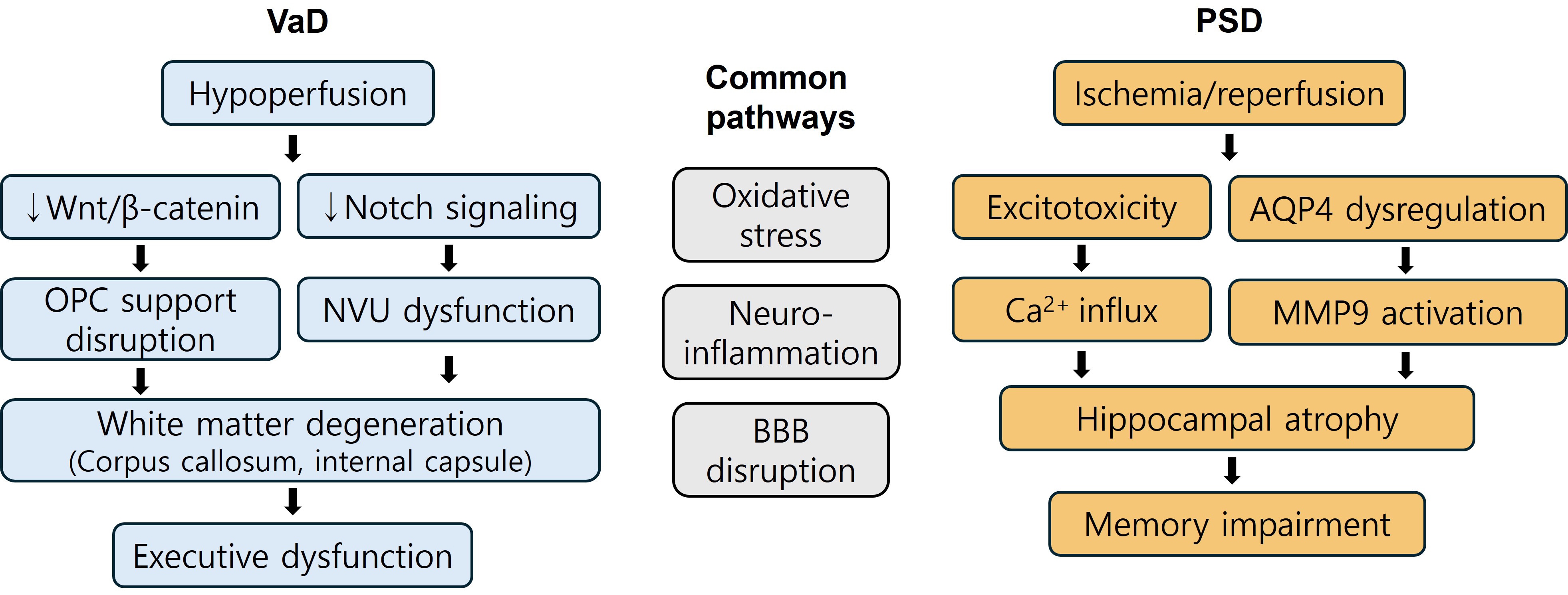 Fig. 1.
Fig. 1.
Molecular mechanisms underlying VaD and PSD. This figure
illustrates the shared and distinct molecular pathways contributing to the
pathogenesis of VaD and PSD. In the central panel, oxidative stress,
neuroinflammation, and blood–brain barrier (BBB) disruption are depicted as
common mechanisms found in both conditions. On the left, VaD-specific pathways
highlight chronic cerebral hypoperfusion-induced alterations in
Wnt/
Oxidative stress is a hallmark of both VaD and PSD, primarily resulting from
cerebral hypoxia, mitochondrial dysfunction, and excessive production of reactive
oxygen species (ROS). These ROS induce lipid peroxidation, DNA damage, and
protein nitration, which contribute to injury of neurons and oligodendrocytes in
vulnerable areas such as the hippocampus and white matter tracts [1, 2].
Neuroinflammation also plays a central role. Activation of microglia and
infiltration of peripheral immune cells elevate proinflammatory cytokines
including TNF-
Taken together, these molecular differences account for the distinct clinical trajectories of VaD and PSD. In VaD, chronic oxidative stress, sustained neuroinflammation, and progressive BBB disruption—along with impaired Wnt/Notch signaling—contribute to gradual white matter degeneration and fronto-subcortical disconnection, manifesting as deficits in executive function and processing speed. In contrast, PSD is characterized by an acute burst of oxidative stress, robust neuroinflammatory activation, excitotoxicity, and aquaporin/MMP-mediated BBB breakdown, which converge on hippocampal and cortical vulnerability. These mechanisms underlie pronounced impairments in memory consolidation, contextual learning, and spatial orientation. Such contrasts highlight VaD as a chronic disconnection syndrome and PSD as an acute circuit failure, providing critical insight for the development of targeted interventions. These shared pathways, alongside disease-specific mechanisms discussed below, are summarized in Table 4 (Ref. [1, 2, 34, 37, 38, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52]).
| Molecular mechanism | VaD | PSD | References |
| Oxidative stress | Chronic ROS elevation due to hypoperfusion and mitochondrial dysfunction | Acute ROS burst during reperfusion exacerbates neuronal injury | [1, 2] |
| Neuroinflammation | Microglial activation sustained by vascular injury and white matter degeneration | Rapid microglial and astrocytic response to ischemic insult | [37, 38, 42] |
| BBB disruption | Gradual endothelial degeneration, tight-junction loss (claudin-5, occludin), pericyte loss | Abrupt MMP-mediated breakdown of BBB post-reperfusion (e.g., MMP-9 upregulation) | [43, 44, 45] |
| Wnt/Notch signaling | ↓ Wnt/ |
Not prominently involved | [46, 47, 48] |
| Excitotoxicity | Not prominent | ↑ Glutamate → NMDA/AMPA overactivation → Ca2+ influx, mitochondrial failure | [41, 49] |
| Aquaporin/MMP activation | Mild or secondary | ↑ AQP4 mislocalization → edema; ↑ MMP-9 → extracellular matrix degradation | [50, 51, 52] |
| White matter integrity | Progressive degeneration (especially in corpus callosum, internal capsule) | Focal secondary degeneration after infarct | [2, 34] |
| Cognitive domains affected | Executive dysfunction, processing speed deficits | Memory consolidation, executive function, spatial orientation deficits | [1, 41] |
VaD pathogenesis is driven by progressive white matter degeneration and impaired
neurovascular repair, notably via dysregulation of developmental pathways.
Suppression of Wnt/
PSD arises from abrupt ischemic insult followed by reperfusion, initiating a
cascade of excitotoxic and inflammatory injury. Excessive glutamate release and
overactivation of N-methyl-D-aspartate (NMDA) and
Despite increasing insight into the molecular basis of VaD and PSD, therapeutic strategies remain largely symptomatic, with limited disease-modifying options. This section compares current clinical interventions with experimental strategies from animal models and highlights key translational hurdles. A comparative overview is presented in Table 5 (Ref. [49, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65]), while Fig. 2 illustrates translational challenges and proposed solutions.
| Therapeutic agent | Mechanism of action | Disease context | Model species (if applicable) | Outcomes | References |
| Donepezil | Cholinesterase inhibitor | VaD ( |
Human | Modest cognitive benefit in VaD with hippocampal atrophy | [56] |
| Rivastigmine | AChE + BuChE inhibition | Subcortical VaD | Human | Mild cognitive improvement | [55] |
| Memantine | NMDA receptor antagonist | PSD | Human | Improves cognition and reduces excitotoxicity | [57] |
| Citicoline | Membrane repair, neurogenesis | VaD, PSD | Human | Enhances cognition, delays progression | [58] |
| Resveratrol | STING/TBK1/IRF3 inhibition, anti-inflammatory, antioxidant | VaD | Rat (2VO) | ↓ microglia & white matter damage; ↑ memory, ↑ cholinergic neurons | [59, 60] |
| Cilostazol | PDE3 inhibition, ↑ NO, ↓ oxidative stress, cAMP/CREB activation | Chronic hypoperfusion | Rat (4VO) | Prevents retrograde amnesia, ↑ working memory | [61, 62] |
| Edaravone-dexborneol | Free radical scavenging + BBB-penetrating anti-inflammatory effects | Small vessel disease | Mouse | ↓ BBB leakage, ↓ microglial pyroptosis, ↑ M2 polarization, ↑ cognition | [63] |
| Minocycline/Curcumin | Anti-inflammatory, glial modulation | PSD, VaD | Rodent | ↓ TNF- |
[64] |
| Erythropoietin | White matter repair, neurovascular protection | Chronic ischemia | Rodent | ↑ remyelination, ↓ gliosis, ↑ cognition | [65] |
| rTMS/BDNF mimetics | Synaptic plasticity enhancement | PSD | Rat (MCAO) | ↑ BDNF, ↑ LTP, ↓ cognitive deficits | [49] |
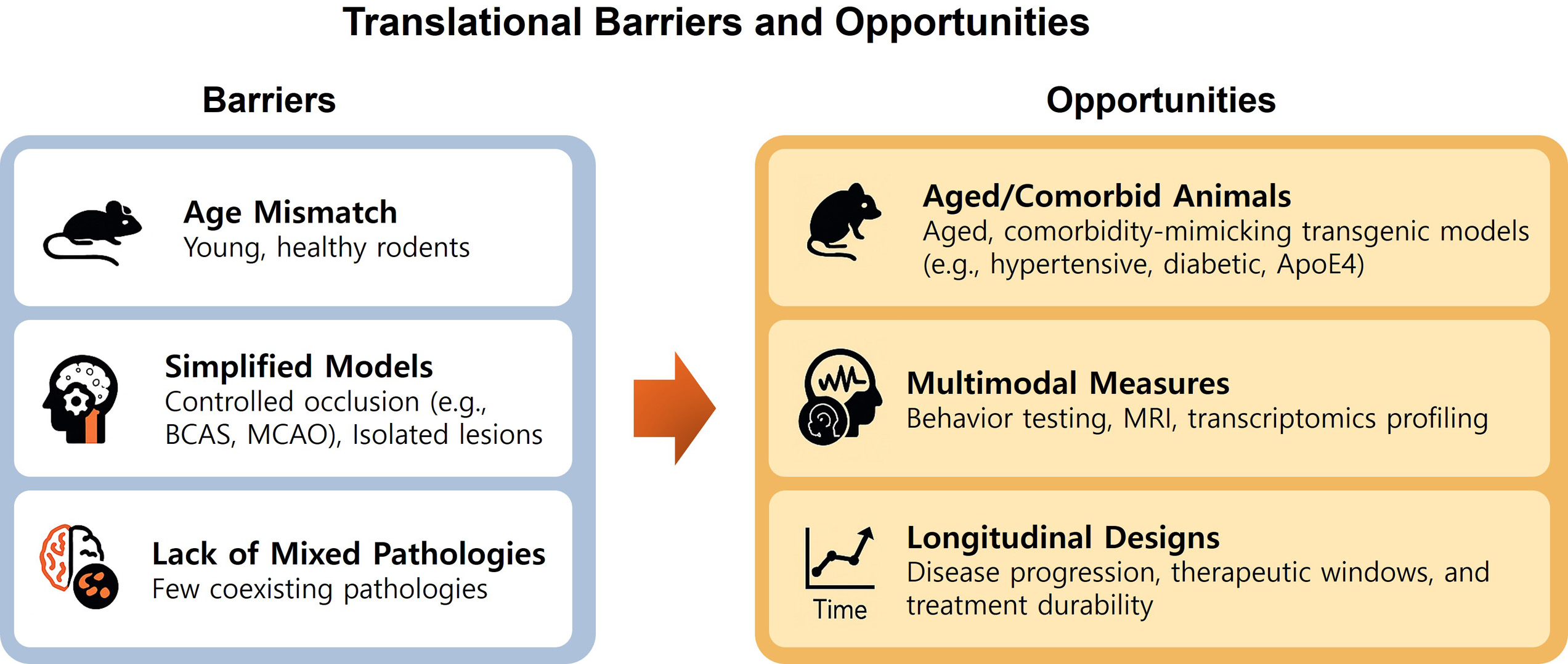 Fig. 2.
Fig. 2.
Translational barriers and opportunities in VaD and PSD research. This infographic summarizes key obstacles and corresponding opportunities for improving translational relevance in preclinical studies of VaD and PSD. On the left, common limitations include the use of young, healthy rodents (age mismatch), reliance on simplified occlusion models (e.g., MCAO, BCAS) that fail to capture human vascular heterogeneity, and the absence of co-pathologies such as amyloid or tau deposition. On the right, corresponding strategies for refinement include use of aged or comorbid transgenic animals, the application of multimodal outcome metrics (e.g., behavior, MRI, transcriptomics), and longitudinal study designs to mimic progressive cognitive decline and therapeutic windows seen in patients. Abbreviations: BCAS, bilateral common carotid artery stenosis.
Cholinesterase inhibitors, such as donepezil and rivastigmine, exert modest cognitive benefits in VaD, particularly in mixed pathologies with concurrent Alzheimer’s disease [55, 56, 66]. Rivastigmine, which inhibits both acetylcholinesterase and butyrylcholinesterase, may be especially beneficial in subcortical VaD. Memantine, an NMDA receptor antagonist, has shown efficacy in PSD by reducing glutamate-mediated excitotoxicity and promoting synaptic plasticity [57, 67]. Citicoline, a cytidine-5′-diphosphocholine derivative, enhances membrane repair and neurogenesis and is used in both VaD and PSD [58, 68]. Equally important is rigorous control of vascular risk factors—including hypertension, diabetes, dyslipidemia, and atrial fibrillation—which remains the cornerstone of disease prevention and management [9, 69]. Nevertheless, these interventions largely alleviate symptoms without reversing molecular or structural neuropathology. Despite these available options, no pharmacological agent has achieved global approval as a disease-modifying therapy for VaD or PSD. Donepezil is approved for VaD in only a few countries, and memantine provides modest benefits in PSD, while citicoline remains supportive rather than curative. Emerging repurposed drugs, such as metformin, are currently under early-phase clinical investigation for vascular cognitive impairment [66, 70]. However, translational failures are frequent, owing to patient heterogeneity, mixed pathologies, underrepresentation of elderly patients with comorbidities, and trial designs that do not fully reflect disease complexity. These challenges highlight why many VaD/PSD drug trials have failed to yield meaningful clinical advances. Looking forward, future therapeutic strategies will require biomarker-guided patient stratification, integration of vascular and neurodegenerative endpoints, and the inclusion of more representative clinical populations. Such approaches will be critical to overcome past limitations and to develop therapies capable of modifying disease trajectories rather than merely alleviating symptoms.
Rodent models of cerebral hypoperfusion and I/R injury have enabled the discovery of candidate compounds targeting inflammation, oxidative stress, and synaptic damage, as well as biologics such as erythropoietin derivatives for white matter repair and non-pharmacological interventions like high-frequency repetitive transcranial magnetic stimulation (rTMS) for BDNF-mediated plasticity.
Resveratrol, a natural polyphenol, has shown consistent neuroprotection in 2VO
models. It suppresses Stimulator of Interferon Genes (STING)/TANK-binding kinase 1 (TBK1)/Interferon Regulatory Factor 3 (IRF3) signaling, attenuates microglial activation
and white matter degeneration, and preserves cholinergic neurons. Behavioral
improvements include better spatial learning in the Morris water maze and Y-maze
[59, 60]. Cilostazol, a selective phosphodiesterase-3 (PDE3) inhibitor, enhances
nitric oxide availability and activates Cyclic Adenosine Monophosphate (cAMP)/cAMP Response Element–Binding Protein (CREB) signaling. In middle-aged rats
subjected to four-vessel occlusion (4VO), it ameliorated working memory deficits
and oxidative stress [61, 62]. Edaravone–Dexborneol, a novel combination of a
free radical scavenger and a BBB-penetrant monoterpene, significantly reduced BBB
leakage, microglial pyroptosis (via NLRP3 inhibition), and cognitive decline in
stroke and small vessel disease models [63]. Minocycline and curcumin, both
broad-spectrum anti-inflammatories, reduced cytokine levels (TNF-
Despite robust preclinical efficacy, clinical translation remains limited. One
major obstacle is age disparity—most rodent models use young, healthy animals,
whereas VaD and PSD primarily affect elderly individuals with comorbidities
(e.g., diabetes, hypertension, immunosenescence) that influence drug
responsiveness [71, 72]. Second, popular models such as MCAO and BCAS simulate
discrete, reproducible injuries, which contrast with the multifocal,
heterogeneous, and progressive pathology in human cerebrovascular disease [73].
Third, pathological overlap with neurodegenerative diseases complicates model
validity. In humans, VaD often coexists with Alzheimer-type changes, including
Despite the continued reliance on symptomatic treatment and vascular risk control in VaD and PSD, emerging molecular insights have paved the way for disease-modifying approaches. These include precision-based interventions tailored to individual genetic and biomarker profiles, strategies to restore BBB integrity, combinatory neurovascular–cognitive therapies, and regenerative cell-based treatments. The following subsections highlight key advances in these domains.
Precision medicine offers the potential for individualized intervention by
integrating a patient’s genetic background, neuroimaging features, and fluid
biomarker profiles. This approach is particularly relevant in VaD and PSD, given
their etiological and clinical heterogeneity. Recent genome-wide association
studies (GWAS) and targeted sequencing have identified several variants
associated with small vessel disease and vascular cognitive impairment. NOTCH3
mutations, traditionally linked to CADASIL, have also been implicated in sporadic
forms of small vessel disease. These mutations impair vascular smooth muscle
integrity and promote perivascular degeneration. Rodent models carrying
Notch3 mutations exhibit white matter vacuolization and reactive gliosis
[76]. Similarly, COL4A1 and COL4A2 mutations compromise basement membrane
stability in cerebral microvessels, leading to spontaneous microbleeds and white
matter lesions. Murine models harboring COL4A1 variants demonstrate
cognitive dysfunction accompanied by vascular fragility [77]. The APOE
Disruption of the NVU—a dynamic interface composed of endothelial cells, astrocytes, pericytes, and the basement membrane—plays a central role in the pathogenesis of VaD and PSD [82]. Among NVU components, BBB breakdown allows serum proteins and peripheral immune cells to infiltrate the parenchyma, initiating glial activation and progressive neuronal dysfunction.
Preclinical studies using rodent models of ischemic injury, such as MCAO or chronic hypoperfusion (e.g., BCAS), have identified several pharmacologic strategies for NVU preservation. Sphingosine-1-phosphate receptor (S1PR) modulators, such as fingolimod, preserve tight junction proteins (claudin-5, occludin), suppress astrocytic and microglial activation, and reduce extravasation of immunoglobulin (Ig) G and albumin in peri-infarct regions [83, 84]. MMP-9 inhibitors and angiopoietin-1 mimetics contribute to endothelial integrity by preventing degradation of the extracellular matrix [85, 86, 87]. These agents are particularly effective during the acute post-ischemic phase, although there is growing evidence of their potential in slowing chronic NVU deterioration in models of VaD.
MSC–derived extracellular vesicles (EVs) have gained attention as a cell-free
regenerative strategy for NVU repair. In aged rat models subjected to bilateral
common carotid artery occlusion, both intravenous and intracerebroventricular
administration of MSC-EVs reduced levels of IL-1
Current NVU-targeted therapies converge on three mechanistic axes: (1) BBB stabilization, through structural reinforcement and inhibition of paracellular leakage [78, 83]; (2) Suppression of neuroinflammation, by modulating astrocytic and microglial reactivity [82, 89]; and (3) Restoration of neurovascular coupling, improving cerebral perfusion to metabolically active neurons [90]. Translational challenges remain significant, notably the lack of aged and comorbid animal models, and the absence of real-time in vivo biomarkers for NVU integrity. Emerging imaging tools such as dynamic contrast-enhanced MRI (DCE-MRI) now allow longitudinal assessment of BBB permeability in human trials [91]. Future research should aim to integrate such imaging with molecular biomarkers and individualized NVU-modifying interventions.
Therapeutic approaches that concurrently target vascular pathology and neuroplasticity deficits are gaining increasing attention in the management of VaD and PSD. This dual strategy reflects the interdependence of cerebrovascular integrity and cognitive function. Several pharmacological agents demonstrate pleiotropic actions on both vascular and neural systems. Citicoline supports phospholipid synthesis and stimulates neurotrophic pathways, leading to cognitive enhancement in patients and improved hippocampal synaptic plasticity in ischemic rodent models [61]. Cilostazol, a phosphodiesterase-3 inhibitor, has been shown to preserve memory function in 2VO rat models by increasing nitric oxide bioavailability and suppressing neuroinflammatory responses [66]. Resveratrol, a polyphenolic compound, modulates the STING/TBK1/IRF3 inflammatory axis and protects cholinergic neurons in models of chronic cerebral hypoperfusion [64, 65]. Non-pharmacological interventions also play a critical role. Aerobic exercise and rTMS have demonstrated cognitive benefits in preclinical settings. In rat models of global ischemia, high-frequency rTMS upregulated brain-derived neurotrophic factor (BDNF) expression in the hippocampus and significantly improved working memory [49]. In summary, combined vascular–cognitive strategies offer synergistic benefits by targeting both underlying pathology and functional decline. Future clinical trials should emphasize early-stage, multimodal implementation guided by individualized risk profiles and supported by molecular and imaging biomarkers.
Regenerative therapies aim to reverse neurovascular degeneration in VaD and PSD by restoring cellular structure and function. Two promising approaches—induced pluripotent stem cell (iPSC)-derived neural precursors and MSC-derived EVs—are gaining traction in preclinical research. iPSC-derived NPCs exhibit multipotency and secrete a broad range of neurotrophic factors, making them attractive for neurorestorative applications in VaD and PSD. In adult rodent models of ischemia, including MCAO and bilateral carotid artery stenosis, transplanted NPCs demonstrated robust survival and integration, particularly within the hippocampal and cortical regions [92].
These NPCs were capable of differentiating into both neuronal and glial lineages and actively secreted vascular endothelial growth factor (VEGF), glial cell line–derived neurotrophic factor (GDNF), and thrombospondins (TSPs), facilitating neurovascular repair and synaptic remodeling [93, 94]. Functionally, NPC transplantation mitigated white matter atrophy and improved cognitive outcomes, as assessed by Y-maze alternation and novel object recognition tasks [95]. In addition, iPSC-derived vascular endothelial cells (iVECs) have been shown to enhance regulatory T cell (Treg) recruitment and suppress microglial activation, thereby supporting remyelination and vascular remodeling [96]. However, translational application of iPSC-derived therapies remains limited by potential tumorigenicity, immunogenicity, and the lack of long-term safety data—particularly in aged or comorbid populations who most frequently develop VaD or PSD.
MSC-derived EVs represent a cell-free, low-immunogenicity alternative to stem cell transplantation. In rat models of chronic cerebral hypoperfusion, EVs enriched in miR-132-3p have been shown to restore tight junction protein expression, enhance cerebral perfusion, and reverse spatial memory deficits [52]. Nonetheless, significant challenges remain, including: (1) Standardization of EV isolation protocols; (2) Optimization of dosage and delivery methods; and (3) Comprehensive characterization of bioactive cargo. Innovative strategies such as scaffold-assisted delivery and engineered synthetic vesicles may help overcome these limitations and improve clinical translation. A summary of regenerative and NVU-targeted strategies is provided in Table 6 (Ref. [52, 61, 64, 65, 66, 88, 92, 93, 94, 95, 96]).
| Therapy | Cell type/Origin | Model type | Mechanism of action | Outcome | Supporting references |
| iPSC-NPCs | Human iPSC-derived neural precursor cells | MCAO, chronic hypoperfusion (rats, pigs) | Neuronal/glial differentiation; VEGF, GDNF, TSP secretion | Improved memory, synaptic density | [92, 93, 94, 95] |
| Endothelial iPSCs | Human iPSC-derived vascular endothelial cells | Chronic hypoperfusion (mice) | Treg cell recruitment, WMI protection | Enhanced white matter recovery | [96] |
| MSC-EVs | Bone marrow– or adipose–derived MSCs | Chronic hypoperfusion (rats, mice) | miRNA transfer (e.g., miR-132-3p), BBB repair, M1→M2 microglia shift | Cognitive rescue, BBB integrity restored | [52, 88] |
| NVU-targeted drugs | Various (resveratrol, citicoline, cilostazol) | Stroke, hypoperfusion (rodents, humans) | BBB stabilization, anti-inflammatory, neurovascular coupling | Improved CBF, reduced inflammation, memory enhancement | [61, 64, 65, 66] |
VaD and PSD represent clinically overlapping but mechanistically distinct
subtypes within the broader spectrum of vascular cognitive impairment. VaD is
predominantly driven by chronic cerebral hypoperfusion, leading to white matter
degeneration, glial activation, and NVU disruption. In contrast, PSD typically
follows acute ischemic events, where excitotoxicity, oxidative stress, and BBB
breakdown dominate the early phase, followed by secondary neurodegeneration and
disconnection of cognitive networks [82, 83, 84]. While both disorders share common
features—such as BBB impairment, neuroinflammation, and synaptic
dysfunction—their temporal evolution, regional vulnerability, and triggering
mechanisms differ, necessitating differential diagnostic and therapeutic
strategies. Despite substantial insights gained from animal models such as BCAS
and MCAO, translational challenges persist. These include species-specific
differences in cerebrovascular anatomy, immune responses, and behavioral
phenotypes [89, 90]. Future preclinical research must increasingly incorporate
models that reflect aging, comorbid conditions (e.g., hypertension, diabetes),
and longitudinal disease progression, to improve clinical relevance. From a
clinical standpoint, there is a pressing need to refine diagnostic criteria by
integrating dynamic molecular biomarkers alongside structural neuroimaging.
Promising candidates include endothelial injury markers (MMP-9, S100
2VO, two-vessel occlusion; 4VO, four-vessel occlusion; AMPA,
All data reported in this paper will also be shared by the lead contact upon request.
JHA and M-HW designed the research study. JHA, MCS, DWK, K-YY, and M-HW wrote the manuscript. All authors contributed to the study’s design, manuscript preparation, and revision. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest. Moo-Ho Won is serving as one of the Editorial Board members of this journal. We declare that Moo-Ho Won had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Bettina Platt.
During the preparation of this work the authors used ChatGPT-3.5 in order to check spell and grammar. After using this tool, the authors reviewed and edited the content as needed and takes full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.