

1 Wuxi School of Medicine, Jiangnan University, 214122 Wuxi, Jiangsu, China
2 Department of Neurosurgery, Jiangnan University Medical Center, 214005 Wuxi, Jiangsu, China
3 Wuxi Neurosurgical Institute, Wuxi School of Medicine, Jiangnan University, 214002 Wuxi, Jiangsu, China
Abstract
Phagocytosis is the process by which certain cells or organelles internalise foreign substances by engulfing them and then digesting or disposing of them. Microglia are the main resident phagocytic cells in the brain. It is generally believed that microglia/macrophages play a role in guiding the brain's repair and functional recovery processes. However, the resident and invading immune cells of the central nervous system can also exacerbate tissue damage by stimulating inflammation and engulfing viable neurons. The functional consequences of microglial phagocytosis remain largely unexplored. Overall, phagocytosis is considered a beneficial phenomenon in acute brain injury because it eliminates dead cells and induces an anti-inflammatory response. Osteopontin (OPN) is a phosphorylated glycoprotein induced by injury in various tissues, including brain tissue. In acute brain injuries such as hemorrhagic stroke and ischemic stroke, OPN is generally believed to have anti-inflammatory effects. OPN can promote the reconstruction of the blood-brain barrier and up-regulate the scavenger receptor CD36. But in chronic diseases such as Alzheimer's disease (AD) and amyotrophic lateral sclerosis (ALS), OPN can cause microglia to engulf neurons and worsen disease progression. We explored the role of OPN in promoting microglial phagocytosis in nervous system disorders.
Keywords
- OPN
- microglia
- phagocytosis
- SPP1
- CD36
In many studies, Osteopontin (OPN) has been shown to enhance the phagocytic ability of
microglial cells [1, 2]. Phagocytosis, defined as the cellular uptake of particles
(
Macrophages consist of resident and migratory cells. Motile macrophages are widely distributed in connective tissue; referred to as microglia in regard to the central nervous system. In the past few decades, the secretory properties and chemotaxis of microglia have been well studied, whereas the phagocytic activity of microglia has received relatively little attention [3].
OPN is a pleiotropic protein encoded by the secreted phosphoprotein 1 (SPP1) gene. It is present in various cells, tissues, and body fluids and has been identified as a multifunctional protein with diverse roles. Upregulation of SPP1 gene expression is often associated with events such as infection, hyper-reaction, autoimmunity, and tissue damage [5]. Most studies of OPN have focused on macrophages in the peripheral immune system [4, 5], but the role of OPN in microglia in the central nervous system has not been fully explored [6]. This review will examine the role of OPN in nervous system diseases, specifically focusing on the effect of OPN on the phagocytic function of microglia.
As of the time this review was written, the research on the involvement of OPN in the phagocytic function of microglia was limited. Nonetheless, it is imperative to conduct further exploration to unravel the potential implications of OPN in central nervous system diseases.
OPN is a highly phosphorylated glycoprotein composed of 314 amino acids with abundant aspartic acid, serine and glutamic acid residues and molecular weights ranging from 44 to 75 kDa [6]. Alternative splicing of OPN precursor RNA produces at least five splicing variants (OPN-SV); OPN-a; OPN-b; OPN-c; OPN-4; and OPN-5. These isoforms retain some of the same domains, including Arg-Gly-Asp (RGD), which binds to the integrin site, the Ser-Val-Val-Tyr-Gly-Leu-Arg (SVVYGLR) domain, which binds to calcium, and the heparin-binding domain for CD44 binding [7]. Additionally, OPN is cleaved by specific matrix metalloproteases and thrombin to form N-terminal and C-terminal peptides. This cleavage produces active fragments and novel receptor binding sites, indicating that hydrolysis of OPN is an important mechanism for regulating OPN function. The recruitment of inflammatory cells by OPN is associated with the RGD domain. The manner in which OPN exerts its effects on immune cells through integrin receptors and intracellular pathways has not been fully elucidated [8]. In a study by Raineri et al. [9], Inducible T cell co-stimulator ligand (ICOSL) served as the receptor for OPN, and their interaction promoted endothelial cell growth. Additionally, macrophages are also known to express ICOSL, suggesting a potential role for this interaction in the phagocytic process [10]. Mature OPN is typically secreted from cells via an N-terminal signal sequence. During this secretion, nascent peptides undergo significant post-translational modifications, including sulfation, phosphorylation, transglutaminase binding, glycosylation, and removal of the signal peptide. The importance of these post-translational modifications for the various functions of OPN has not been fully explored [11]. Translation of an OPN mRNA with a different start codon, results in the generation of two OPN isoforms. The shorter isoform, iOPN, remains within the cell and lacks the N-terminus, and the longer isoform, sOPN, is secreted by vesicles [12]. These processes may represent potential control over the various functions of OPN.
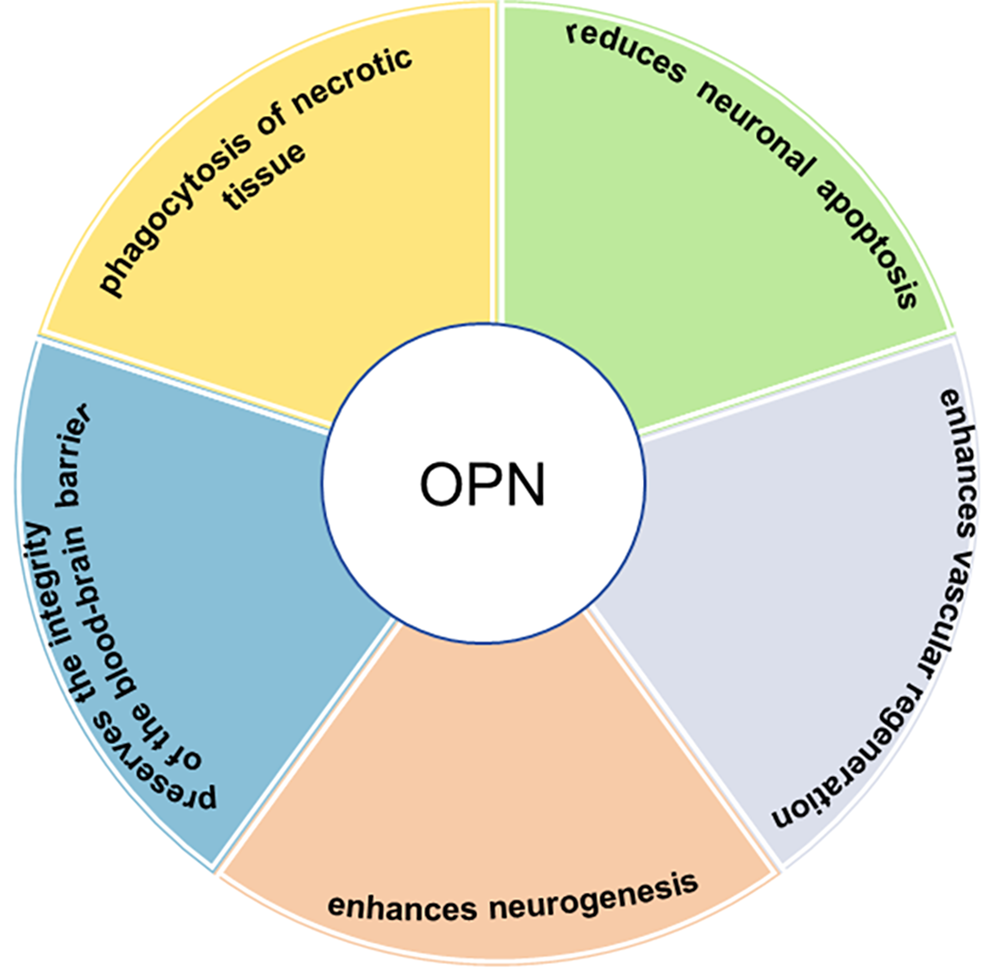
OPN can be produced by a variety of cells, including immune cells (e.g., macrophages, B cells, natural killer cells, dendritic cells (DCs), and T cells), bone cells, epithelial cells, nerve cells, endothelial cells, fibroblasts, and smooth muscle cells [13]. The regulation of OPN expression is highly precise, occurring in a spatiotemporal and cell-specific manner, and is influenced by environmental factors, age, and the specific brain region. This degree of control significantly affects the immune cells’ response to injury [14]. OPN plays multiple roles in the reconstruction of the blood-brain barrier, apoptosis, neuroinflammation, proliferation, chemotaxis, differentiation, and survival of various cells [15]. In vitro, OPN can influence microglia function by transforming proinflammatory microglia into an anti-inflammatory state, indicating a shift from M1 to M2 [16]. OPN promotes the phagocytosis of necrotic tissue, reduces neuronal apoptosis, preserves the integrity of the blood-brain barrier, enhances neurogenesis and vascular regeneration, and thereby facilitates tissue repair [17] (Fig. 1). There is increasing evidence that OPN plays a significant role in neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and multiple sclerosis (MS), as well as in acute brain injuries including traumatic brain injury (TBI), stroke, and hypoxic-ischemic brain injury [17].
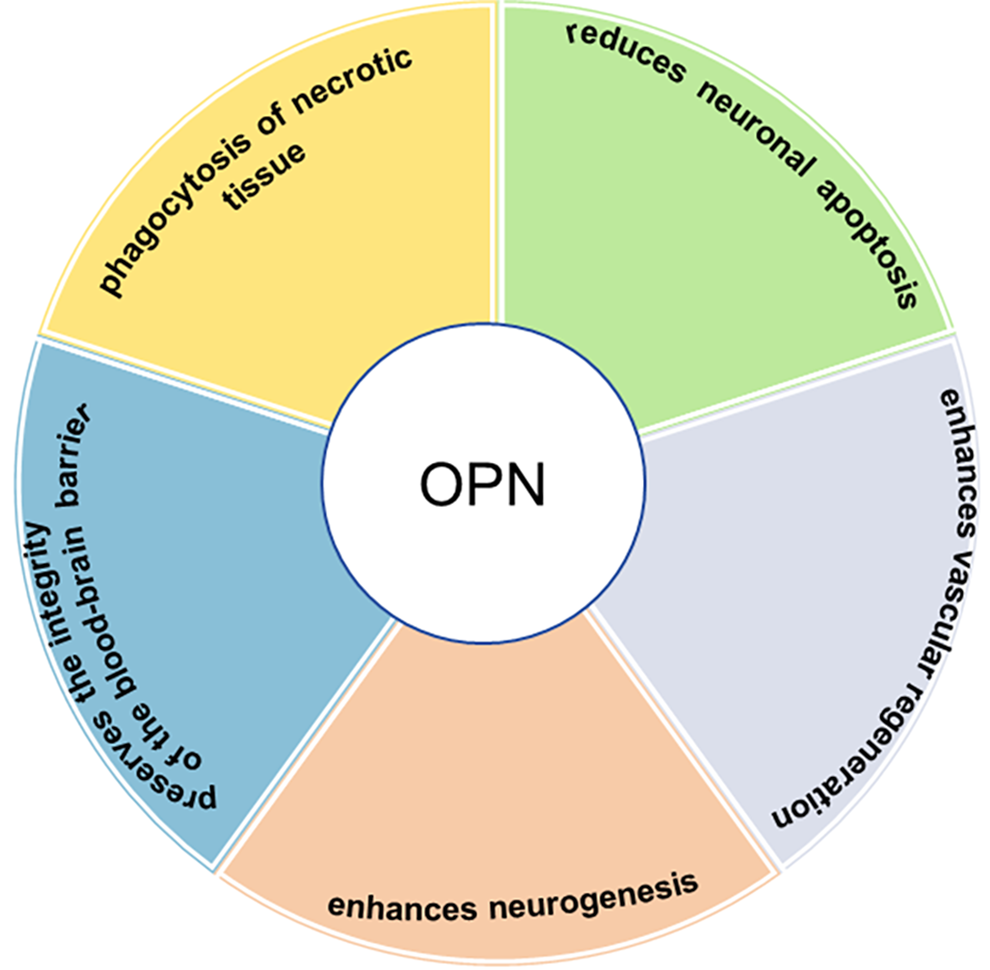 Fig. 1.
Fig. 1.
OPN has a protective effect on the nervous system. OPN promotes the phagocytosis of necrotic tissue, reduces neuronal apoptosis, preserves the integrity of the blood-brain barrier, enhances neurogenesis and vascular regeneration, thereby facilitating tissue repair. OPN, Osteopontin.
The regulation of microglial phagocytosis by OPN in vitro remains a topic of controversy. According to Christensen et al. [1], microglia treated with 1 nM OPN showed nearly twice the uptake of beads than did controls, with a significant increase in phagocytic activity, stimulation of microglial proliferation, and inhibition of superoxide production. However, in experiments by Rabenstein et al. [16], treatment with OPN at either 6.25 µg/mL or 12.5 µg/mL for 24 hours did not produce a significant difference on the cytopathic activity of microglia from that of untreated control cells. Many studies lean toward the idea that OPN can indeed regulate microglial cell phagocytosis in vitro [5, 18].
On a global scale, stroke, particularly ischemic stroke, is a significant cause of morbidity and mortality within the nervous system. There is increasing evidence suggesting that the immune system plays a complex role in the pathophysiology of stroke [19]. Schilling et al. [20] demonstrated in vivo that both resident microglia and hematogenous macrophages contribute to debris clearance after cerebral infarction, with microglia playing a more pivotal role than macrophages. When activated, microglia produce inflammatory mediators and often engulf dead or even normal neurons, leading to acute aseptic inflammation that induces additional neuronal damage. Microglia play a crucial role in neural interactions with the immune system during post-infarction recovery [21].
The importance of microglial phagocytic activity in ischemic brain damage has been emphasized in research [8]. The phagocytosis of dead neurons by microglia is crucial for promoting axonal regeneration and facilitating the recovery of the microenvironment [22]. Shin et al. [23], suggested that OPN protein accumulated on the surface of dead neurons in cerebral infarctions. These fragments were then encapsulated by OPN and ingested by microglia. Their experiments found that 4 weeks after ischemia, only some OPN-labeled fragments remained scattered in the infarct core, whereas most of the remaining fragments were ingested by microglia. Dashdulam et al. [24], showed that OPN treatment increased the expression of CD36, a marker of the microglial M2 phenotype and a scavenger receptor, indicating that OPN promotes microglial phagocytosis. Similarly, in the study by Davaanyam et al. [25], OPN was found to have the ability to induce phagocytosis in microglia, which is RGD structure-dependent. Inhibition of the extracellular regulated protein kinases (ERK), protein kinase B (AKT), and focal adhesion kinase (FAK) signaling pathways significantly inhibits the phagocytic induction of OPN. Further experiments are required to elucidate the mechanisms involved in the phagocytic induction of OPN. Currently, it is believed that OPN can promote microglial phagocytosis and protect the nervous system. The role of OPN in acute inflammation and nerve repair in ischemic brain injury by modulating microglial phagocytosis warrants further investigation.
The incidence of cerebral hemorrhage is higher in men, and the disease is more prevalent in winter, with the sickness rate increasing with age. From 1985 to 2011, the 30-day mortality rate after cerebral hemorrhage decreased from 40% to 30%, while the 48-hour case fatality rate remained unchanged [26]. The primary injury caused by cerebral hemorrhage is often irreversible. The recovery from secondary cerebral injury stemming from hemolysis is a crucial aspect. Microglia are recruited to the site of cerebral hemorrhage and may assist in the elimination of hematomas by phagocytizing erythrocytes. The key aspect of phagocytosis lies in whether microglia can effectively clear the large number of red blood cells before irreversible hemolytic brain damage occurs [27]. Therefore, the altering of phagocytosis of erythrocytes by microglia represents a feasible target for the development of new therapies. In the experiment reported by Zheng et al. [26] sustained depletion of microglia in the early stages of cerebral hemorrhage led to dysplastic astrocytic scarring, increased neutrophil infiltration, and impaired tissue repair. Replenishment of microglia attenuated those adverse effects, and OPN was found to be both sufficient and necessary for the reloaded microglia to function effectively. In the late stages of cerebral hemorrhage, the overall net effect of astrocytic scarring shifts from “protective” to “detrimental”, but this can be partially reversed by late repopulation of microglia [26]. OPN is essential for the function of microglia in cerebral hemorrhage. But further studies investigating whether OPN can promote phagocytosis in cerebral hemorrhage are needed.
Subarachnoid hemorrhage (SAH), accounting for 5–10% of all strokes worldwide,
can be incidentally discovered in healthy individuals during physical
examinations or manifested through various symptoms. If SAH remains undetected,
it can lead to catastrophic outcomes upon rupture of the vascular lesion, often
resulting in fatality. Currently, there is no effective treatment for the brain
injury caused by SAH [28]. The pathophysiological mechanisms of SAH encompass
early brain injury (EBI) and delayed cerebral ischemia (DCI), with increasing
focus on EBI in recent studies [29]. Within 72 h after SAH onset, a series of
pathological changes are collectively defined as early brain injury (EBI),
including reduced cerebral blood flow, cerebral edema, blood-brain barrier
damage, extravasation of blood components, and accumulation of breakdown
products. EBI is strongly associated with poor prognosis [30]. Microglia are
capable of expressing heme oxygenase and presenting CD36 on the cell surface,
thereby facilitating the phagocytosis of dead neurons and red blood cells, and
consequently promoting the rapid resolution of hemorrhage. Blocking CD36 or heme
oxygenase can result in worsened consequences of SAH [31]. Sarosiek et al.
[32] observed that the Overexpression of OPN can reduce the protein expres- sion of Tumor necrosis factor alpha (TNF-
AD is the most common cause of dementia globally, accounting for approximately
60–80% of all dementia cases and affecting over 50 million people worldwide. AD
is characterized by the accumulation of extracellular amyloid beta (A
Hypoxic-ischemic encephalopathy (HIE) in newborns is one of the leading causes of disability and death in infants. HIE present with conditions such as mental retardation, cerebral palsy, visual impairment, hyperactivity, and epilepsy. In rodents, inflammation is induced within hours after HIE injury, with microglia playing a significant role [37]. During brain development, microglia are responsible for synaptic pruning, which involves the removal of excess neurons and weak synapses, thereby controlling brain connectivity through phagocytosis and playing a crucial role in the postnatal development of brain circuits [38]. Xin et al. [39] found that mesenchymal stem cell extracellular vesicles can reduce neuroinflammation, synaptic damage, and microglial phagocytosis after hypoxic-ischemic injury by inhibiting OPN expression. Hypoxic-ischemic exposure was associated with increased microglial phagocytosis of viable neurons, and mesenchymal stem cell extracellular vesicles were able to inhibit microglial phagocytosis and reduce neuronal loss [39]. Inhibiting OPN expression in neonatal HIE may have beneficial effects by inhibiting microglial phagocytosis. However, due to the limited research in this area, further investigation is necessary to explore this potential therapeutic approach.
PD is a rapidly growing neurodegenerative condition and represents the second
most prevalent neurodegenerative disease after AD. The loss of dopaminergic
neurons in the substantia nigra leads to nigrostriatal insufficiency, with tremor
and bradykinesia being characteristic manifestations of the disease [40]. Another
distinct feature of PD is the often observed accumulation of the neuronal protein
alpha-synuclein (
Amyotrophic Lateral Sclerosis (ALS) is a fatal disorder characterised by the degeneration of upper and lower
motor neurons, resulting in progressive paralysis, respiratory failure, and
ultimately death. It is the most common adult motor neuron disease, with an
incidence rate of 5 cases per 100,000 individuals [45]. In the early stages of
ALS, microglia may impair nearby motor neurons by secreting proinflammatory
cytokines; after clinical onset, they may exert neuroprotective effects through
the phagocytosis of degenerated debris [46]. In a mouse ALS study by Morisaki
et al. [47], genetic ablation of OPN delayed disease onset but
accelerated disease progression. The researchers also observed that mouse OPN
promoted microglia-dependent phagocytosis by OPN [3]. It is also interesting that
they found that the promotion of OPN on microglial engulfment activity was not
affected by anti-CD44 or anti-integrin
MS is a chronic disorder of the central nervous system characterised by demyelination and the loss of axons. It typically manifests between the ages of 20 and 40 and is the most common cause of neurological disability in young adults [48]. The phagocytic ability of microglia is crucial for remyelination, and a decrease in this ability can lead to reduced clearance of inhibitory myelin fragments by microglia. The diminished expression of the scavenger receptor CD36 in senescent microglia is associated with decreased phagocytic activity [49]. Notably, the overexpression of CD36 in cultured microglia has been shown to rescue defects in the phagocytosis of myelin fragments [49]. Davaanyam et al. [25] observed that OPN expression increased the CD36 expression in microglia. Additionally, Dong et al. [50] showed that down-regulation of OPN attenuated demyelination and axon loss in a middle-aged mouse model of MS, wherein oxidised phosphatidylcholine was found to mediate demyelination and later axonal loss [50]. However, in another study, OPN was found to promote myelination and remyelination in vitro [51]. Despite those findings, many findings support the view that OPN may play an adverse role in MS. Consequently, OPN may be a potential drug target for MS treatment in the future by virtue of its impact on regulating microglial phagocytosis.
The occurrence of neurodegenerative diseases is often accompanied by the migration of local macrophages, astrocytes, and microglia, all of which play a role in the clearance of cellular debris. OPN may also be secreted by other cell types, including neurons and astrocytes. OPN may have diverse effects in various diseases. This variability in effects may be associated with its multiple isoforms, significant post-translational modifications, and its regulation of microglial phagocytosis, which is essential for the clearance of cellular debris, foreign substances, and reduction of cytotoxicity induced by harmful agents. However, phagocytosis may also lead to cellular death in living tissues, as evident in previous research that indicates that neurodegenerative diseases and other neuropathologies can result from microglial phagocytosis of stressed neurons. Nearly all central nervous system disorders are accompanied by microglial pathology, along with concurrent homeostatic microglial phenotypes. The involvement of OPN in both promoting tissue damage and participating in repair and regenerative mechanisms highlights its versatile role in complex biological processes that follow injury. Its functions in extracellular matrix remodeling, tissue repair, angiogenesis, cell migration, and proliferation position it as a critical factor in tissue repair and regeneration. The dual roles of OPN suggest that it may play a balancing act in the healing process, facilitating tissue recovery and functional restoration after injury.
Research has demonstrated that OPN can shift microglia from the proinflammatory M1 state to the anti-inflammatory M2 state, indicating the significant role of OPN in regulating disease-associated microglial status, which is important for the treatment of nervous system diseases. Further investigations focusing on the different isoforms of OPN, cleaved fragments, and post-translational modifications are necessary to understand why OPN plays varied roles in different diseases. Nonetheless, it is undeniable that OPN can regulate microglial phagocytosis and plays a crucial role in the pathogenesis of various nervous system diseases. Understanding the mechanisms by which OPN induces microglial phagocytosis could represent a crucial approach for modulating the inflammatory microenvironment in the brain. However, the precise mechanism by which OPN induces microglial phagocytosis remains unclear and controversial, warranting further research. Nevertheless, modulating microglial phagocytosis via OPN may constitute a potential therapeutic strategy for nervous system diseases.
XDZ, PPL and ZXT mainly conceptualized the notion of this review manuscript. PPL and ZXT revised the manuscript and contributed to literature review. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.