
 , Danyang Chen 4,*
, Danyang Chen 4,*1 School of Sports Medicine and Rehabilitation, North Sichuan Medical College, 637000 Nanchong, Sichuan, China
2 College of Sports Medicine, Wuhan Sports University, 430079 Wuhan, Hubei, China
3 School of Medicine, Taizhou University, 318000 Taizhou, Zhejiang, China
4 Rehabilitation Medical Center, Taizhou Hospital of Zhejiang Province Affiliated to Wenzhou Medical University, 317000 Taizhou, Zhejiang, China
†These authors contributed equally.
Abstract
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by the
onset of symptoms, typically occurring later in life, and significant deficits in
cognitive functions including learning, memory, speech, and behavior. Ongoing
research endeavors seek to explore methods for preventing and treating AD, as
well as delving into the molecular mechanisms underlying existing and novel
therapeutic approaches encompassing exercise, diet, and drug regimens for
individuals with AD or those at risk of developing AD. Among these interventions,
dietary interventions have garnered increasing attention due to their potential
in addressing the disease. Eating is among the most fundamental of human daily
activities, and controlled dietary practices, such as fasting, have gained
prominence as essential clinical methods for disease prevention and treatment.
Research findings indicate that fasting holds promise in effectively alleviating
and improving the cognitive decline associated with age or as consequence of
disease. The clinical efficacy of fasting in addressing AD and related disorders
might be grounded in its influence on various molecular mechanisms, including
neuroinflammation, glial cell activation, insulin resistance, autophagy
regulation, nerve regeneration, the gut microbiome, and accumulations of
amyloid-
Keywords
- fasting
- Alzheimer's disease
- neuroinflammation
- insulin resistance
- gut microbiome
- brain-derived neurotrophic factor (BDNF)
- A
- tau protein
Alzheimer’s disease (AD), ranked as the fifth-leading global cause of mortality,
represents a neurological disorder with a growing incidence among the elderly.
Its prevalence has more than doubled since 1990, reached 43.8 million in 2016,
and is expected to exceed 152 million by 2050 [1]. While advancements in science
and technology have significantly extended human life expectancy, they have also
contributed to the growing incidence of AD. The strategies for preventing and
treating AD, as well as many other diseases, involve a combination of prescribed
medications, exercise, and dietary regimens. Among these, a class of drugs known
as anti-amyloid-
In 1907, Alois Alzheimer made a significant observation regarding a 51-year-old
woman who exhibited memory impairments and severe difficulties in reading and
pronunciation. Upon examining brain tissue from this individual post-mortem,
Alzheimer conducted silver staining and observed the presence of neural plaques
and neurofibrils when examining the tissue under a microscope. These distinctive
features later became the defining pathological characteristics of the newly
identified disease, which was subsequently named AD [15]. AD is characterized by
a progressive decline in cognitive function, including learning, memory,
emotional regulation, and behavior, and the potential to lead to fatal outcomes
[16]. Research has identified two main forms of AD; early-onset and late-onset,
each associated with distinct genetic profiles. Early-onset AD is associated with
mutations in the genes governing presenilin (PS)1/2 and amyloid precursor protein
(APP) expression, while late-onset AD is associated with the presence of
apolipoprotein E4. Regardless of the onset type, AD is characterized by
A
More than a century has passed since the initial discovery of AD, yet its
specific pathogenesis remains controversial. Currently, there are several
prevailing theories, namely the A
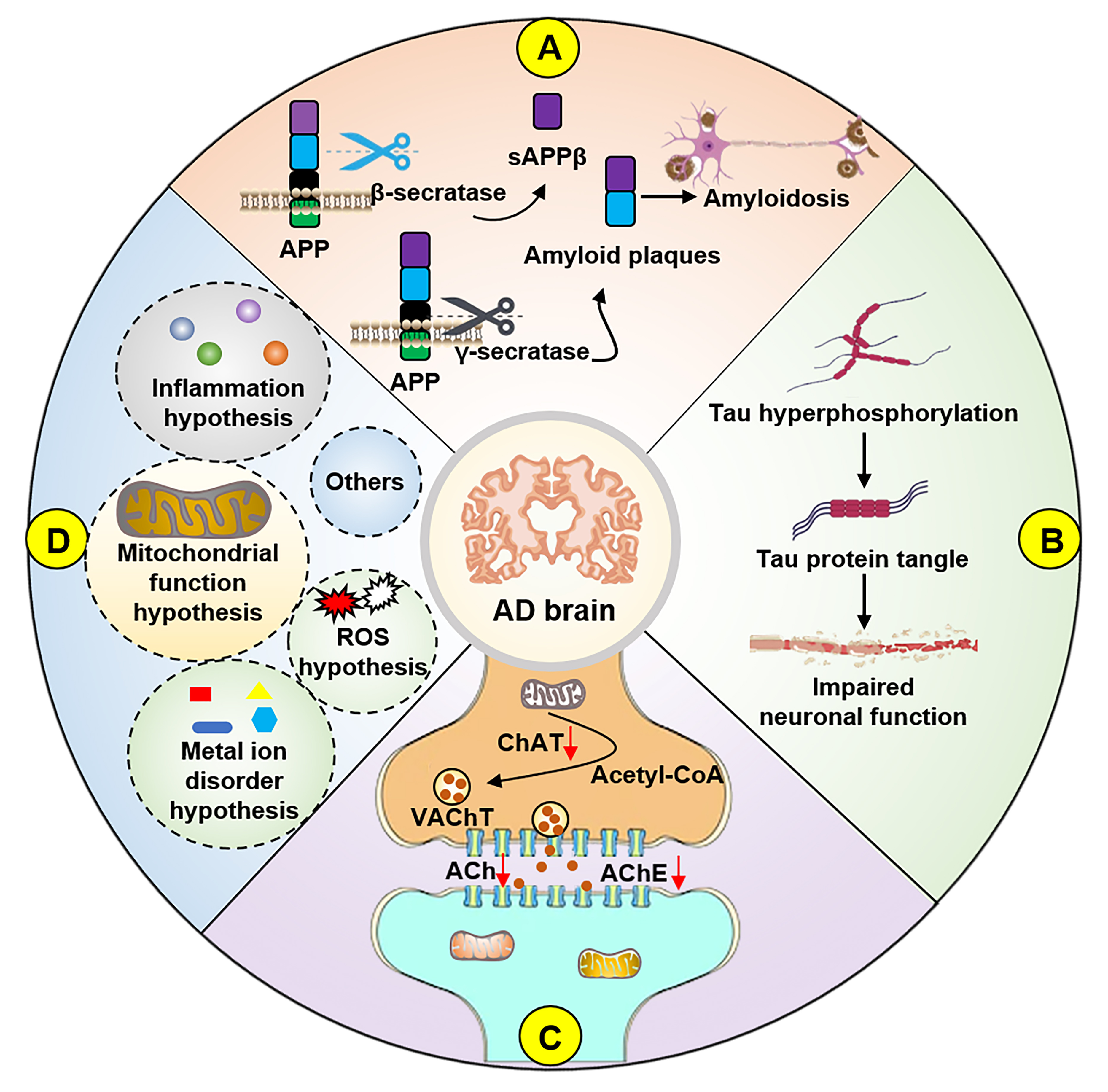
The cholinergic hypothesis posits that AD is linked to a reduction in cholinergic neurons and changes in the activity of acetylcholine transferase and cholinesterase. These pathological changes result in decreased acetylcholine levels, a neurotransmitter crucial for synaptic transmission, learning, memory, and other advanced cognitive functions. The tau protein abnormal phosphorylation hypothesis proposes that the hyperphosphorylation of tau protein leads to the formation of neuronal tangles. Tau protein plays a vital role in stabilizing microtubules within neurons. Dysregulation of protease activity, including protein kinases and protein phosphatases, causes tau to malfunction, resulting in tangle formation [21] and impaired axonal transport. The resulting loss of communication and neuronal death might cause and aggravate AD development.
In addition to these theories, a deeper understanding of AD has given rise to other hypotheses, including the neuroinflammation hypothesis [22], metal ion disorder hypothesis [23], mitochondrial cascade hypothesis [24], and oxidative stress hypothesis (Fig. 1) [25]. The presence of multiple theories suggests that the pathogenesis of AD is multifaceted and cannot be attributed to a single mechanism. These theories, while addressing AD symptoms and informing potential therapeutic strategies, might collectively contribute to an intricate mechanism whose dominant pathogenic factor, if one such exists, remains uncertain [26, 27, 28, 29]. Currently, the clinical diagnosis of AD involves a battery of behavioral, learning, and memory assessments, along with evaluations of blood and cerebrospinal fluid biomarkers, brief mental state examinations, brain magnetic resonance imaging, and fluorodeoxyglucose positron emission tomography [30]. As mentioned earlier, prevention and treatment strategies for AD encompass a combination of exercise, drug, and dietary interventions.
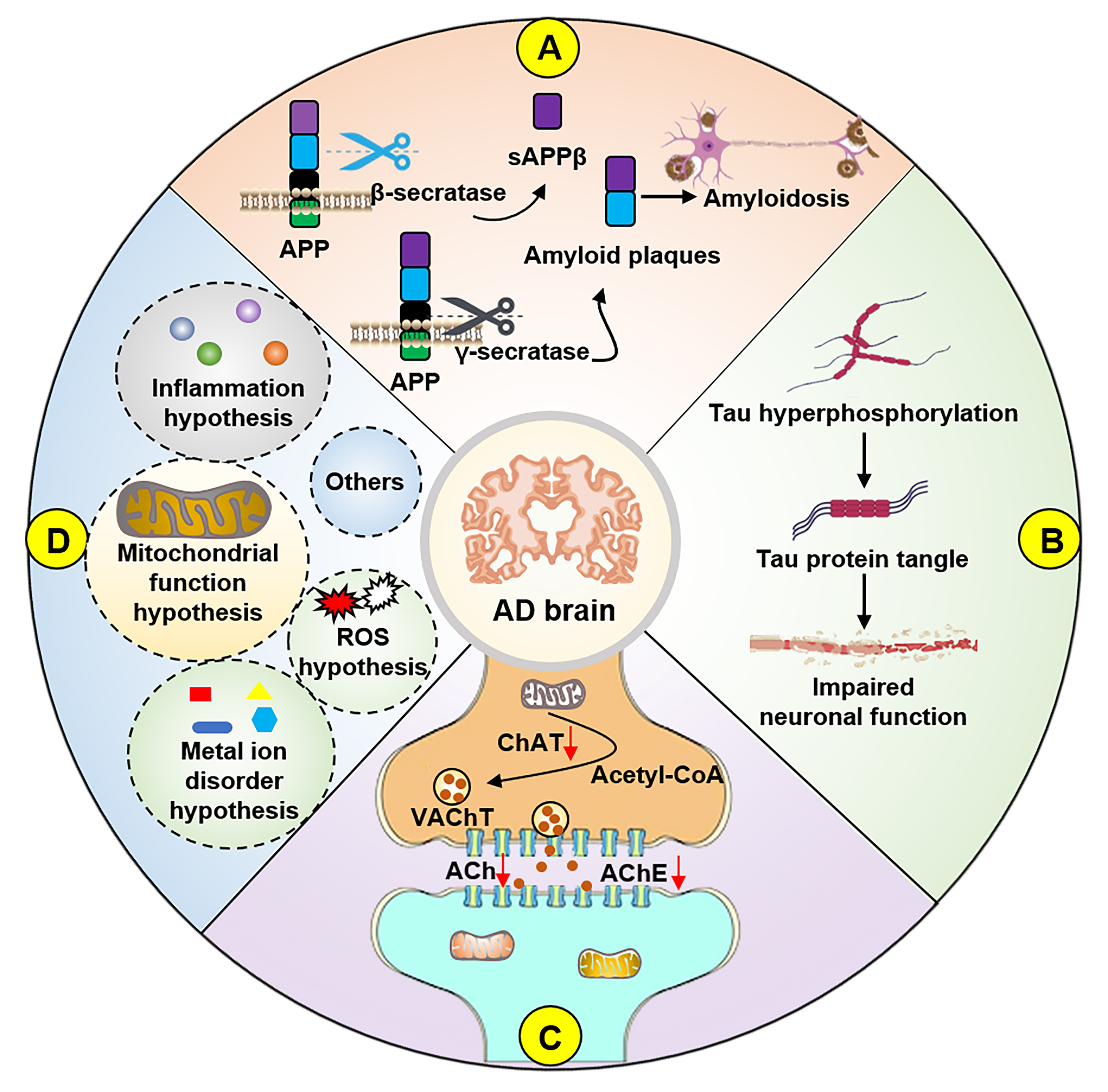 Fig. 1.
Fig. 1.Hypotheses for the pathogenesis of AD. (A) APP is cleaved by
Fasting represents a type of calorie restriction involving the subjective reduction or temporary suspension of specific or partial food intake. It has been demonstrated to yield favorable health outcomes when incorporated into routine clinical care and strategies for disease prevention. Additionally, fasting simulates the patterns of reduced material and energy intake that humans encountered during evolution. Importantly, scientific evidence, both at the molecular and clinical levels, supports fasting as a catalyst for improving health [31]. Across diverse cultures and influenced by various religious beliefs and lifestyle choices, fasting has evolved into distinct established modes. These include time-restricted eating, which involves limiting daily eating to a 6–8 h window (often achieved by skipping breakfast or dinner) with meal intervals of less than 8 h. Alternate-day fasting entails fasting every other day, with fasting periods exceeding 36 h. Periodic fasting involves maintaining minimal energy intake on two consecutive or intermittent days per week. Long-term fasting extends for more extended durations, surpassing 2 days and even weeks with reduced caloric intake. Additionally, fasting mimetics, such as metformin, spermidine, and rapamycin, replicate the effects of fasting [31, 32, 33, 34]. In large number of healthy and overweight individuals, long-term fasting has exhibited positive outcomes, including a longer lifespan, enhanced quality of life, weight management, and potential benefits in countering cognitive decline [35, 36, 37].
Mild cognitive impairment (MCI) is a state of progressive memory and cognitive function decline that does not significantly effect daily life functioning and does not meet the diagnostic criteria for dementia. Given the heightened prevalence of AD within the MCI population, it is often considered a pre-AD manifestation. Timely implementation of fasting can be a beneficial intervention for preventing cognitive decline and the onset of AD [5]. This relationship might be closely associated with the regulation of material energy intake. An associated study revealed that the proportion of MCI cases in the elderly population with high nutritional status was significantly higher than those with good nutritional status [38]. Numerous human studies have demonstrated the positive impact of intermittent dietary interventions on cognitive performance in older adults with symptoms of MCI [39, 40]. Furthermore, fasting has proven effective in improving cognitive impairment associated with various conditions, including radiotherapy [41], cognitive impairment caused by chronic cerebral hypoperfusion [42], cognitive dysfunction caused by hyperglycemia [43], and cognitive impairment caused by traumatic brain injury [44]. These findings collectively underscore the favorable role of dietary intervention in the prevention and amelioration of MCI.
As the condition of individuals with MCI continues to decline, there is a substantial likelihood of progression to AD. A study involving older adults who practiced time-restricted fasting revealed a significant positive association between caloric restriction and cognitive ability [45]. Additionally, it was observed that daily extended fasting had a mitigating effect on cognitive impairment resulting from excessive carbohydrate consumption [46]. Notably, in 17-month-old mouse models of AD, fasting demonstrated improvements in age-related behavioral deficits [47]. In oestrogen-deficient rat models of AD, fasting proved effective in ameliorating memory impairments and mitigating symptoms related to metabolic disorders [14], indicating that the effects of fasting might vary between sexes. Despite counterarguments against the therapeutic use of fasting [48], the aforementioned body of evidence supporting the therapeutic advantages of fasting suggest its considerable potential in preventing and treating AD.
Several studies have indicated that fasting helps alleviate symptoms of
neurodegenerative diseases, including AD, Parkinson’s disease, epilepsy, and
multiple sclerosis; possibly by regulating neuroinflammation, insulin resistance,
autophagy, and the gut microbiome [49]. Experiments involving individuals with
MCI, AD, and healthy individuals have discovered that fasting might improve
cognitive function by influencing processes such as the synthesis and degradation
of ketone bodies, the gluconeogenesis pathway, and the up-regulation of Homer
protein homolog 1 protein expression [4, 40]. In the context of AD models,
fasting has been found to reduce inflammation, mitigate insulin resistance,
regulate intestinal microbes, and lower Tau protein phosphorylation. It can also
reduce A
Currently, increasing research conducting clinical trials involving fasting interventions for patients with AD, including in-depth exploration of the effects of fasting through low-protein diets, time-restricted diets, and the use of rosiglitazone XR (which might mimic the effects of fasting). Furthermore, the potential clinical benefits of dietary restrictions have prompted new investigations aimed at scientifically quantifying fasting strategies according to individual physical conditions, thus improving their clinical adaptability.
Fasting, as an economical, convenient, and non-invasive intervention, holds the
potential for widespread application in clinical settings to delay the onset of
AD [51]. The molecular mechanisms underlying the therapeutic benefits of fasting
in patients with AD encompass factors such as neuroinflammation, insulin
resistance, mitochondrial integrity, gut microbiome composition, nerve
regeneration, autophagy, and the regulation of A
Excessive inflammation within the body is regarded as an indicator of disease.
While a certain level of inflammation is typically maintained in a healthy state
without causing harm, an increasing body of evidence implicates both peripheral
and central acute and chronic inflammation in the onset and progression of AD
[52, 53]. Fasting has demonstrated its capacity to mitigate inflammation, thereby
serving as a preventive and therapeutic approach to disease management [54].
Inflammation has emerged as a primary target for treating neurodegenerative
diseases. AD, in particular, results in a significant increase in
neuroinflammation, and efforts to alleviate this excessive inflammation are
anticipated to yield improvements in disease symptoms [55]. Intermittent fasting
has exhibited a significant reduction in phosphorylated (p)-nuclear factor kappa
B (NF-
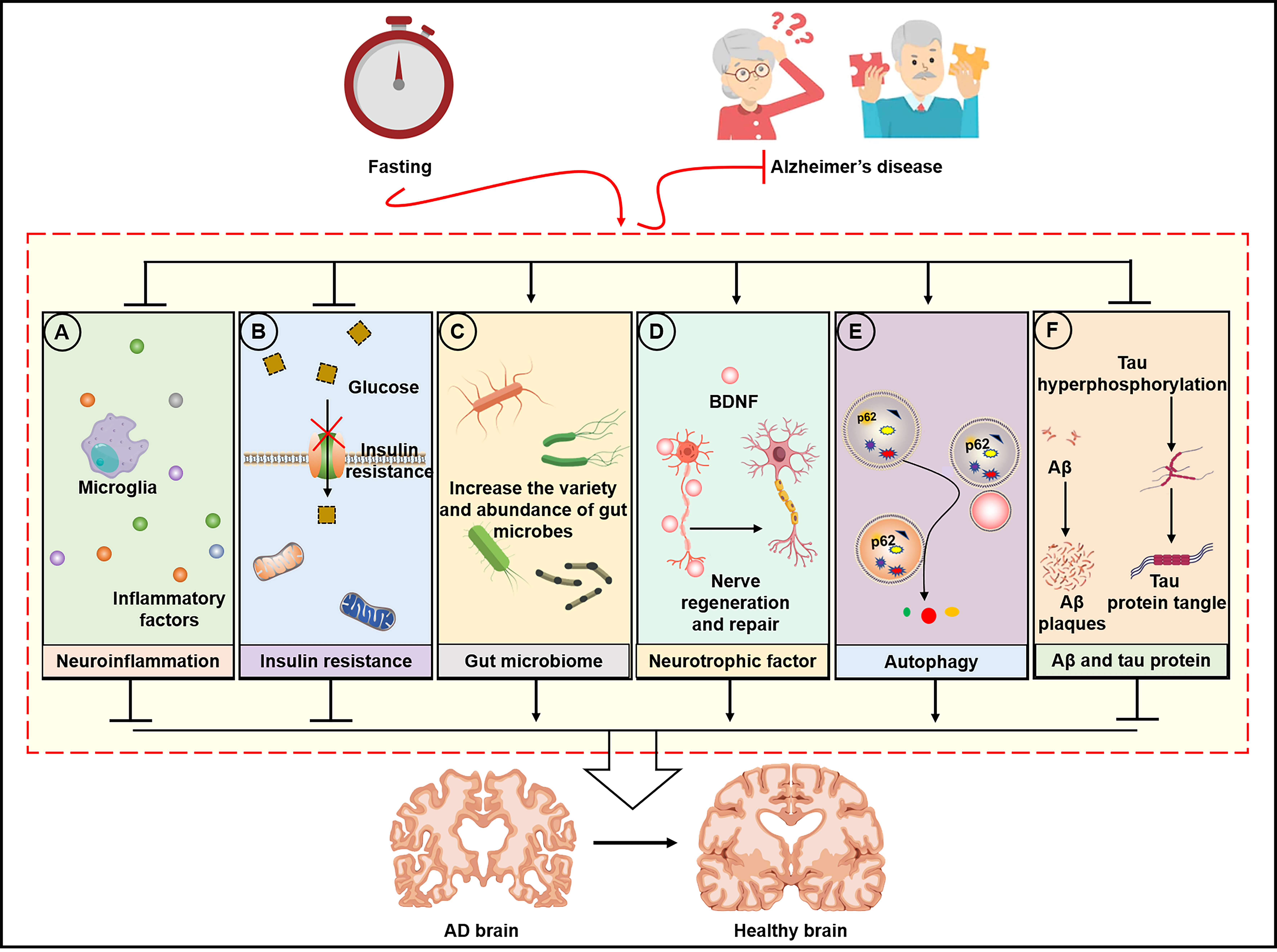
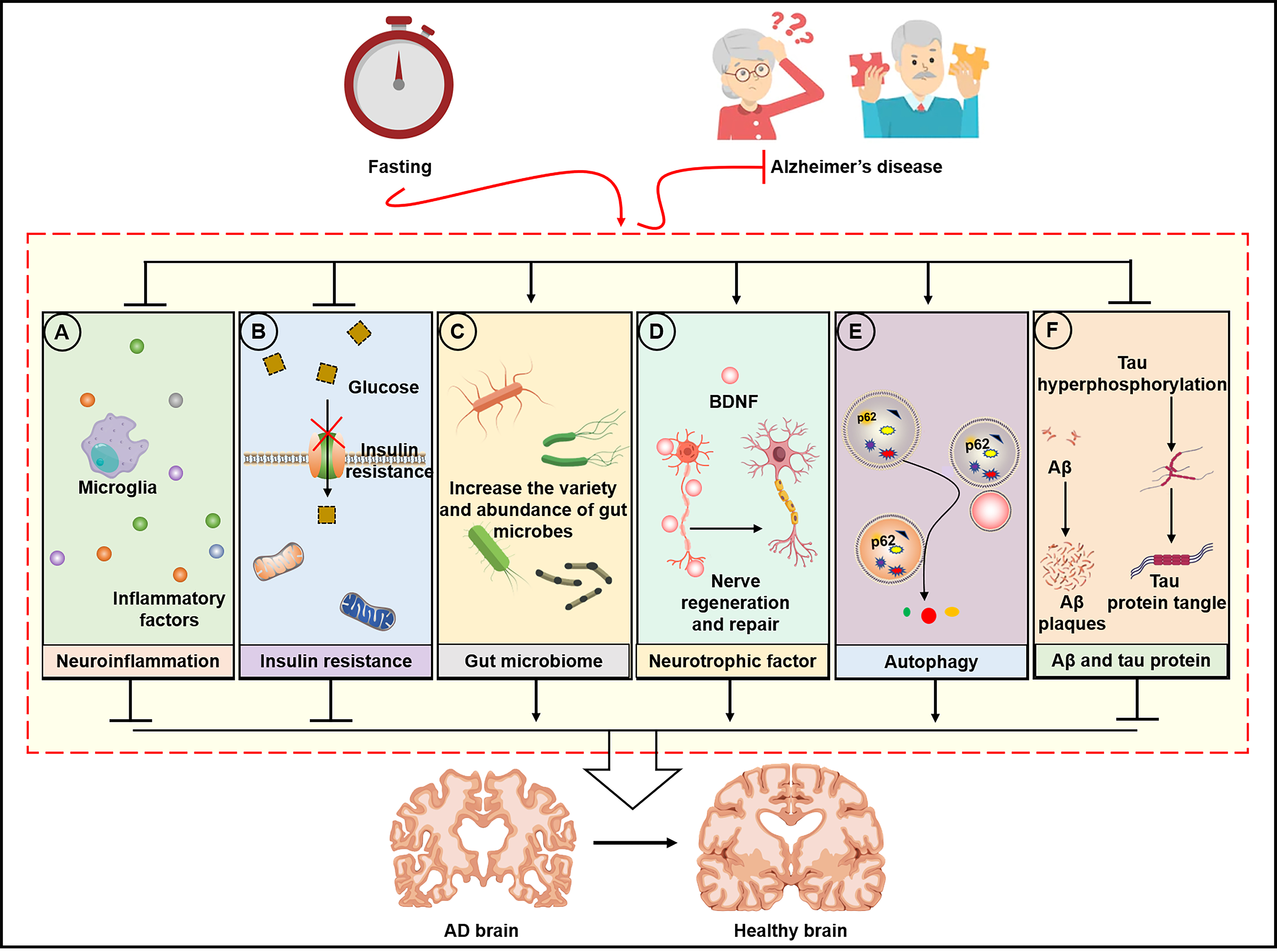 Fig. 2.
Fig. 2.Molecular mechanism of fasting intervention in AD. (A) Fasting
improves neuroinflammation by reducing inflammatory factors, (B) reduces insulin
resistance and increases glucose transport in neurons, (C) regulates gut microbes
to improve the “gut-brain” axis, (D) improves neurotrophic factors to induce
nerve regeneration and repair, (E) activates autophagy, and (F) reduces
A
AD is sometimes referred to as “type 3 diabetes” because of its frequent
co-occurrence with diabetes. This strong association is partly attributed to
insulin resistance in the brain [59, 60]. AD often coincides with a decrease in
the body’s insulin resistance and a subsequent increase in blood sugar, which
contribute to the onset and development of AD [61]. Fasting interventions have
demonstrated their ability to ameliorate insulin resistance [62]. In an AD model
involving the injection of A
The microorganisms that parasitize the intestinal tract form the gut flora, a
vital contributor to human health. The medical and healthcare fields increasingly
acknowledge the significance of regulating the diversity and abundance of the gut
microbiome as an effective means to prevent and control diseases. A 13-month
longitudinal study investigating the effects of time-restricted feeding and
dietary macronutrient regulation on the cognitive abilities of 8-month-old mice
revealed that differences in gut microbiome diversity and composition were
consistent with variations in dietary intervention, with time-restricted feeding
demonstrating the most substantial improvement in cognitive function [66]. These
results highlight the influence of the gut microbiome on brain function,
indicating that the gut microbiome’s health plays a crucial role in the
progression of neurodegenerative diseases. This association is supported by
evidence showing that fasting-mediated regulation of the gut microbiome can
mitigate cognitive impairment in a murine model of AD [67]. Moreover,
administering 200 mL probiotic supplements containing Lactobacillus
acidophilus, Lactobacillus casei, Bifidobacterium, and
Lactobacillus fermentum to older adults with AD for 12 weeks was found
to elevate plasma malondialdehyde and high-sensitivity C-reactive protein levels
while enhancing
AD is associated with structural and functional abnormalities in neuronal
synapses [70], along with reduced neurotrophic signaling that exacerbates AD
progression [71]. Elevating BDNF has been shown to mitigate neuronal loss and
promote nerve regeneration and repair [72, 73]. While strategies like sleep and
exposure to hypoxia can up-regulate BDNF, they come with inherent risks when
applied to older patients [74, 75]. Exercise is another approach that elevates
BDNF, presenting significant potential for patients with AD [76, 77]. However,
the symptoms and age of onset associated with AD might render exercise
impractical and possibly dangerous. Fasting positively affects BDNF levels,
offering a more viable alternative without the limitations of the a forementioned
interventions [78, 79, 80]. In the 3xTg AD mouse model, intermittent fasting promoted
the differentiation and maturation of hippocampal neurons by activating glycogen
synthase kinase 3
Autophagy is the cellular process through which ageing and damaged organelles,
as well as misfolded proteins, can be eliminated. This process involves
encapsulating these components in a phospholipid bilayer and then binding the
vesicle to lysosomes, where they are broken down into reusable amino acids,
glucose, and other essential substances [84]. Given the implication of abnormal
autophagy levels in the pathophysiology of AD, the regulation of autophagy has
emerged as a potential target in AD treatment [85]. Both exercise and fasting,
even when implemented in the short term, have been shown to induce the regulation
of autophagy in neurons [34]. In PDAPP-J20 transgenic mice, a 6-week fasting
regimen yielded a neuroprotective effect by significantly increasing the number
of LC3-positive glial cells and reducing intracellular A
Basic experiments and the development of pharmacotherapies for AD have strongly
implicated A
| Model | Fasting method | Tissue, sample, or evaluation | Molecular mechanism | Refs |
| AD model (express human APOE4 [E4FAD] and triple-transgenic [3xTg] mice) | The fasting-mimicking diet (FMD); 4 days of FMD and 10 days of refeeding/time, 5 times in total | Hippocampus | phosphorylated (p)-tau/tau, microglia level and activation, AT8 |
[7] |
| AD model (injection of amyloid- |
3 h feeding and 16 h fasting/day, 7 weeks in total | Serum and joint histology | Tumour necrosis factor (TNF)- |
[58] |
| AD model (5xFAD mice) | every other day, 4 months in total | Cortex | glutamic acid decarboxylase 67↓; TNF- |
[59] |
| AD model (injection of A |
3 h feeding and 16 h fasting/day, 4 weeks in total | Hippocampus | A |
[14] |
| AD model (injection of A |
Intermittent fasting diet, 8 weeks | Faecal | p-tau/tau, TNF- |
[68] |
| Ageing mice (senescence-accelerated mouse-prone 8) | Alternate day fasting, 8 weeks | Brain | sirtuin 1, brain-derived neurotrophic factor (BDNF), heat shock protein 70↑; p-glycogen synthase kinase 3 |
[56] |
| Ageing mice (Fisher 344x Brown Norway F1, 22 months) | 51 kCal standard or ketogenic diet once daily for 8 to 21 months | Faecal | Allubaculum, [Eubacterium] ventriosum group↓; Intestinimonas, [Ruminococcus] gauvreauii group, memory↑ | [67] |
| Ageing rats (24 months Wistar rat) | Alternate day fasting, 3 months | Cortex, hippocampus, and hypothalamus; Morris water maze test | Learning and memory function, synaptic and cell adhesion molecule expressions↑; Calcineurin expression, protein carbonyl content ↓ | [92] |
| Ageing human (age over 50 years) | Eating time window less than 10 h, 6 months | Questionnaire evaluation | Cognitive status↑ | [45] |
| Patients with AD | Modified ketogenic diet or their usual diet supplemented with low-fat healthy-eating guidelines and optional recipes, 12 weeks | Questionnaire evaluation | Alzheimer’s Disease Cooperative Study – Activities of Daily Living Scale, Quality of Life–AD↑ | [93] |
| AD model (Amyloid-beta precursor protein [APP] |
Alternate day fasting, 1, 4, or 12 months | Elevated plus maze testing | Spatial learning and memory↑ | [94] |
| AD model (APP/presenilin 1 [PS1]) | Alternate day fasting, 5 months | Cortex | lipoprotein lipase messenger ribonucleic acid and protein↑ | [95] |
| AD model (5xFAD mice) | Intermittent fasting diet (every other day), 10–12 weeks | Hippocampus, and faecal | Bacteroidetes, Bacteroidia, A |
[10] |
| AD model (3xTg mice) | Intermittent fasting | Hippocampus | Insulin, adenosine monophosphate-activated protein kinase, and protein kinase A signalling, BDNF, memory↑ | [81] |
| AD model (PDAPP-J20 transgenic mice), and C6 and BV2 cells | Dietary restriction (40% dietary restriction in dietary restriction (DR), 5 days DR followed 9 days feeding, each cycle lasting 2 weeks, 3 cycles in total) | Hippocampus | A |
[87] |
| Green fluorescent protein-LC3 mice, and N2a cells (Human APP Swedish mutation [N2aSwe], treated with butyric acid [5 µmol/L]) | Intermittent fasting for 48 h followed 24 h feeding, 3 weeks; 12, 24, 36, and 48 h | Hippocampus, or N2a cells | Oxidative damage and apoptosis, amyloid precursor protein [APP]↓; LC-3II, lysosome-associated membrane protein 2A, autophagy↑ | [88] |
| AD model (5xFAD mice) | Fasting 48 h | Hippocampus | Macroautophagy↑ | [89] |
| AD model (APP/PS1 mice), and U251 cells | Intermittent fasting diet (every other day), 5 months | Cortex | AQP4-M1/M23, A |
[50] |
| AD model (Tg2576 mice) | Dietary restriction (30% dietary restriction) | Hippocampus | A |
[91] |
While an increasing body of evidence supports the positive role of fasting in promoting overall health, disease prevention, and recovery, the literature is not entirely in agreement. Long-term and intermittent fasting might have adverse consequences, including reduced reproductive capacity and a significant decline in short-term memory and verbal expression [96]. Long-term fasting can also disrupt the body’s energy levels, metabolism, hormonal balance, immunity, and body weight, resulting in deleterious effects such as muscle atrophy and low immunity [97]. For instance, a single night of fasting was observed to impair handgrip strength and reduce muscle mass in hospitalized adult males [98]. Furthermore, the safety of fasting for critically ill patients remains uncertain and requires additional clinical data [99]. Migraineurs might experience more severe symptoms and disruptions to their daily lives during fasting [100], and fasting can reportedly trigger hypoglycemia-induced coma and other adverse symptoms [101]. Although fasting may have positive effects on health maintenance, it may also have adverse consequences.
However, it is essential to recognize that fasting regimens should not come at the expense of compromising one’s health, as they may pose risks for specific groups of individuals. It is important to exercise caution and consider the following groups when contemplating fasting: (1) severely ill patients; (2) individuals with low blood sugar levels; (3) individuals with compromised digestion and absorption; (4) rapidly growing adolescents; (5) pregnant women. These groups exhibit unique physiological conditions, and it remains uncertain whether reduced energy and nutrient intake resulting from fasting could lead to greater damage. Furthermore, it is important to note that fasting methods have only been tested in a limited number of individuals. The suitability of fasting for the elderly population, who are at a higher risk of chronic diseases, might require confirmation through larger-scale studies. Patients in these groups should engage in a systematic assessment of their health status, involving nutritionists, physicians, and rehabilitation specialists. A carefully planned approach, including establishing fasting tolerance limits, monitoring nutritional status, assessing adverse reactions, and having emergency self-rescue measures in place, should be implemented step by step to ensure their well-being.
Fasting, a dietary intervention approach characterized by severe regulation and
restriction of food intake, has demonstrated therapeutic potential in preventing
and treating AD. The primary underlying mechanism for these clinical benefits
might involve neuroinflammation, insulin resistance, the gut microbiome, neuronal
nutrient factors, autophagy, A
JS, DC and ZZ designed and constructed this article, ZZ conceptualized the study, ZZ and HZ wrote the manuscript, HZ and XW contributed the design and drawing of the figures, and JS and DC completed final editing and revision of the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We thank Bullet Edits Limited for the linguistic editing and proofreading of the manuscript.
This work was supported by Construction of Sports Rehabilitation Center at North Sichuan Medical College (NO. 22SXFWDF0002).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.