1. Introduction
1.1 Parkinson’s Disease Forms and Relation to -Synuclein
Aggregation
Parkinson’s disease (PD) manifests in two primary forms: familial and sporadic.
The familial variant is linked to genetic mutations, particularly in the
-synuclein (-syn) gene, such as A30P [1],
A53T [2], E46K [3], H50Q [4, 5], and G51D [6], which account for about 15% of all cases [7]. The exact cause of sporadic
PD remains elusive, although both genetic and environmental factors are believed
to play a role. Certain pesticides like rotenone and paraquat [8], as well as
toxins like 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) [9], have been
associated with sporadic PD. Recent population-based studies indicate that air
pollutants generated by urban transportation significantly increase the risk of
disease [10, 11, 12]. Moreover, variations in PD incidence across different
ethnic groups also hint at genetic influences [13]. Recent research underscores
the significance of environmental toxins in sporadic PD development [14]. The
interplay between genetic and environmental factors, along with processes like
neuroinflammation, oxidative stress, and -syn misfolding, likely
contributes to sporadic PD pathogenesis. Addressing -syn aggregation
has emerged as a potential therapeutic strategy, indicating the complex nature of
sporadic PD etiology.
Research efforts in PD rely heavily on understanding its prodromal stages and
identifying accessible markers for staging. The Braak hypothesis, proposed by
Braak et al. [15] in 2006, offers a conceptual framework that outlines
the potential sequence of PD development. According to this hypothesis, PD
pathology may initially emerge in olfactory structures and enteric nerves in the
gut, possibly years or even decades before affecting the substantia nigra pars
compacta (SNpc). Subsequent neurodegeneration in the SNpc causes dopaminergic
(DA) neuron loss, and motor symptoms appear. Research conducted in 2019 by Kim
et al. [16] seems to confirm this theory, and the vagus nerve has been
proposed as a potential conduit of -syn aggregates (-syn AGs)
from the enteric nervous system to the brain. This research also found that
truncal vagotomy prevented the transmission of pathological -syn as
well as behavioral and motor deficits in mice. While the hypothesis has sparked
debate due to the diverse nature of clinical PD presentations and contradictory
pathological findings, it suggests a compelling idea: in some PD cases, the
disease may originate in the peripheral nervous system. Moreover, it implies that
many non-motor symptoms observed in PD patients could be inherent to the
disease’s early development and natural progression.
The onset of PD symptoms is linked to neuropathological changes in basal ganglia
structures (BG) that manifest as motor and non-motor symptoms. Considering the
intricate pathophysiology of PD, two phenotypes are currently distinguished,
known as “brain-first” and “body-first” [17]. These subtypes suggest
contrasting routes of -syn pathology propagation [18, 19]. In the
“body-first” subtype, initial pathology may emerge in the enteric or peripheral
autonomic nervous system before spreading to the medulla oblongata via the vagus
nerve. This ascending pathology affects the pons, leading to rapid eye movement
(REM) sleep behavior disorder (RBD) before involving the SNpc [20]. In the
“brain-first” subtype, pathology may originate in the amygdala or olfactory
bulb and then spread to the brainstem and cortex [21]. A chronic inflammatory
state in the central nervous system (CNS) appears to be a common feature linking
both subtypes of PD. Neuroinflammation is believed to accompany progressive
neurodegeneration triggered by abnormal -syn accumulation in neurons.
Glial cells, particularly microglia and astrocytes, actively participate in
PD-related neuroinflammation, displaying diverse pro- and anti-inflammatory
functions. They contribute to both the spread of neuroinflammation within the CNS
and the protection of DA neurons. Glial cells maintain constant communication
with each other, with neurons, and with peripheral immune cells.
1.2 -Syn in PD
Alpha-synuclein is a leucine-rich, 14 kDa protein encoded by the synuclein alpha
(SNCA) gene, of which the aforementioned variants (A30P, A53T [22], E46K [23], H50Q, and G51D) are associated with
familial PD [7, 24]. In pathological states, -syn may aggregate (which
is equivalent to toxic gain of function and results in neurotoxicity in PD), be
secreted from neurons into the extracellular space, transferred as free protein
or by extracellular vesicles, and detected in the blood and cerebrospinal fluid
(CSF) of PD patients [7, 25]. Increased -syn expression may be a result
of SNCA gene multiplications [26, 27]. Different structural forms of
-syn can occur, including monomeric (which is nontoxic and soluble),
fibrillar, and oligomeric. They are recognized by microglial membrane receptors,
resulting in pro-inflammatory microglial activation. This can indicate that
-syn not only exists as a pathological marker of PD but also
contributes to inflammation [7, 28]. It is thought that, overall, aggregated
forms of -syn are involved in a number of pathologies, e.g.,
neuroinflammation [29], mitochondrial dysfunction [30], and endothelial
degeneration [31, 32]. Monomeric -syn induces microglial phagocytosis
and, conversely, -syn AGs can inhibit these processes [7].
Some strains of -syn are considered to be mediators for spreading
pathological forms of aggregates in the brain, being able to pass through the
blood-brain barrier (BBB) and between different brain cells [33, 34]. The BBB is
a natural barrier formed by astrocytes, pericytes, microglia, and also
metabolically active endothelial cells (ECs), which is connected with junctional
proteins [35] and basement membranes (BMs), creating parenchymal (astroglial) and
endothelial BMs [36].
Aggregated forms of -syn can interact with neuronal membranes, causing
their disruption [37], and reduce microglial phagocytosis by binding to Fc gamma
receptor IIB (FcRIIB). This possibly impairs the clearance of
aggregated molecules, which further exacerbates neuroinflammation and, thus,
neurodegeneration [38]. However, research has also shown that a fibrillar form of
-syn has a higher potential to elevate levels of the pro-inflammatory
cytokines secreted by BV-2 microglial cells compared with other forms of
-syn [39, 40, 41]. This research also indicated that the fibrillar form
of -syn preferentially underwent phagocytosis by these cells. In its
fibrillar forms, -syn can alter autophagy processes, as well as the
functionality of mitochondria in microglial cells [42]. Fibrils of -syn
might also impair communication between ECs and neurons as, in the study of Kuan
et al. [43], pathological forms of -syn in EC-neuron
co-cultures lead to endothelial dysfunction.
Change in -syn structure acts as a damage-associated molecular pattern
(DAMP) that leads to the activation of microglial immune receptors, e.g.,
toll-like receptor 2 (TLR2). -Syn seems to interact with TLR2
receptors, while these, in turn, appear to induce the polarization of
pro-inflammatory phenotype (M1) microglia [28, 44, 45]. However, TLR2 is not the
only receptor in the family that interacts with -syn, resulting in
microglial activation. In 2011, Stefanova et al. [46] showed that TLR4
mediates the phagocytosis of recombinant -syn (via translocation of
nuclear factor kappa-light-chain-enhancer of activated B (NF-B) cells).
Fibrillar -syn can also activate the NF-B pathway in
microglia [47], as well as nod-like receptor protein 3 (NLRP3) inflammasomes in
human microglial cells [48]. NLRP3 can be found in astrocytes and microglia, and
drives the extracellular secretion of pro-inflammatory interleukin-1
(IL-1), leading to exacerbated autophagy and neuronal damage [48, 49].
-Syn can also bind to CD11b integrin, resulting in Rho signaling
pathway activation and, subsequently, to the activation of nicotinamide adenine
dinucleotide phosphate (NADPH) oxidase 2 (NOX2), which is a critical factor for
microglial activation [50]. Overall, pathological misfolded -syn acts
both as an effector and a regulator of glial functionality and is the main factor
contributing to neuronal death in PD. In patients with PD, CNS inflammation is
aggravated by the induction of glial cell activation, pro-inflammatory cytokines,
reactive oxygen species (ROS), and nitric oxide (NO) secretion and release,
leading to ongoing neurodegeneration.
In this article, we explore the immunomodulatory roles of glial cells, with a
focus on their involvement in inflammation progression caused by -syn
accumulation, as well as their potential activation of repair and neuroprotective
mechanisms. In addition, we compile scientific evidence confirming the dual role
played by microglia and astrocytes in the propagation of inflammation within the
CNS in PD. Furthermore, we discuss scientific data highlighting the crucial role
of -syn in the mechanisms involved in recruiting peripheral immune
responses in PD.
2. Role of Astrocytes
The role of astrocytes in the CNS includes anti-inflammatory and neuroprotective
functions [51]. These cells have numerous delicate processes that extend from
around 80% of their cell membrane surface and maintain constant contact with
blood vessels, neuronal synapses, and other glial cells. Two primary types of
astrocytes exist – fibrous and protoplasmic – both of which are present in the
white and gray matter of the spinal cord and brain, though they differ in
morphology [51]. Astrocyte morphology can vary depending on the phenotype they
assume, either pro-inflammatory or anti-inflammatory. Moreover, astrocytes differ
between brain regions, e.g., striatal astrocytes differ in morphology from those
in the hippocampus [52, 53], and their functional capabilities decrease with age
[54]. The differences in functionality include electrophysiological properties,
astrocyte-synapse proximity, and Ca2+ signaling [55]. Such differences could
be a reason that specific brain regions are vulnerable to damage in PD, as
demonstrated by the latest research conducted by Bondi and collaborators [56].
This study confirmed the age- and location-dependent density and morphology of
astrocytes in mouse SNpc. From the perspective of neuronal survival and
maintaining the integrity of the BBB, the involvement of astrocytes in the
release of neurotrophic factors, such as mesencephalic astrocyte-derived
neurotrophic factor (MANF), cerebral dopamine neurotrophic factor (CDNF) [57, 58], brain-derived neurotrophic factor (BDNF), and glial cell-derived
neurotrophic factor (GDNF) [59], is crucial. In response to CNS damage or
inflammation, astrogliosis occurs, resulting in a change of morphological forms
among astrocytes [e.g., radial, fibrous, and protoplasmic] [60]. The neurotoxic
activity of astrocytes, which contributes to the death of neurons and
oligodendrocytes, can also be triggered by classically activated microglial
cells, particularly by IL-1, tumor necrosis factor (TNF), and
complement component 1q (C1q) secretion [61], as well as by the release of
pathological -syn, even though its expression in astrocytes is less
pronounced than in neurons [62].
Astrocytes and microglia are in constant contact, mutually influencing each
other’s functions. Astrocytes can respond to inflammatory signals by regulating
the activation of microglia, but they can also react to inflammation resulting
from microglial activation and adopt neurotoxic rather than neurotrophic
functions. Astrocytes are found in the post-mortem brains of patients
with PD and other neurodegenerative disorders, as well as in -syn
pre-formed fibril (PFF) injected mice [61, 63].
While astrocytes secrete pro-inflammatory factors like chemokines (e.g., C–C
motif chemokine ligand 2 (CCL2), CCL5, C-X-C motif chemokine ligand 1 (CXCL1),
CXCL10, and CXCL12 [64]), it is essential to note that their anti-inflammatory
properties outweigh the pro-inflammatory properties [65]. Astrocytes primarily
support brain homeostasis by upregulating channels, like Kir4.1 (inwardly
rectifying K+ 4.1) [66], and transporters, like glutamate-aspartate
transporter (GLAST), excitatory amino acid transporter 1 (EAAT1) [67], to remove
potassium and glutamate ions from damaged neurons. Additionally, they regulate
oxidative stress by producing glutathione (GSH), which helps counteract the
neurodegeneration induced by ROS [68, 69]. Animal study suggest that transient
GSH depletion in the SNpc triggers an inflammatory reaction with a response from
the astroglia [70]. Thus, the weakened protective role of astrocytes, which may
result from their age-related decline in cell number in the SNpc, could promote
inflammatory processes and neurodegeneration. Astrocytes encircle endothelial
cells directly, secreting factors that boost and maintain the integrity of the
BBB [71] and can promote barrier properties and functionality, even in non-neural ECs [72].
Astrocytes serve as energy suppliers for neurons by acting as the brain’s
glycogen reservoir and providing energy substrates such as lactate and ketone
bodies [73]. Due to the progressive neurodegeneration seen in PD, neurons require
more energy, and the relatively low density of astrocytes in the SNpc makes DA
neurons in this region particularly vulnerable [73]. Astrocytes also demonstrate
neuroprotective properties through their influence on -syn spreading.
Evidence suggests that astrocytes can endocytose neuron-derived -syn
AGs and transport them to lysosomes for degradation, as is seen with microglia
[74, 75]. Astrocytes degrade -syn AGs more effectively than neurons,
probably due to the higher abundance of lysosomes in astrocytes [74].
Apart from their neuroprotective roles, astrocytes activate genes associated with immune
functions in response to the transfer of -syn from neuronal cells [76].
To delve into this phenomenon, Lee et al. (2010) [76] exposed primary
astrocyte cultures to a conditioned medium from -syn-expressing SH-SY5Y
neuronal cells and analyzed gene expression changes via microarray analysis. They
found a significant upregulation in genes associated with pro-inflammatory
cytokines and chemokines upon exposure to extracellular -syn,
indicating the induction of an inflammatory response in astrocytes.
-Syn, which is typically confined within neurons, can be released
through unconventional exocytosis, particularly under stress [77]. This release
is accompanied by an increased presence of -syn AGs outside neuronal
cells. These aggregates, once in the extracellular space, are capable of being
internalized by both neurons and glial cells via endocytosis [78, 79]. Research
conducted by Lee et al. [76] showed that -syn formed inclusion
bodies in astrocytes that take up the protein, indicating that astrocytes could
be involved in the accumulation of -syn released from nearby neurons.
Such aggregation can disrupt the functions of astrocytes and increase microglial
activation (especially in the mid-brain, spinal cord, and brainstem), thereby
contributing to neuroinflammation [80]. Expanding upon these observations, recent
studies have demonstrated the transfer of -syn between neurons
through successive exocytosis and endocytosis events [81, 82]. This transfer process
results in the formation of Lewy body-like structures within recipient neurons,
ultimately leading to their demise [83]. -Syn accumulation inhibits
autophagy by microglia, and, at the same time, intracellular transportation of
-syn is intensified by disrupted autophagic processes [84]. Such
intercellular transmission may serve as a fundamental mechanism driving the
spread of Lewy body pathology during the progression of PD. More than that,
non-neuronal cells can also take part in -syn propagation as astrocytes
can contain -syn inclusions and are possibly able to mediate the
transfer of this protein between the cells of the neurovascular unit (NVU) [85].
Astrocytes exposed to neuron-derived -syn induce the production of the
pro-inflammatory cytokines (IL-1, IL-1, IL-6, tumor necrosis
factor- (TNF-)) and chemokines that can activate microglia
[76, 86, 87, 88, 89]. In addition, astrocytes play a pivotal role in amplifying
neuroinflammation by responding to pro-inflammatory signals, such as
IL-1, TNF-, and C1q, as well as to fragmented mitochondria
from activated microglia [61, 90]. In MPTP-treated monkeys, elevated expression
of interferon (INF)- receptor was observed on astroglia, along with an
increased TNF- immunoreactivity associated with astroglia. This
suggests that overactivation of astroglial cells might have a significant impact
on the advancement of PD [91]. In an inducible mouse model expressing the mutant
A53T -syn variant, specifically in astroglial cells, the primary
observation was the combination of microgliosis and rapidly progressing
paralysis, as well as widespread astrogliosis. Additionally, the overexpression
of the mutant -syn in astroglial cells disrupted the normal functions
of astrocytes. This disruption resulted in compromised integrity of the BBB,
disturbed homeostasis of extracellular glutamate, and ultimately, led to
significant loss of DA neurons in the midbrain and motor neurons in the spinal
cord [80]. Astrocytes derived from induced pluripotent stem cells (iPSCs) of PD
patients and containing -syn AGs exhibit a heightened reactive state
and secrete increased levels of pro-inflammatory cytokines, including IL-6 and
CCL-5, when subjected to inflammatory stimuli [92]. Upon exposure to transmitted
-syn, astrocytes engage in both receptor-mediated endocytosis and
interactions with receptors on the cell membrane. TLR2, recognized as a pattern
recognition receptor, plays a role in the uptake of -syn in astrocytes,
while TLR4 does not participate in this process [88, 93, 94, 95]. Both TLR2 and
TLR4 are implicated in mediating the pro-inflammatory effects of transmitted
-syn. In a separate investigation, a PD mouse model that overexpresses
mutant -syn, exhibited both structural and functional changes in
astroglial mitochondria, along with disrupted secretion of factors crucial for
neuronal differentiation [96]. These findings imply that the buildup of
-syn in astroglial cells may play a role in the onset of PD.
A recent study demonstrated that PD mutant mice that overexpress both human
-syn and transglutaminase 2 (TG2) exhibited increased -syn
aggregation and heightened astroglial activation compared with mice that
overexpress only -syn. This suggests that TG2 may play a significant
role in the accumulation of -syn and the development of PD and related
disorders, offering a novel target for therapeutic interventions [97].
Although astrocytes secrete factors that promote BBB integrity and maintenance,
they also produce factors that can disrupt these. For example, vascular
endothelial growth factor (VEGF), typically known for promoting vascular growth,
impairs BBB integrity under unfavorable conditions. In pathological states,
astrocytes secrete VEGF and act adversely towards endothelial integrity [98].
Moreover, the elevated reactive astrocyte count observed in some diseases is
considered to be associated with BBB disruption [72]. In the human induced pluripotent stem cells (iPSC)-derived BBB co-culture model, BBB dysfunctions may
arise as a result of TNF triggering astrocytes to adopt an inflammatory reactive
state. This occurs, among other mechanisms, through the activation of signal
transducer and activator of transcription 3 (STAT3), which is correlated with
vascular inflammation in post-mortem human tissue [72]. Vascular changes have
been observed in both males and females in the post-mortem substantia nigra (SN)
[99]. Another study by Jackson et al. [100] demonstrated that
astrocyte-derived apolipoprotein E4 (APOE4) is associated with BBB disruption,
leading to BBB leakage and impaired tight junctions. The removal of
astrocyte-derived APOE4 alleviated these phenotypes [100]. In vitro
modeling of the BBB with astrocytes carrying the leucine rich repeat kinase 2
(LRRK2) G2019S mutation showed modified function and morphology of vessels,
altered possibly through mitogen-activated protein kinase kinase 1 and 2
(MEK1/2). Importantly, inhibiting MEK1/2 improved vessel integrity. Overall,
astrocytes carrying this mutation have a pro-inflammatory profile and reduce the
integrity of the BBB [99].
While microglia are traditionally recognized as the primary cells for activating
inflammasomes in the brain, astrocytes can also express and activate these
inflammatory signaling complexes [101, 102, 103]. In PD model mice overexpressing
mutant human A53T -syn, NLRP3 inflammasomes are activated within the
midbrain to produce IL-1, although this process largely relies on the
microglial uptake of -syn [102]. Furthermore, astrocytes engage in
crosstalk with various other cell types, including oligodendrocytes, endothelial
cells, and peripheral immune cells, during neuroinflammatory responses [49, 104].
Overall, astrocytes play a crucial role in maintaining neuronal balance,
mitigating neuroinflammation, and supporting neuroregeneration and neuronal
renewal.
3. Role of Microglia
Microglial cells occur most densely in regions such as the SN, hippocampus, BG
nuclei, and olfactory bulb; the SNpc contains a notably high concentration of
microglia, which makes DA neurons particularly susceptible to degradation [105].
Research conducted by De Biase et al. [106] in 2017 suggests that
microglial cells in the SNpc specifically may be predisposed to malfunctioning in
pathological states and may differ from those within the BG, including in such
features as anatomy, transcriptomes, lysosome content, and membrane properties.
This study also showed that neuronal phenotypes differ between the SNpc and the
ventral tegmental area (VTA), which seems to confirm the specific susceptibility
of neurons in the SNpc to damage [106]. Microglial activity changes with aging as
these cells become more predisposed to transition into inflammatory phenotypes
[107, 108].
Within the CNS, microglia serve as an innate immune system and first line of
defense against harmful factors such as viruses and bacteria, which may be
removed by phagocytosis [7]. During phagocytosis, foreign particles and cells are
recognized, engulfed, and digested by microglia. This represents one of the major
pathways for clearing misfolded -syn in PD, with the involvement of,
for example, TLRs [109] and the complement system [7, 110]. Microglia display
diverse morphological variations within a heterogeneous population and possess
both pro- and anti-inflammatory characteristics [111, 112].
Microglial cells account for about 7% of non-neuronal brain cells of different
species, depending on the brain region [113]. They exhibit distinct morphological
forms and are categorized into two primary phenotypes. The M1 phenotype is
activated via the classical pathway, e.g., by lipopolysaccharide (LPS) [114, 115], which prompts the microglia to adopt a pro-inflammatory stance [28].
Activation of microglia can also be induced by -syn [116], which leads
to ROS production and results, in turn, in neurotoxicity [28].
Conversely, alternative activation, e.g., by IL-4 [114, 115], leads to the
anti-inflammatory (M2) phenotype that is characterized by anti-inflammatory
properties. Microglia take on a branched morphology, featuring a small, elongated
cell body and numerous extended processes that contract and stretch, thereby
facilitating exploration of the surrounding environment. The M2 phenotype can be
divided further into M2a, M2b, and M2c types [114, 117].
When exposed to abnormal -syn, pathogens, or remnants of damaged
neurons, microglia transition from a resting state to an active state, resulting
in alterations in cell morphology, gene expression, and the production and
expression of various factors [116, 118].
Microglia are also observed in embryos [119] where they secrete growth factors
to promote the survival of newly formed neurons, but can also eliminate
abnormally formed nerve cells [120]. Importantly, major histocompatibility
complex class II (MHC class II) molecules are expressed on microglial cells,
enabling them to present antigens to other microglial cells, as well as to immune
cells (such as T lymphocytes), which leads to the initiation of immune response
mechanisms [115, 120]. For this reason, microglia are considered to be
antigen-presenting cells (APCs). Sporadic PD with late onset is correlated with
polymorphism in the MHCII locus – human leukocyte antigen – DR isotype (HLA-DR)
[121]. MHCII molecules are also expressed on macrophages [122] and monocytes
[123] (whereas MHCI are expressed on neurons), and are responsible for
antigen-presenting, T cell recruitment, and promoting immune responses to
specific stimuli. MHCI and II molecules are considered to be the bridge between
innate and adaptive immunity [124].
Microglia are in constant contact with other cells from their environment,
including neurons. An example of communication between a nerve cell and microglia
is that which is associated with type I membrane glycoprotein (CD200) – a
glycoprotein expressed on neurons, ECs, and astrocytes, which has a specific
receptor (CD200R) located on microglia [125, 126]. Proper communication between
CD200 and its receptor maintains the microglia in a resting state (under
physiological conditions) [126, 127, 128]. Damaged neurons (e.g., under
inflammatory conditions) may not be able to secrete adequate CD200, with IL-4
being a key component in this mechanism [120, 129]. The abnormal interaction
between CD200 and CD200R leads to exacerbated DA neuronal death, increased
microglial activity and increased production of the pro-inflammatory cytokines
IL-6 and TNF-. This was demonstrated in a 2011 study by Zhang
et al. [127] on a rat model of PD induced by 6-hydroxydopamine (6-OHDA)
administration. Thus, it can be concluded that, via their contact with microglia,
neurons can regulate microglial function and activation. Microglia can also
phagocytose components of the NVU, such as endothelial cells [130].
Furthermore, microglia that exhibit the M1 phenotype impact BBB permeability
[131], leading to the infiltration of peripheral immune cells [132] and contribute to the release of a plethora of pro-inflammatory cytokines, ROS
[133], and NO [134]. They also contribute to the elimination of pathogens and/or
damaged neurons [135], and produce pro-inflammatory factors that can alter BBB
integrity [99]. There is evidence indicating that classical microglial activation
can trigger the infiltration of peripheral immune cells into the CNS, thereby
exacerbating and prolonging the inflammatory state within the brain [136]. A
study by Haruwaka et al. [130] suggests the presence of an accumulation
of microglial cells in the proximity of cerebral vessels in the very early stages
of inflammation, preceding any alterations to BBB integrity. Interestingly, it
seems that such contact, with ECs as well, leads to BBB protection, while an
extended state of systemic inflammation is associated with higher activation of
microglia and weakened BBB functioning [130]. Moreover, microglial activation has
been shown to exist even before the death of DA neurons in a rat 6-OHDA model of
PD [137], indicating that microglia are activated and assume pro-inflammatory
functions prior to the degeneration of DA neurons, as observed in PD.
Neuroinflammation within the CNS can yield positive outcomes and aid in
neutralizing threats. Microglial cells exhibiting the M2 phenotype play a crucial
role in this process by secreting anti-inflammatory factors and expressing
specific genes that facilitate the repair and regeneration of impaired neurons
[28, 138]. Microglia take part in releasing anti-inflammatory cytokines, growth
factors like transforming growth factor (TGF)-1 [139], and
neurotrophins, all of which are crucial for promoting neuroregeneration within
the SNpc [118]. Microglial cells also express FcR [134, 140], which
enables them to eliminate pathological forms of -syn that are
associated with immunoglobulin G (IgG) complexes [141]. Anti-inflammatory IL-4 can stimulate
microglial cell proliferation and induce genes prompting microglia to activate in
an alternative manner, thus promoting neuroprotective actions and inhibiting
pro-inflammatory properties [115, 142]. In order to shield nerve cells from
toxicity and inflammation, it is vital to maintain proper microglial function and
CNS homeostasis, along with the interaction between microglia and neurons [141].
Recent research by Stoll et al. [143] on rats treated with
-syn pre-formed fibril (PFF) injections presents a new perspective on
glial gene expression in the case of -synucleinopathy. Several gene
expressions were analyzed, associated with astrocytes of both pro-inflammatory
(e.g., C3, Gbp2, Ggta1, Serping1) and anti-inflammatory functions
(Tm4sf1, Emp1, S100a10). Even though the experiment showed that the
upregulation of pro-inflammatory-associated genes was far greater than that of
anti-inflammatory-associated genes, the expression of genes associated with all
different phenotypes was increased. The authors highlight how context plays a
huge role in glial phenotype changes; astrocytes and microglia work together and
swap their functions in response to inflammatory signals. For example, the
expression of C3 (complement component 3) differed between microglia and
astrocytes, localizing initially in microglia at 2–4 months post PFF injection
and then shifting to astrocytes at 6 months post injection. Researchers concluded
that the main pathways recognized by gene expression upregulation in
-synucleinopathy are complement, phagocytosis, T cell recruitment and
activation, and cytokine/chemokine and inflammasome signaling [143]. This appears
to be the latest and broadest view and also confirms the involvement of the
innate immune system in PD synucleinopathy. The latest study by Leandrou
et al. (2024) [144] identifies specific molecular pathways involved in
microglial and astrocytic responses to -syn oligomers, such as the p38
mitogen-activated protein kinases/activating transcription factor 2 (p38/ATF2)
signaling pathway in microglia and the NF-B pathway in astrocytes, that
opens new possibilities of influence on both glial cell types in PD. Table 1
(Ref. [32, 34, 38, 39, 40, 41, 43, 45, 47, 48, 49, 63, 74, 75, 76, 79, 80, 86, 88, 89, 92, 93, 94, 95, 97, 99, 102, 116, 128, 143, 144, 145, 146, 147, 148, 149, 150, 151, 152]) shows the results of experimental studies using in vivo
and in vitro methods, as well as brain post-mortem analyses in PD
patients, highlighting changes in the function and phenotype of astrocytes,
microglia, and endothelial cells induced by -syn aggregation.
Table 1.
The involvement of astrocytes, microglia, and endothelial cells
in alpha-synuclein aggregation-dependent neurodegeneration in Parkinson’s
disease. The presented compilation arises from studies using in vitro
(cell cultures), in vivo (animal models), and post-mortem (tissue
samples from PD patients) methods.
| Cell type |
Function/phenotype |
Alpha-syn-mediated changes |
In vitro/in vivo/post-mortem model |
References |
| Astrocytes |
Ability to form functionally proper BBB |
Deterioration of structural integrity |
Human PD donors, cell culture |
[99] |
|
|
decreased expression of the water channel protein aquaporin-4 |
iPSC-derived astrocytes |
[89] |
|
Cooperation with endothelial cells during maintenance of the BBB |
Abnormal accumulation of Glut1 and vWF, and the redistribution of aquaporin-4 to the soma of astrocytes |
-Syn transgenic mice |
[80] |
|
Neurotoxic phenotype |
Microglia-mediated conversion |
-Syn PFF mouse model |
[63] |
|
Pro-inflammatory phenotype |
NLRP3 inflammasome-dependent; increase in caspase-1 and IL-1 levels, ASC protein levels, and the number of GFAP+ cells |
Mouse model; primary culture |
[49] |
|
|
TLR4-dependent gene expression; cylooxygenase-2, NO synthase mRNA; phosphorylation of p38 mitogen-activated protein kinase and c-Jun N-terminal kinase, and NF-B1 nuclear translocation |
Primary TLR4+/+ and TLR4−/− mouse astrocytes |
[88] |
|
|
Induction of pro-inflammatory genes for IL-1, IL-6, TNF-; secretion of CCL2, CCL20, CXCL1, CX3CL1; involvement of TLR-2 signalling, NF-B1, Fc receptor signalling, antigen processing (TAPBP and CD74) |
Cell line SH-SY5Y; primary astrocytes from rat |
[76] |
|
|
Increased IL-6, CXCL1, and IL-8 secretion |
iPSC-derived astrocytes |
[89] |
|
|
Increased IL-6 and CCL5 secretion |
iPSCs from PD |
[92] |
|
|
Increased IL-6 and TNF- secretion |
Cell culture |
[40, 41] |
|
|
TG2-dependent pro-inflammatory phenotype; increased number of GFAP cells |
TG2KO/SynTg double-modified mice |
[97] |
|
Antigen-presenting phenotype |
Elevated expression of HLA-DMA and MHC class II proteins |
Human iPSC-derived astrocytes |
[86] |
|
TLR2 – dependent pro-inflammatory responses |
Overexpression of TLR2; increased IL-6, TNF-, and IL-1 secretion |
-Synuclein transgenic mice |
[94] |
|
Uptake of -syn oligomer/fibrils by cells |
Pronounced |
Human iPSC-derived astrocytes |
[74, 75] |
| Microglia |
Production and secretion of pro-inflammatory cytokines |
Increased TNF- and IL-1 production and secretion |
BV2 cell culture |
[39] |
|
TLR4 – dependent pro-inflammatory responses |
Increased phagocytic activity, pro-inflammatory cytokine release (TNF-, IL-6); CXCL and ROS production |
TLR4 deficient (TLR4−/−) mice, microglial; cell culture |
[93] |
|
|
Increased phagocytic activity, activated NF-B and p38 pathways |
Cell culture |
[116] |
|
TLR2 – dependent pro-inflammatory responses |
Increased proportion of microglia displaying an ameboid and reactive morphology |
-Syn transgenic mouse model |
[95] |
|
|
Increased IL‐1 secretion dependent on NLRP3 inflammasome assembly and caspase‐1 activity |
Human PD donors, cell culture |
[48] |
|
|
Increased percentage of microglial cells with amoeboid morphology and production of IL-6, IL-1, and TNF- |
Primary rat microglia and TLR2−/− mice |
[45] |
|
Pro-inflammatory gene induction |
Elevated expression of TNF-, IL-1, IL-6, and COX-1 |
-Syn transgenic mice |
[80] |
|
|
Elevated expression of CXCL10, Rt1-a2, Grn, Csf1r, C3, C1qa, Tyrobp, Serping1, and Fcer1g in Cd74+; i.e., genes involved in complement protein expression, phagocytosis, T cell recruitment and activation, cytokine and chemokine release, and inflammasome signalling |
-Syn PFF injected rats |
[143] |
|
NLRP3 inflammasome-dependent pro-inflammatory phenotype |
Increased level of cleaved caspase-1 and adaptor protein ASC; microgliosis |
Post-mortem brains of patients with PD; -syn PFF-injected mice |
[102] |
|
Pro-inflammatory MHCII response |
Infiltration of CCR2+ and Ly6C+ peripheral monocytes into the SNpc |
AAV2-SYN transduced mice |
[151, 152] |
|
Uptake of -syn aggregates by cells; phagocytosis |
Pronounced |
BV2 microglial cells; rat microglia cells |
[79] |
|
|
Inhibition of the uptake of -syn aggregates in the absence of T lymphocytes |
BV2 microglia |
[147] |
|
|
Inhibition of phagocytosis |
A53T -syn transgenic mice |
[38] |
|
Infiltration of peripheral T CD4 lymphocytes into the brain |
MHCII and IL-4-dependent; induction of MHCII+ on microglia; increased IL-4 production by CD4 lymphocytes |
-Syn overexpressing mouse model |
[148] |
|
|
MHCII-dependent microgliosis and peripheral blood T lymphocyte transfer into striatum and SNpc |
-Syn PFF injected immunocompromised (NSG) mice |
[150] |
|
Pro-inflammatory phenotype |
FcR-mediated conversion |
Primary cultures from mice |
[47] |
|
Astrocyte-mediated anti-inflammatory phenotype |
Activation of the p38/ATF2 (microglia) and the NF-B (astrocytes) pathways |
Transgenic A53T mice, mouse primary microglia |
[144] |
|
Microglia and astrocyte interaction |
Increased number of astroglia and microglia in close proximity to CD3 positive cells during T cell infiltration; higher IFN- and significantly higher TNF- mRNA level |
-Syn TG transgenic mice |
[149] |
|
|
Downregulation of CD200-CD200R1 and CX3CL1-CX3CR1 pathway |
rAAV-hSYN-injected mice |
[128] |
| Endothelial |
BBB structure |
Deterioration of BBB integrity |
Brain tissue from PD patients |
[32] |
|
Deterioration of BBB integrity |
Downregulated expression of tight junction proteins |
hCMEC/D3 human brain endothelial cells; brain tissue from PD patients |
[43] |
|
|
Upregulation of LRP1-ICD |
Mouse model |
[34] |
|
|
Pathological activation of pericytes |
Transgenic mice |
[145] |
|
|
Elevated release of IL-1, IL-6, MCP-1, TNF-, and MMP-9 by pericytes |
RBECs co-cultured with rat brain pericytes |
[146] |
The list of abbreviations: ASC, apoptosis-associated speck-like protein
containing a caspase recruitment domain; ATF2, activating transcription factor-2;
BBB, blood-brain-barrier; CCL2, C-C motif chemokine ligand 2; CCL20, C-C motif chemokineligand 20; CCL5, C-C motif chemokine ligand 5; CCR2, C-C chemokine
receptor type 2; CD200, type I membrane glycoprotein; CD200R1, CD200
receptor 1; COX-1,
cyclooxygenase-1; CX3CL1, C-X3-C motif chemokine ligand-1; CX3CR1,
C-X3-C motif chemokine receptor-1; CXCL1, C-X-C motif chemokine ligand 1; FcR, Fc gamma receptor; GFAP, glial
fibrillary acidic protein; Glut1, glucose transporter 1; HLA-DMA, major
histocompatibility complex, class II, DM alpha; IFN-, interferon gamma;
IL-1, interleukin 1; IL-1, interleukin 1 beta; IL-4, interleukin 4; IL-6,
interleukin 6; IL-8, interleukin 8; iPSC, induced pluripotent stem cells; LRP1-ICD,
low-density lipoprotein receptor-related protein-1; Ly6C, lymphocyte antigen 6
complex, locus C1; MCP-1, monocyte chemotactic protein-1; MHC, major
histocompatibility complex; MMP-9, matrix metalloproteinase-9; NF-B,
nuclear factor kappa-light-chain-enhancer of activated B cells; NLRP3, NLR family
pyrin domain containing 3; PD, Parkinson’s disease; RBECs, rat brain endothelial
cells; ROS, reactive oxygen species; SNpc, substantia nigra pars compacta;
TAPBP, TAP-associated glycoprotein, tapasin; TG2, transglutaminase-2; TLR2,
toll-like receptor 2; TLR4, toll-like receptor 4; TNF, tumor necrosis factor;
vWF, von Willebrand factor; -syn, alpha-synuclein; PFF, preformed fibril; Rt1-a2,
mature alpha chain of major histocompatibility complex class I antigen; Grn, progranulin; Csf1r,
macrophage colony-stimulating factor 1 receptor; AAV2-SYN, adeno-associated virus serotype 2
vector-mediated -syn expression; rAAV2-hSYN, recombinant adeno-associated viral
vector-mediated human -syn expression; hCMEC/D3, human cerebral microvascular endothelial cell line.
By assuming both pro- and anti-inflammatory phenotypes, microglia play a pivotal
role in initiating and perpetuating neuroinflammation, as well as in the
neuroprotective and regenerative processes within the CNS. The interactions
between DA neurons, microglia, and astrocytes during -syn-induced
neurodegeneration in the SNpc are summarized in Fig. 1.
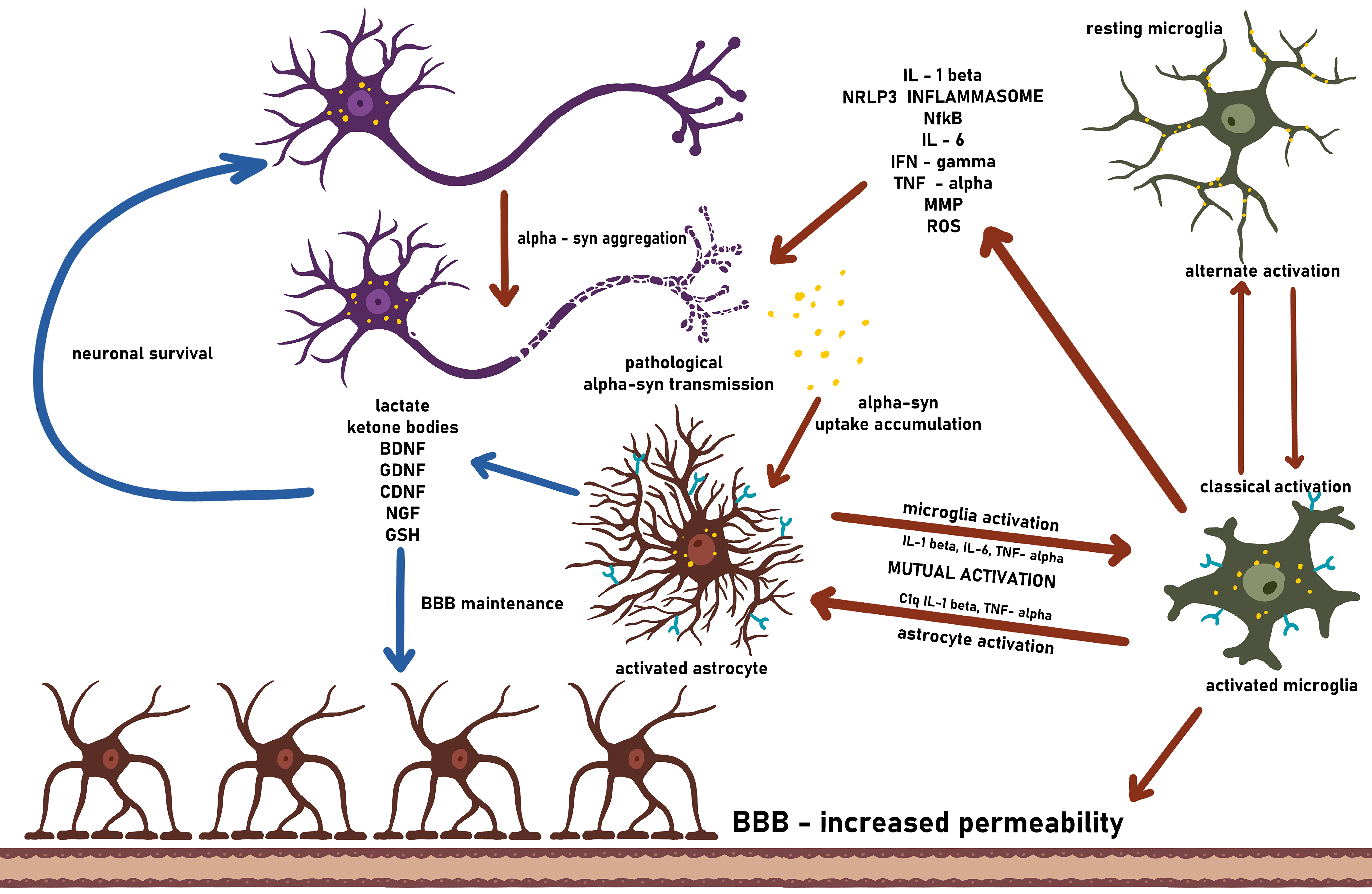 Fig. 1.
Fig. 1.
Cooperation between astrocytes and microglia leads to
neurodegenerative processes that are triggered by the aggregation of
pathologically altered -syn (depicted by red arrows), while also
involving glial cells in neuroprotective mechanisms (shown by blue arrows). The
diagram illustrates the interplay among neurons, microglia, and astrocytes, with
detailed explanations provided above. All abbreviations have been explained in
the text. The figure was created using Clip Studio Paint Pro v 2.0 (license
number: L6136027487). BDNF, brain-derived neurotrophic factor; GDNF, glial
cell-derived neurotrophic factor; CDNF, cerebral dopamine neurotrophic factor;
GSH, glutathione; BBB, blood-brain barrier; TNF, tumor necrosis factor; MMP,
matrix metalloproteinase; NF-B, nuclear factor
kappa-light-chain-enhancer of activated B cells; ROS, reactive oxygen species;
NGF, nerve growth factor; NRLP3, NOD-like receptor protein 3; IFN, interferon; IL, interleukin; C1q, component 1q.
4. Involvement of Glial Cells in Recruiting Peripheral Immune
Mechanisms in PD
When the body’s internal balance is maintained, the CNS enjoys a state of
immunological privilege. However, conditions like those observed in PD lead to
ongoing inflammation in the CNS and compromise the integrity of the BBB. This
breach allows activated pro-inflammatory lymphocytes and monocytes to infiltrate
regions of the CNS affected by neurodegeneration [28]. It is noteworthy that ECs
are also known to be responsible for trafficking immune cells [153]. The
disruption of the BBB integrity and functionality of its components leads to
enhanced neuronal loss as a result of severe neuroinflammation and -syn
accumulation. In addition, the permeability of the BBB in PD has been shown to be
significantly increased [145, 154]. BBB dysfunction can also be caused by changes
in the functionality of ECs, to which -syn-induced inflammation
contributes. This leads to the activation of ECs and the release of
pro-inflammatory molecules. The ECs appear to shrink, and gaps appear between
them, increasing the permeability of the EC layer. Ultimately, this results in
the recruitment of leukocytes, which are involved in the progression of
neuroinflammation [155]. EC structure and functionality can also be modulated by
vascular endothelial growth factor A (VEGFA), which is released by astrocytes and
stimulated by microglia that produce IL-1 in chronic inflammation states [156, 157]. It is thought that VEGF can mediate BBB breakdown [158]. In addition,
-syn-activated pericytes lead to BBB dysfunction by releasing
pro-inflammatory factors [146]. Elabi et al. [145] showed that the
activation of pericytes at an early stage of the disease in the human
-syn overexpression mouse model resulted in vascular alterations
associated with BBB altered permeability.
Research suggests that, under conditions of chronic CNS inflammation, peripheral
immune cells can migrate into the CNS [147, 159]. Various communication
mechanisms between microglia and neurons are implicated in this process. The
shift in microglial profile to an active state also contributes to this
phenomenon [136]. Effective interaction between neurons producing fractalkine
glycoprotein (C-X3-C motif chemokine ligand-1, CX3CL1) and its receptor on microglia (C-X3-C motif chemokine receptor-1, CX3CR1) [160] typically
modulates microglial cell phenotype [161]. However, dysfunction in this mechanism
can lead to the increased release of pro-inflammatory factors and
neuroinflammation [162]. The pro-inflammatory microglia profile is considered to
take part in immune cell recruitment [163].
Damaged neurons release the pathological form of -syn into the
environment, prompting microglia to adopt a pro-inflammatory response to counter
the threat to the CNS. Exogenous -syn can induce this microglial
response directly, which, in turn, leads to the release of pro-inflammatory
cytokines, increases phagocytic activity, and further aggravates
microglial-induced inflammation. This may result in the transmission of
-syn in a prion-like way [7]. Thus, -syn contributes to BBB
disruption indirectly – by promoting inflammation [164]. The aberrant protein
may possibly enter cervical lymph nodes, initiating the activation of macrophages
[165] and thus effector T cell responses. Subsequently, T helper (TCD4) and T cytotoxic (TCD8) lymphocytes
breach the BBB and migrate to the site of inflammation. -Syn peptides
bound to MHCI on neurons and MHCII on microglia re-stimulate peripheral immune
cells, which incites inflammatory reactions [148, 166]. Helper T cells (CD3/CD4+)
are found near activated astrocytes, blood vessels, and places of high expression
of pro-inflammatory cytokines, with CD3+ cells co-expressing interferon gamma (IFN-)
[149]. While pro-inflammatory factors released by lymphocytes and microglia aid
in removing pathological proteins, they also harm neighboring neurons, which
leads to the further release of -syn. These interconnected mechanisms,
which perpetuate the continuous presence of the protein in the extracellular
space and sustain inflammatory processes, are referred to as a self-propelling
loop of neuroinflammation [167].
A recent study by Hourfar et al. [164] was conducted to explore the
impact of -syn AGs on BBB integrity in the human cerebral microvascular
endothelial cell line (hCMEC/d3). The results indicated that -syn AGs
have a direct and damaging impact on ECs in the BBB (causing their dysregulation
and mitochondrial dysfunction), with astrocytes playing a shielding role due to
their ability to interact with pathological aggregates. However, in inflammatory
states, it seems that the functions are reversed and, together with microglia,
astrocytes take part in BBB disruption [164].
Outside the CNS, Th2 lymphocytes prompt B cells to generate anti--syn
antibodies. These antibodies cross the BBB, bind to -syn epitopes on
neuron surfaces, and activate the complement system, resulting in
antibody-dependent cytotoxicity and acute inflammation [166]. However, with
disease progression, the T cell subsets change, resulting mainly in an imbalance
between Th (Th2 and Th17) and Treg cells, and contributing to neurodegeneration
by pushing the pro-inflammatory phenotypes [124]. The infiltration of peripheral
immune cells into the CNS not only triggers the secretion of pro-inflammatory
factors by activated immune cells, but also exerts anti-inflammatory effects
[150]. Treg cells that are able to release, for example, IL-10 and TGF-,
can modulate both the activity of M1 microglia (since Treg depletion has been
shown to result in activation of microglia via the STAT3 pathway, enhancing
inflammation after spinal cord injury [168]) and other immune cells as well
[169]. Additionally, factors secreted by Th2 cells, such as IL-5 and IL-4, prompt
pro-inflammatory microglia to transition to the anti-inflammatory/resting
phenotype – M2 [166].
The advancement of PD is closely tied to the activity of glial cells, including
astrocytes and microglia. These cells can take on different phenotypes that
determine their pro- or anti-inflammatory behaviors. When glial cells become
pro-inflammatory, they release cytokines and chemokines that fuel the onset and
continuation of neuroinflammation in the CNS. Conversely, shifting to an
anti-inflammatory phenotype aids in shielding neurons and other brain cells by
expressing factors that promote their protection and regeneration. Astrocytes,
microglia, and neurons interact with each other, influencing their functioning
through various forms of intercellular communication. Several different pro- and
anti-inflammatory factors are found in the CSF and/or blood of PD patients,
including TNF-, IFN-, TGF-, IL-1, IL-2,
IL-4, IL-6, IL-8, and IL-10 [170, 171, 172].
Although neuroinflammation in the CNS is generally associated with
pro-inflammatory microglial activity, research conducted by Harms et al.
[151] cast the neuroinflammatory properties previously attributed to microglia in
a new light. Researchers suggested that the activation of resident immune cells
in the CNS depends strongly on peripheral monocytes infiltrating the brain in
pathological states [151, 152]. The study showed the recruitment of
pro-inflammatory peripheral monocytes with C-C chemokine receptor type 2 (CCR2) (induced by -syn
exacerbated expression) as a main and crucial component in the response of
-syn-dependent CNS cells in a mouse model of PD [151]. CCR2 is a chemokine
receptor, located on myeloid cells from the periphery, and the interaction
between CCR2 and its ligand CCL2 seems to be fundamental for monocytes entering
brain tissue. CCL2-CCR2 signaling induces monocyte infiltration through the BBB,
where they facilitate anti- and pro-inflammatory reactions as macrophages [151]. The
study also showed that inhibiting such infiltration of CCR2+ monocytes reduces
the inflammation mediated by -syn. CCR2-knockout mice exhibited
attenuated MHCII expression in the SNpc [152]. As previously shown by the researchers,
modulation of MHCII diminished -syn neurotoxicity [152]. High
expression of MHCII (HLA-DR) in the CNS, in the proximity of -syn and
thus of dying neurons, seems to indicate that APCs and adaptive immune processes
play pivotal roles in neurodegeneration in PD [124, 173].
Monocytes are divided into classical (CD14+/CD16–; differentiating into
dendritic cells and macrophages), non-classical (CD14–/CD16+), or integral
(CD14+/CD16+) types, and they vary in terms of the particles released, which
include IL-6, IL-10, CCL2, IL-1, IL-8, and TNF [124, 174, 175, 176]. In
PD patients, monocytes undergo changes in their functionality, including their
activation process, proliferation, and phagocytosis, with the duration of the
disease as an affecting factor [124].
An overview of the mechanism of lymphocyte recruitment involving microglial
cells through a compromised BBB to sites affected by neurodegeneration resulting
from -syn aggregation is shown in Fig. 2.
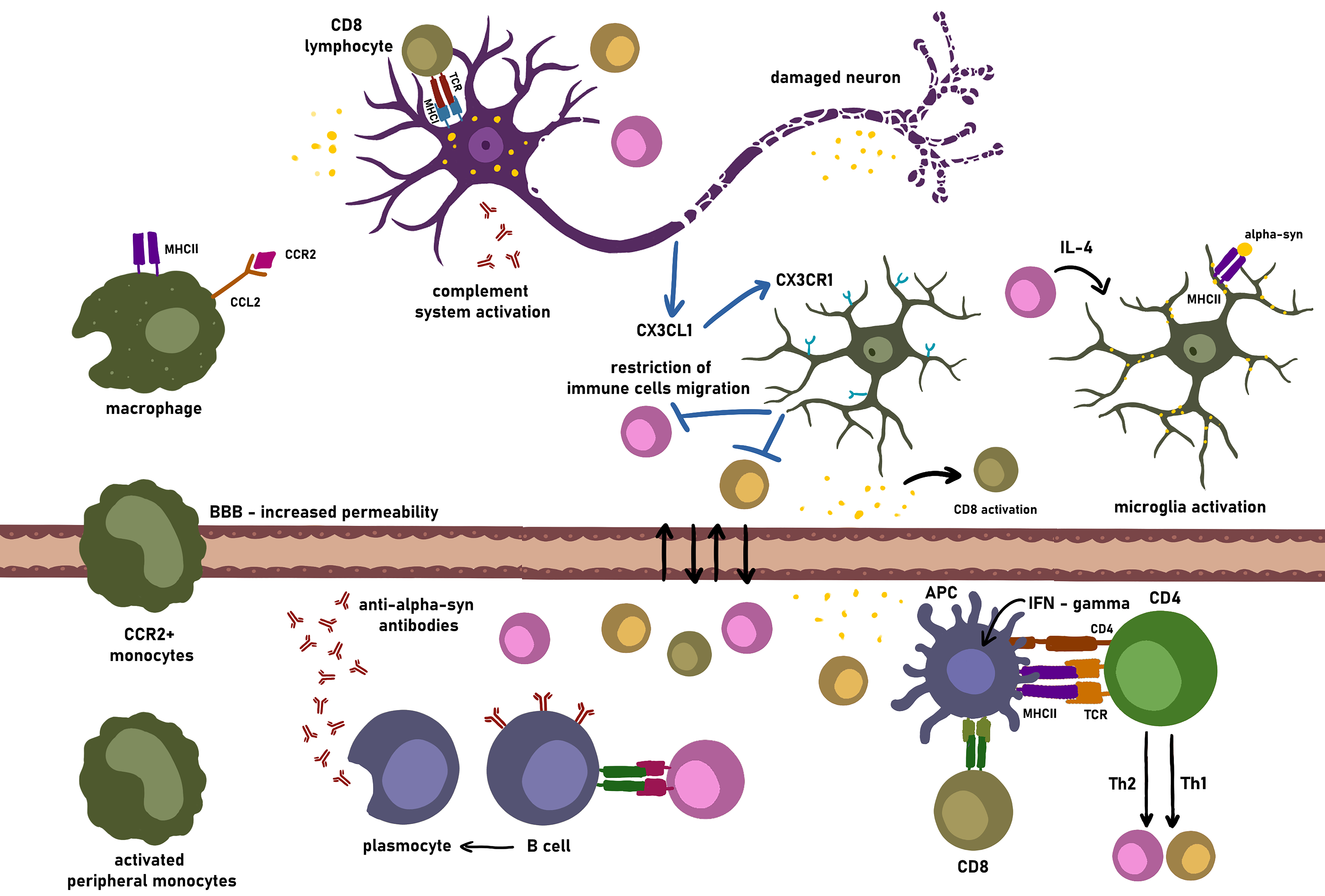 Fig. 2.
Fig. 2.
Peripheral immune system cells are recruited through a
compromised BBB because of -syn presentation to peripheral
lymphocytes. Depending on their activation type, microglial cells can either
intensify or mitigate (blue lines) the inflammatory response. Further details are
discussed in the accompanying text. All abbreviations have been explained in the
text. The figure was created using Clip Studio Paint Pro v 2.0 (license number:
L6136027487). APCs, antigen-presenting cells;TCR, T-cell
receptor; Th1, type 1 helper T cells; Th2, type 2 helper T cells.
5. Conclusions
The latest data indicates that PD does not only impact the SNpc dopaminergic
neurons but also affects other brain structures and the peripheral nervous
systems, often manifesting as non-motor symptoms before motor signs appear. The
inflammation triggered by activated microglia and astrocytes is believed to drive
these diverse symptoms. The aggregation of -syn exacerbates
neuroinflammation in PD, worsening the loss of neurons. However, crucial details
are still missing about the specific structural forms of -syn that
activate microglia and astrocytes. Neuroinflammation in the CNS compromises the
integrity of the BBB, increasing its permeability and allowing peripheral immune
cells to infiltrate and migrate towards the inflamed areas. Components of the
immune system, along with glial cells, work to neutralize pathological
-syn, thus clearing the extracellular space. In addition, they trigger
antibody-dependent immune responses and activate other cells, which exacerbates
inflammation. Understanding the factors that can alter the immunomodulatory
properties of glial cells, especially in connection with their reparative
abilities, offers promising avenues for developing therapies that can slow down
the progression of PD.
Author Contributions
OH, BG equally contributed to the analysis of selected articles for the text, manuscript, table and figures preparation. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Ethics Approval and Consent to Participate
Not applicable.
Acknowledgment
We would like to express our gratitude to Agata Ledóchowska for her technical support in creating the figures.
Funding
The cost of proofreading the publication was covered by statutory of Department of Animal and Human Physiology fund (531-D080-D248-24).
Conflict of Interest
The authors declare no conflict of interest.



 , Beata Grembecka 1,*
, Beata Grembecka 1,*