
 , Hui-juan Li 1,3, Jian-lin Liang 1, Qian-qian Ren 1, Chang-xin Li 1,*
, Hui-juan Li 1,3, Jian-lin Liang 1, Qian-qian Ren 1, Chang-xin Li 1,* , Sui-yi Xu 1,*
, Sui-yi Xu 1,*1 Department of Neurology, Headache Center, The First Hospital of Shanxi Medical University, 030001 Taiyuan, Shanxi, China
2 Department of Geriatrics, General Hospital of TISCO, 030001 Taiyuan, Shanxi, China
3 Center for Medical Genetics & Hunan Key Laboratory of Medical Genetics, School of Life Sciences, Central South University, 410000 Changsha, Hunan, China
Abstract
The aim of this study was to investigate the possible molecular mechanisms underlying cerebral small vessel disease caused by a missense mutation in the high-temperature serine peptidase A1 gene, HtrA1 (NM_002775.4, Exon4, c.905G>A, p.Arg302Gln). Stable strain models were constructed using wild-type and mutant HtrA1 overexpression lentiviral vectors to infect mouse brain microvascular endothelial cells (bEnd.3 cells).
HtrA1 mRNA and protein expression were analyzed by Western blot and quantitative real-time polymerase chain reaction. Western blot technique was also used to evaluate the expression of transforming growth factor (TGF)-β/Smads-related signaling pathway proteins and the oxidative stress pathway protein nicotinamide adenine dinucleotide phosphate oxidase 4 (NOX4). The level of reactive oxygen species (ROS) was evaluated using dichloro-dihydro-fluorescein diacetate (DCFH-DA) fluorescent probes.
HtrA1 mRNA and protein expression levels were found to be decreased in mutant cells, whereas the levels of ROS, the TGF-β/Smads proteins, and the caspase3 and cleaved-caspase3 apoptotic proteins were increased.
Lentivirus-mediated missense mutation in HtrA1 leads to activation of the TGF-β/Smads pathway and to increased apoptosis of mouse brain microvascular endothelial cells via the oxidative stress pathway. Further in vivo studies are required to explore the connections between different signaling pathways in animals, and to identify potential molecular targets for clinical therapy.
Keywords
- HtrA1
- bEnd.3
- TGF-β/Smads
- oxidative stress
Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL) is an inherited cerebral small vessel disease (CSVD) caused by homozygous mutations in HTRA1 [1]. CARASIL is characterized by non-hypertensive CSVD, early-onset subcortical infarcts in adults, progressive motor and cognitive impairments, alopecia, and vertebral disease [2]. HTRA1 was first recognized as a causative gene for CARASIL in 2009 [1]. Subsequently, CARASIL cases were reported in China [3], India [4], North America and Africa [5]. Symptomatic HTRA1 variant carriers, also known as cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy 2 (CADASIL2), have heterozygous HTRA1 mutations related to CSVD [6, 7].
HtrA1 encodes a serine protease that is widely expressed in blood vessels [8, 9]. It has important roles in cell proliferation, migration, and apoptosis, in
addition to its role in controlling protein aggregation through refolding,
translocation, or degradation [10]. HtrA1 has dual activity as a chaperone
protein and serine protease, and is involved in numerous physiological processes
such as extracellular matrix remodeling [11, 12] and transforming growth
factor-
Nozaki and colleagues first discovered that the heterozygous HTRA1 gene
missense mutation was p.Arg302Gln, and patients presented with stroke, early
onset cognitive impairment, gait abnormalities, and alopecia. By using PolyPhen2
(http://genetics.bwh.harvard.edu/pph2/), and sorting intolerant from tolerant
(http://sift.jcvi.org/), the mutations were predicted to be damaging, and
considered to be heterozygous HTRA1 mutations related to CSVD [16]. It
was shown earlier that HtrA1 is associated with cell oxidative stress [19] and
apoptosis [20]. Endothelial cell dysfunction is a key mechanism in
cerebrovascular structural and functional alterations in patients with CSVD [21].
To further understand the pathogenicity caused by HtrA1 mutations, we
generated homozygous (Hom) and heterozygous (Het) HtrA1 mutant cell
lines by infecting bEnd.3 cells with wild-type (WT) and mutant HtrA1
overexpression lentiviral vectors. The expression of TGF-
The construction of WT and mutant overexpression lentiviral vectors
for HtrA1 were carried out by GenePharma (https://www.genepharma.com, Shanghai, China).
LV8N (EF-1a/mCherry&Puro) and LV5 (EF-1a/GFP & Puro) (GenePharma, Shanghai,
China) are overexpression lentiviral vectors that differ only in fluorescence
expression, with the remaining structures being identical. The coding sequence
(CDs) region and translated protein sequences of mouse WT and mutant (c.905G
bEnd.3 cells were obtained from Procell Life Science (Procell, Wuhan, Hubei, China). The cell line was certified by species identification. The cell line was
certified and tested to ensure it was free of mycoplasma contamination. The
culture medium was high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM;
HyClone, Logan, UT, USA, SH30022.01) containing 10% fetal bovine serum (Boster,
Wuhan, Hubei, China, PYG0001). Cells were grown in an incubator at 37 °C with 5%
CO2, and passaged at a ratio of 1:4 once the cell density had attained
70–80% confluence. Cells with a stable growth status in the P4–P6 generations
were selected for the subsequent experiments. When cells were approximately 50%
fused, bEnd.3 cells were infected with serum-free, high-glucose DMEM plus the
viral solution. The serum-containing medium was replaced after 24 h of incubation
and the fluorescence in each group was observed after 72 h. Cells were screened
using 5 µg/mL of puromycin (Melone, Dalian, Liaoning, China,
MA0318), and stable transfection was determined when
Cells from each group were collected and total RNA was obtained using TRIzol (Mei5 Biotech, Beijing, China, MF034-01), chloroform, and isopropanol extraction. The concentration and purity of RNA was determined using Take3 microtiter plates (Biotek, New Castle, DE, USA), and cDNA was obtained with the PrimeScriptTM RT reagent kit and gDNA Eraser (Takara, Osaka, Japan, RR047A). The RNA concentration was adjusted to 500 ng/µL. The products were amplified using a quantitative real-time polymerase chain reaction (qRT-PCR) instrument (LightCycler 480II, Roche, Basel, Switzerland) with a reaction mixture of 20 µL comprised of 10 µL of TB Green Premix Ex TaqII (Takara, Osaka, Japan, RR820A), 2 µL of cDNA template, 6.4 µL of sterile enzyme-free water, and 0.8 µL each of upstream and downstream primers (10 µmol/L). The amplification programs were as follows: predenaturation (95 °C for 30 s); PCR reaction (95 °C for 5 s, 60 °C for 30 s, 40 cycles); melting curve analysis (95 °C for 5 s, 60 °C for 1 min, 95 °C, 1 cycle, 50 °C for 30 s). (Gapdh) served as the reference for mRNA, and expression levels of HtrA1 mRNA were determined with the 2-ΔΔCt method. Primers were designed and synthesized by Sangon Biotech (Shanghai, China), with the sequences shown in Table 1.
| Gene | Sequence | |
| Forward | Reverse | |
| WT-HtrA1 | 5′-tgacggcgggcatctccttc-3′ | 5′-tcttggtgacagctttccctttgg-3′ |
| Mut-HtrA1 | 5′-gctgaagaatggagctacctat-3′ | 5′-ggtcaatcttgataagcgcaat-3′ |
| Gapdh | 5′-ccctggccaaggtcatccat-3′ | 5′-tcacgccacagctttccaga-3′ |
WT, wild-type; PCR, polymerase chain reaction; Mut, mutation; HtrA1, high temperature requirement factor A1; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Culture medium from each group of cells was centrifuged to remove the
supernatant, and a mouse enzyme-linked immunosorbent assay (ELISA) kit was used to evaluate the concentration of
HtrA1 as recommended by the manufacturer (Meimian, Yancheng, Jiangsu, China,
MM-45445M1). A multifunctional microtiter plate enzyme labeling instrument was
used to measure optical density (OD) at 450 nm. The standard curve was plotted
based on the OD value of the standard (correlation coefficient
bEnd.3 cells were lysed using enhanced radio immunoprecipitation assay (RIPA)
lysis buffer containing a broad-spectrum protease inhibitor (Boster, Wuhan,
Hubei, China, AR0102-100). The resulting supernatants were centrifuged to obtain
protein extracts from each group. The protein concentration of lysates was
measured with a bicinchoninic acid (BCA) protein kit (Boster, Wuhan, Hubei,
China, AR0146). Proteins (30 µg) from each sample were
electrophoresed on 10% sodium dodecyl
sulfate (SDS)-polyacrylamide gels (Solarbio, Beijing, China, P1200-50T) and
subsequently transferred to 0.22 µm polyvinylidene
fluoride membranes (Millipore, Belmont, MA, USA, ISEQ00010). These were incubated
with protein dry powder at a concentration of 5% (Boster, Wuhan, Hubei, China,
AR0104) for 2 h before incubation with the following primary antibodies: rabbit
anti-HtrA1 (1:250, Boster, Wuhan, Hubei, China, A01801-1), anti-Smad2 (1:1000,
Abclonal, Wuhan, Hubei, China, A11498), anti-Smad3 (1:1000, CST, Boston, MA, USA,
9523T), anti-Smad4 (1:1000, Abclonal, Wuhan, Hubei, China, A19116), anti-nicotinamide adenine dinucleotide phosphate oxidase 4 (anti-NOX4)
(1:1000, Boster, Wuhan, Hubei, China, BM4135), anti-caspase3 (1:1000, Abclonal,
Wuhan, Hubei, China, A2156), anti-cleaved-caspase3 (1:1000, abcam, Cambridge, UK,
ab32042), and anti-GAPDH (1:5000, Abways, Shanghai, China, AB0037). The membranes
were then washed with tris-buffered saline with Tween solution (TBST) and labeled
with goat anti-rabbit IgG-horseradish peroxidase (1:5000; Boster, Wuhan, Hubei,
China). Luminescent solution was prepared using an ultrasensitive enhanced
chemiluminescence (ECL) substrate kit (Boster, Wuhan, Hubei, China, AR1197) and dripped
onto the membrane, which was then visualized with an all-purpose imaging analysis
system (Bio-Rad, Hercules, CA, USA). Image J software (V1.8.0, University of
Wisconsin, Madison, WI, USA) was used to determine grayscale values of the target
proteins, and their relative expression level was assessed using glyceraldehyde phosphate dehydrogenase (GAPDH) or
Cells were cultured for 24 h in 6-well plates at 1.5
Data was analyzed using SPSS 26.0 software (IBM Corp., Chicago, IL, USA).
GraphPad Prism 8 (GraphPad Software, Inc., San Diego, CA, USA) was used for
graphing, and quantitative information was presented as the mean
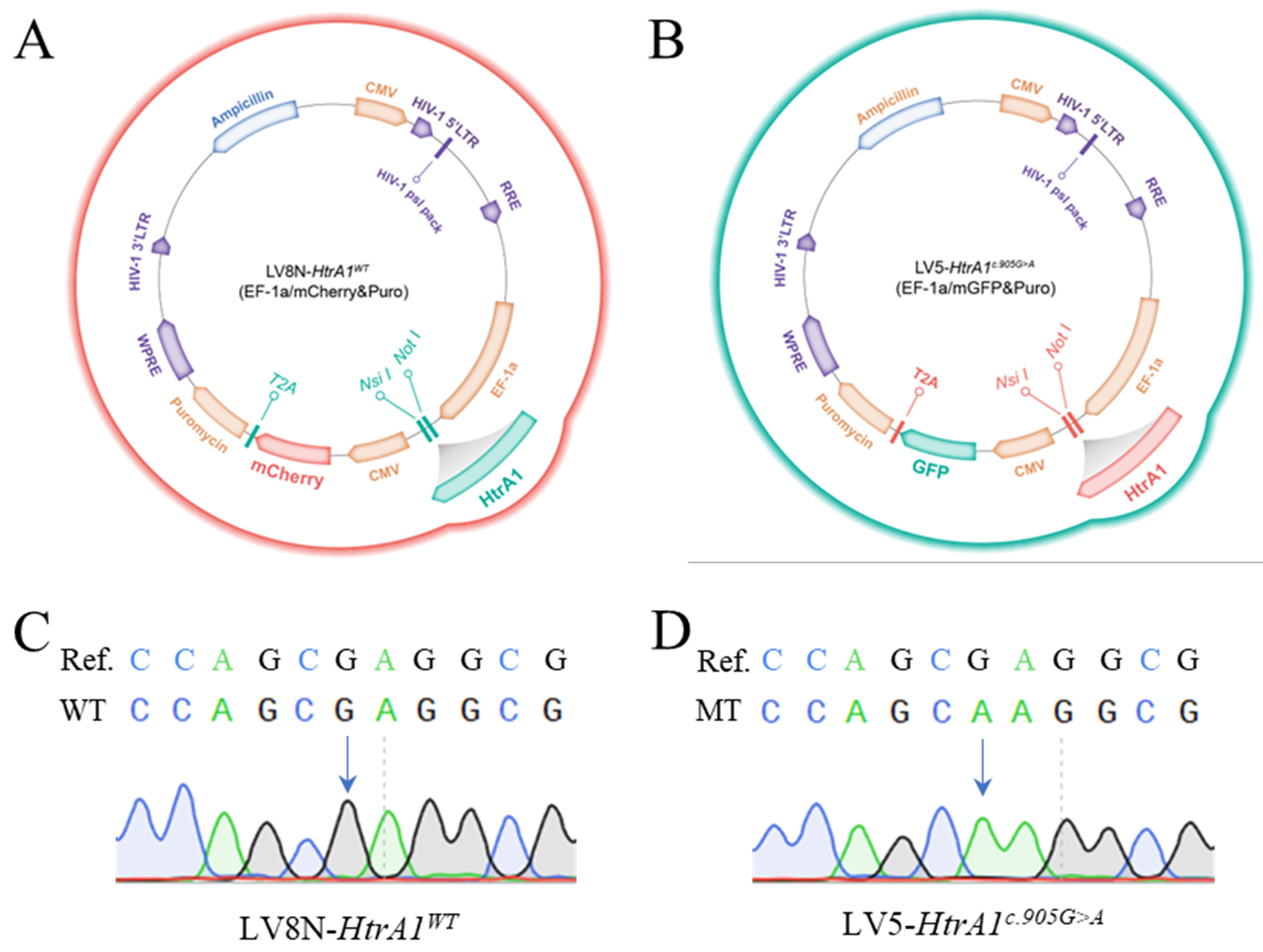
WT and mutant HtrA1 fragments were cloned into the lentiviral vectors
NotI/NsiI of LV8N (EF-1a/mCherry&Puro) and LV5 (EF-1a/GFP&Puro), respectively.
WT LV8N-HtrA1 and mutant LV5-HtrA1 were verified by Sanger
sequencing. The sequencing results for WT LV8N-HtrA1 perfectly matched
the mouse HtrA1 gene sequence alignment, while those for mutant
LV5-HtrA1 were consistent with the HtrA1 gene mutation site
(c.905G
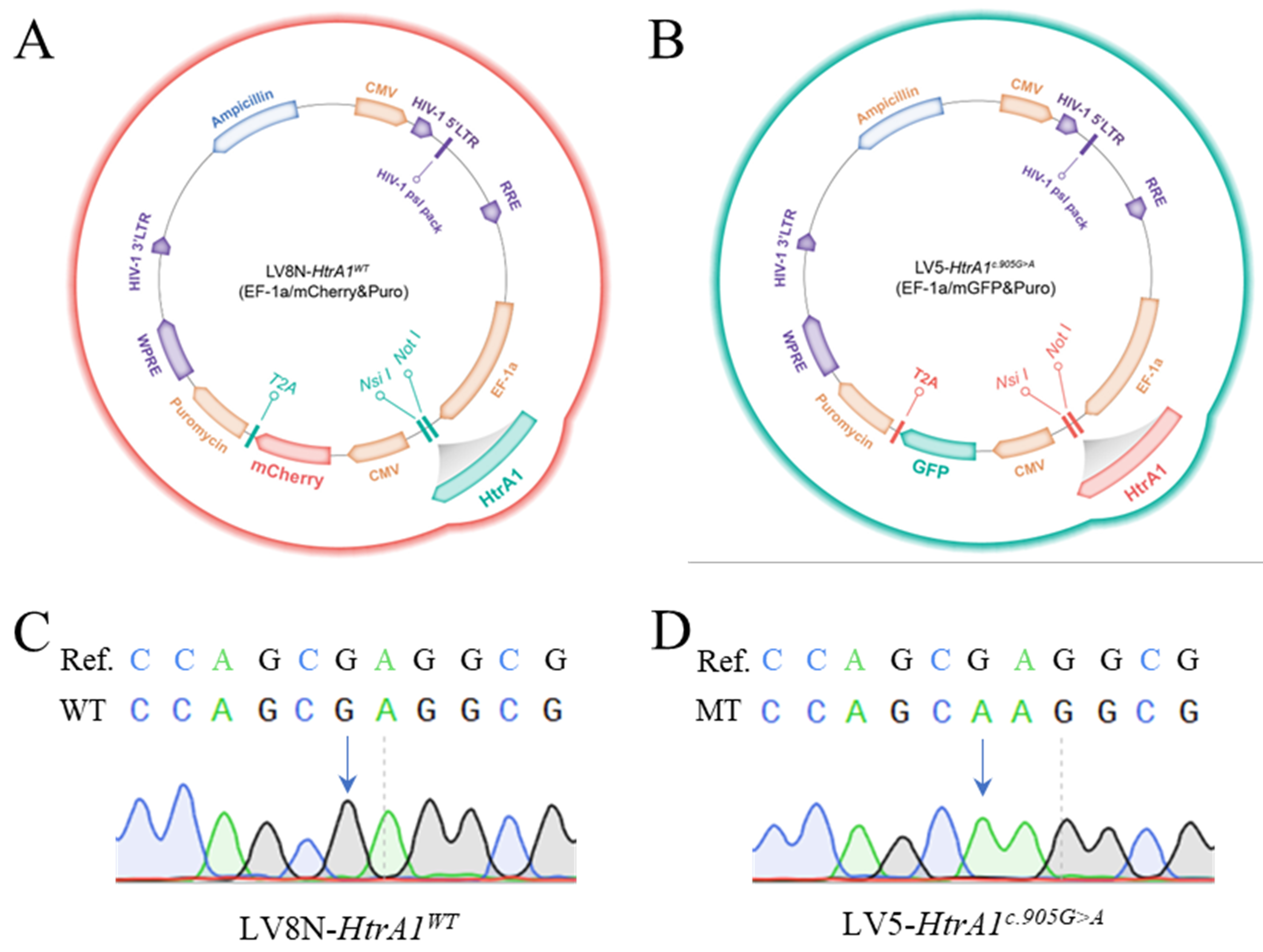 Fig. 1.
Fig. 1.
Design and construction of HtrA1WT and
HtrA1c.905G>A overexpression vectors. (A) Lentiviral vector LV8N
overexpressing HtrA1WT and containing mCherry and
puromycin. (B) Lentiviral vector LV5 containing GFP and puromycin
overexpressing HtrA1c.905G>A. (C) Sanger sequencing of
LV8N-HtrA1WT. (D) Sanger sequencing of LV5-
HtrA1c.905G>A. CMV, cytomegalovirus; EF-1
After confirming the correct sequences for the WT LV8N-HtrA1 and mutant
LV5-HtrA1 lentiviral vectors, lentiviral titer tests were performed for
LV8N, LV5, WT LV8N-HtrA1, and the mutant LV5-HtrA1. Lentiviral
titers exceeded 1
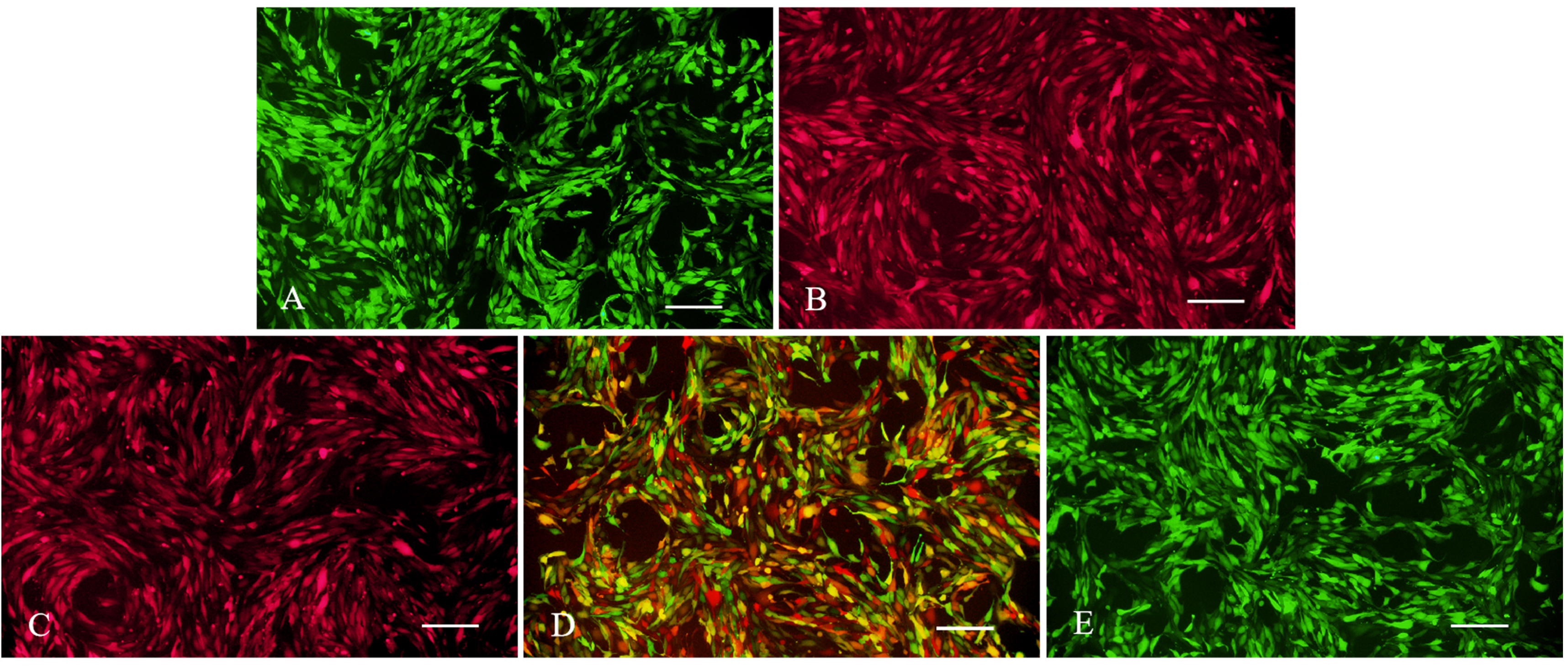
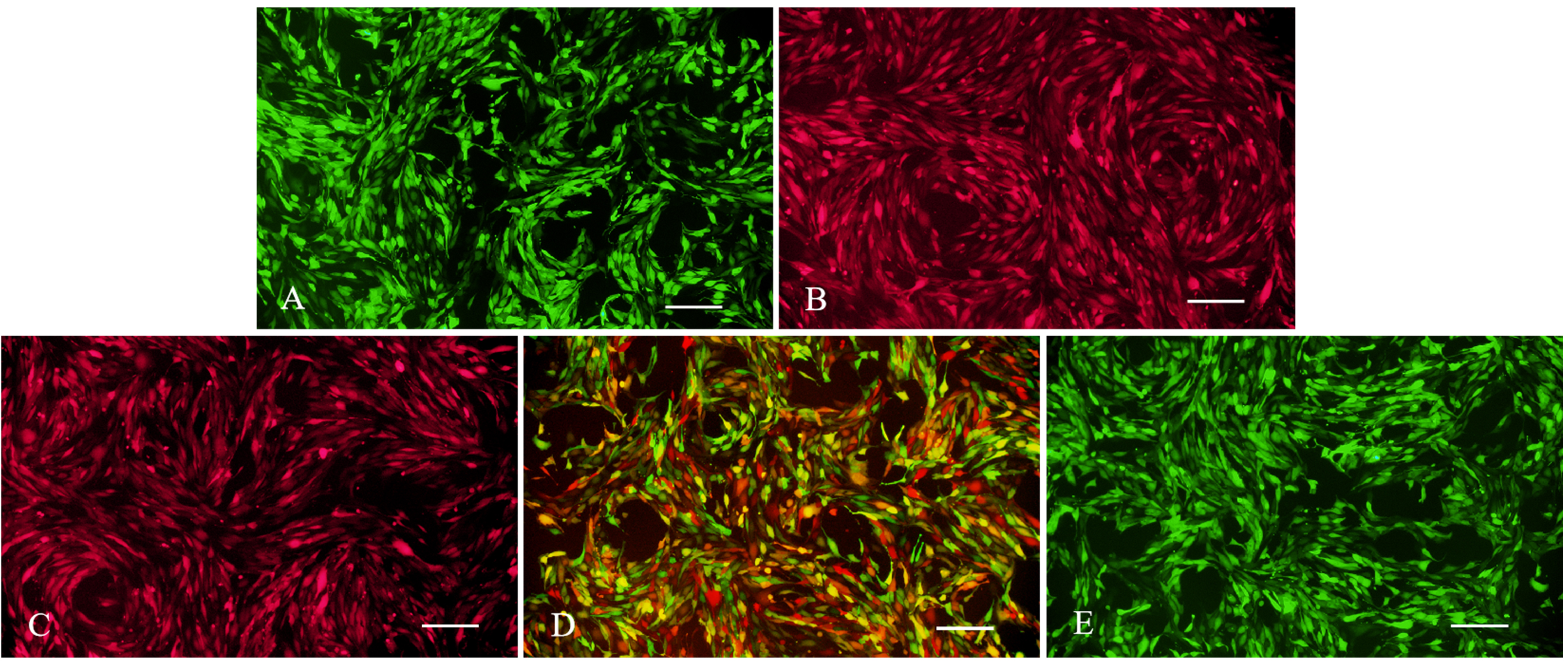 Fig. 2.
Fig. 2.
Fluorescence expression of stable strains of negative control (NC), wild-type (WT), heterozygous (Het), and homozygous (Hom) cells. (A) NC cells (LV5-NC with GFP). (B) NC cells (LV8N-NC containing mCherry red fluorescence). (C) WT cells (wild-type LV8N-HtrA1 containing mCherry red fluorescence). (D) Het cells (wild-type LV8N-HtrA1 containing mCherry red fluorescence; mutant LV5-HtrA1 containing GFP). (E) Hom cells (Mutant LV5-HtrA1 containing GFP). Bar = 200 µm.
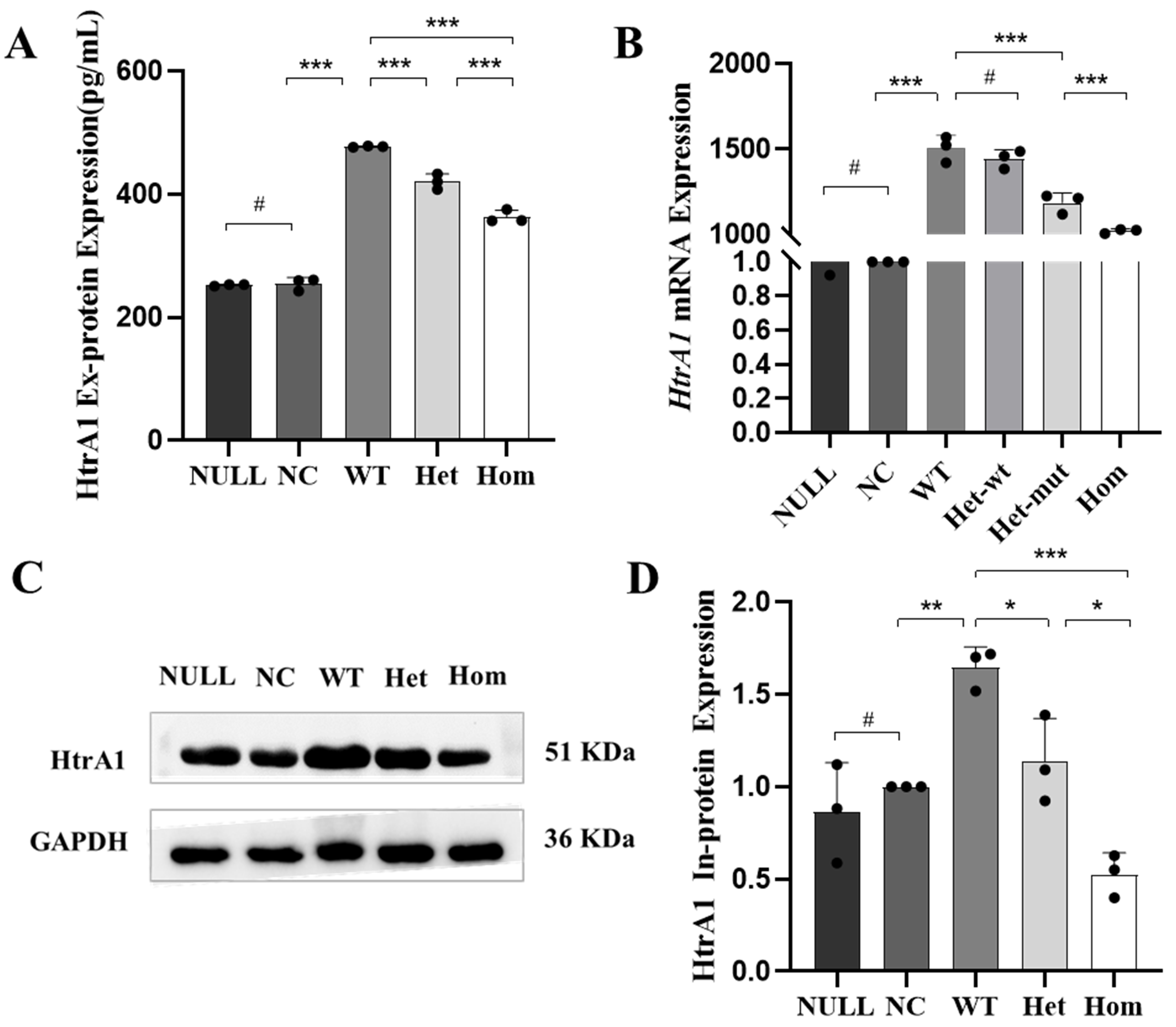
Extracellular and intracellular levels of HtrA1 mRNA and protein expression were
evaluated in each group using ELISA, qRT-PCR, and Western blot analysis. The
HtrA1 protein concentration in the supernatant of each cell group was evaluated
by ELISA. The difference in HtrA1 protein expression between NC and NULL cells
was not statistically significant (p
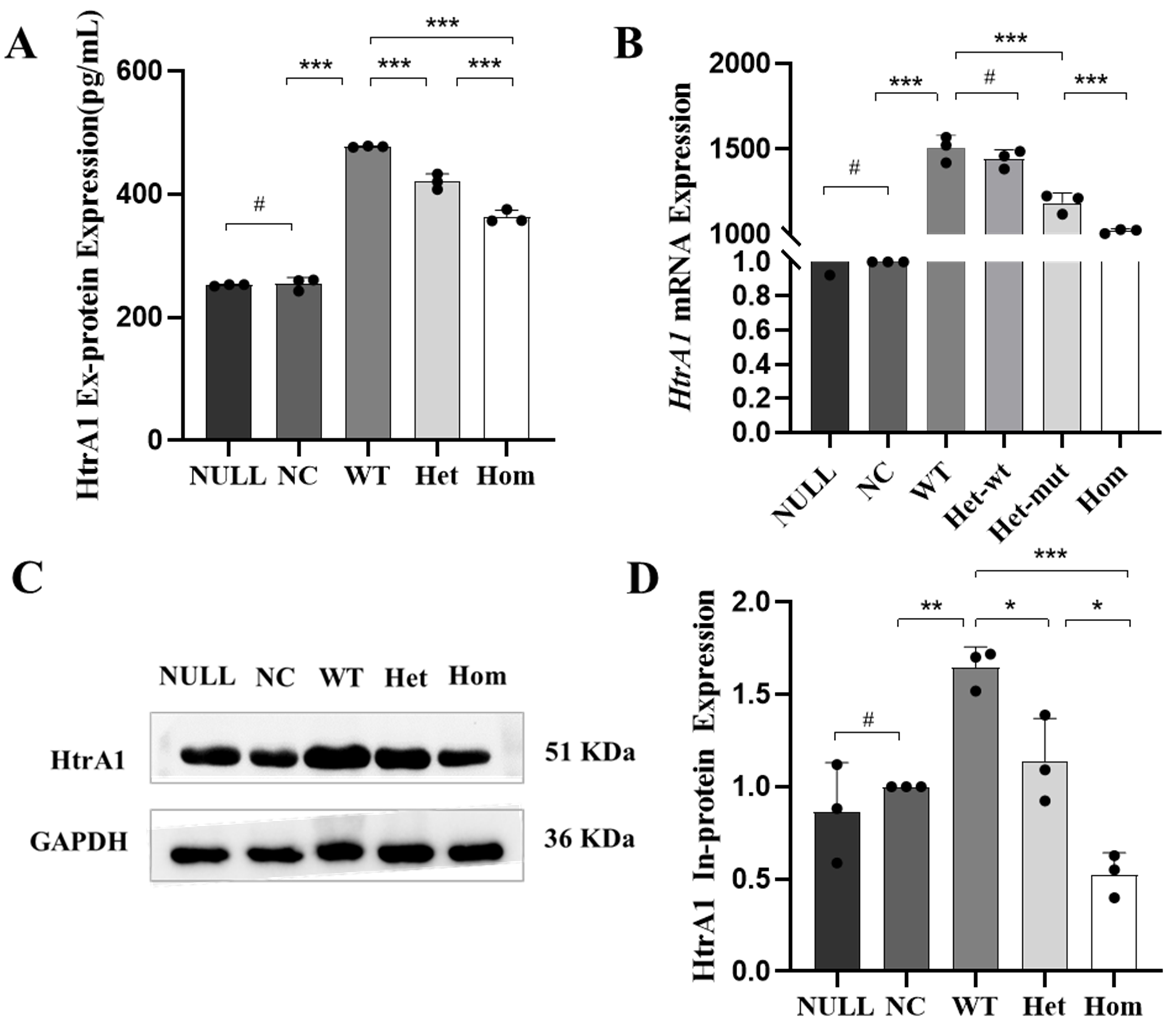 Fig. 3.
Fig. 3.
HtrA1 expression in bEnd.3 cells and in supernatants. (A) HtrA1
expression in bEnd.3 cell culture medium supernatants as measured by ELISA. (B)
qRT-PCR determination of HtrA1 mRNA expression in bEnd.3 cells. (C,D)
Measurement of HtrA1 protein expression levels in bEnd.3 cells by Western blot
analysis. #p
The above results showed that HtrA1 overexpressing cell lines were successfully constructed, and that expression of HtrA1 mRNA and protein was reduced after mutation of HtrA1. The reduction in HtrA1 mRNA and protein expression was even greater in cells with homogeneous mutation of HtrA1.
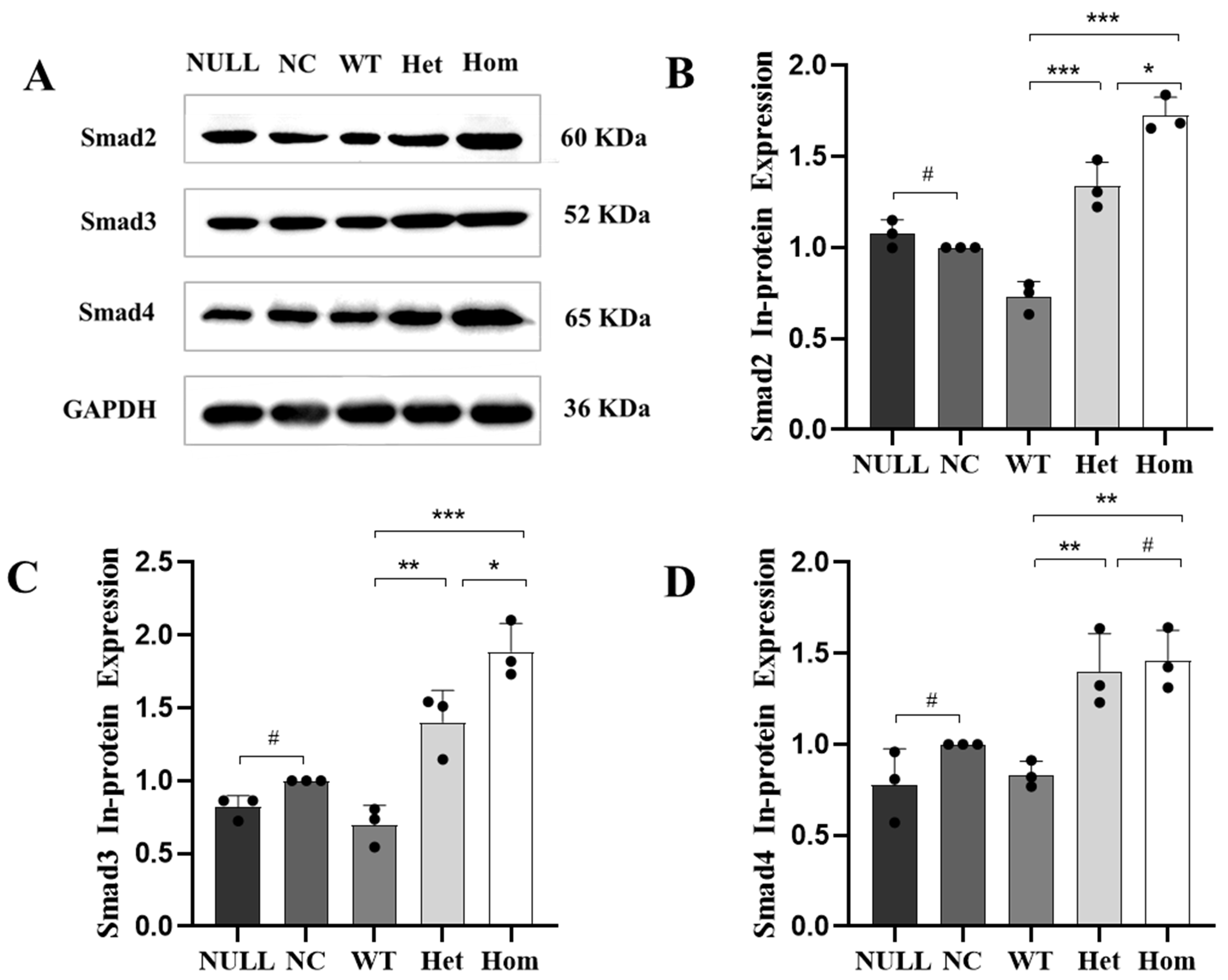
Intracellular levels of Smad2, Smad3, and
Smad4 protein expression in each group were evaluated by Western blot analysis
(Fig. 4A). No significant differences in expression were apparent between NULL
and NC cells (p
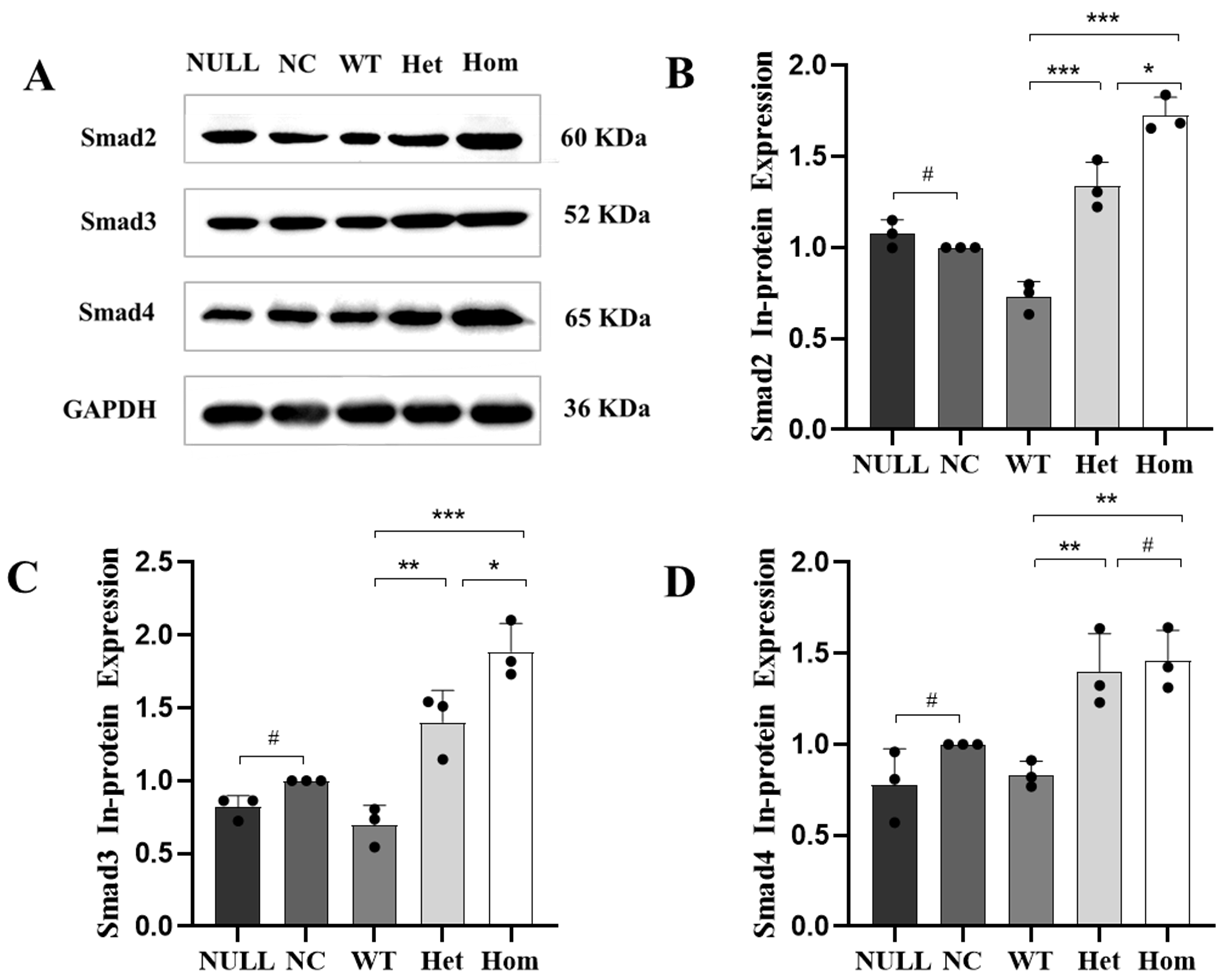 Fig. 4.
Fig. 4.
Expression of Smad2, Smad3,
and Smad4 proteins in bEnd.3 cells. (A)
Western blot analysis was used to determine the expression levels of Smad2,
Smad3, and Smad4 in bEnd.3 cells. The relative content of (B) Smad2, (C) Smad3,
and (D) Smad4 protein is shown for each cell group. #p
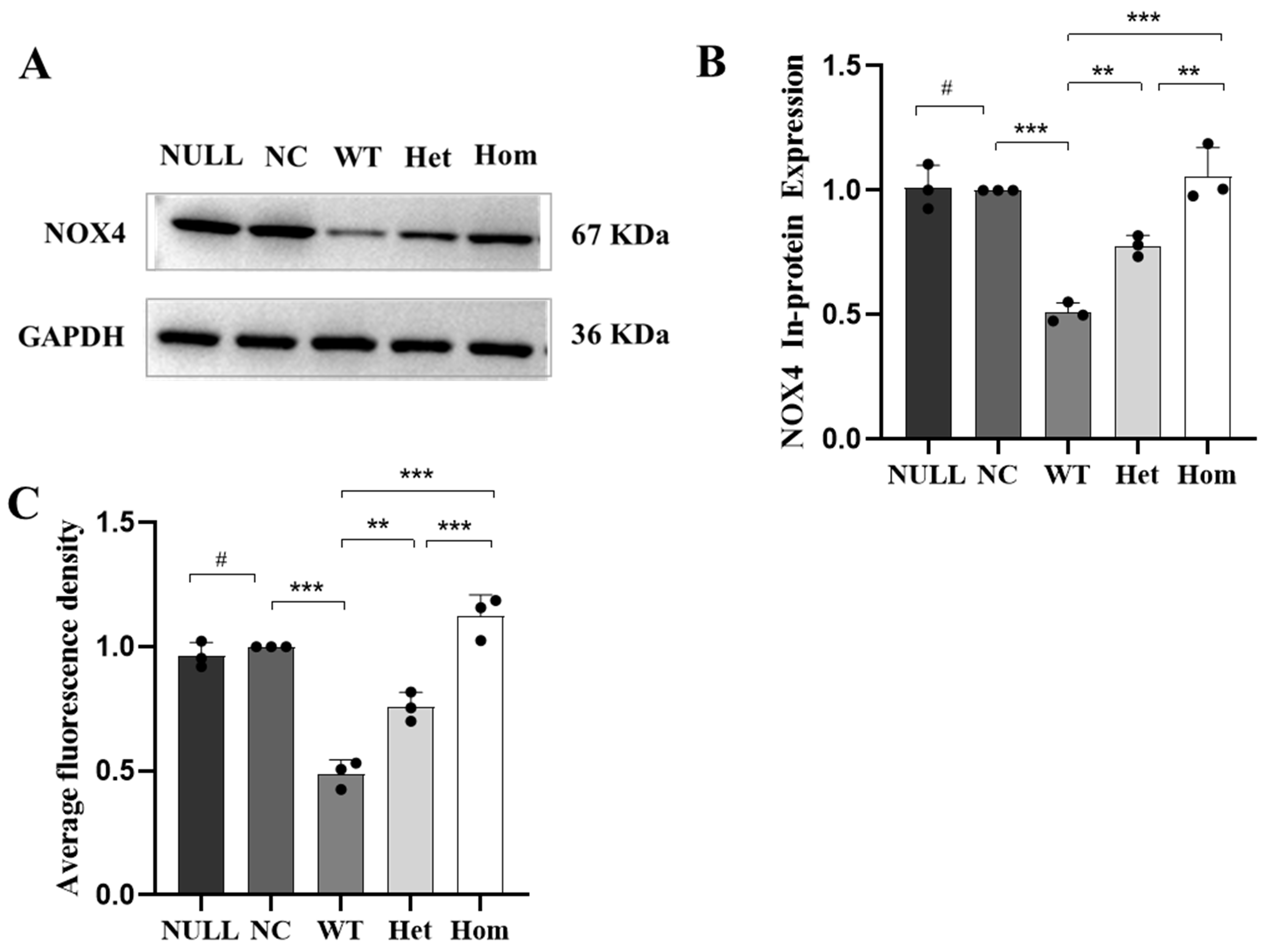
Western blot analysis was used to determine intracellular nicotinamide adenine
dinucleotide phosphate oxidase 4 (NOX4) protein expression in each cell group
(Fig. 5A,B). NOX4 protein expression was not significantly different between NULL
cells and NC cells (p
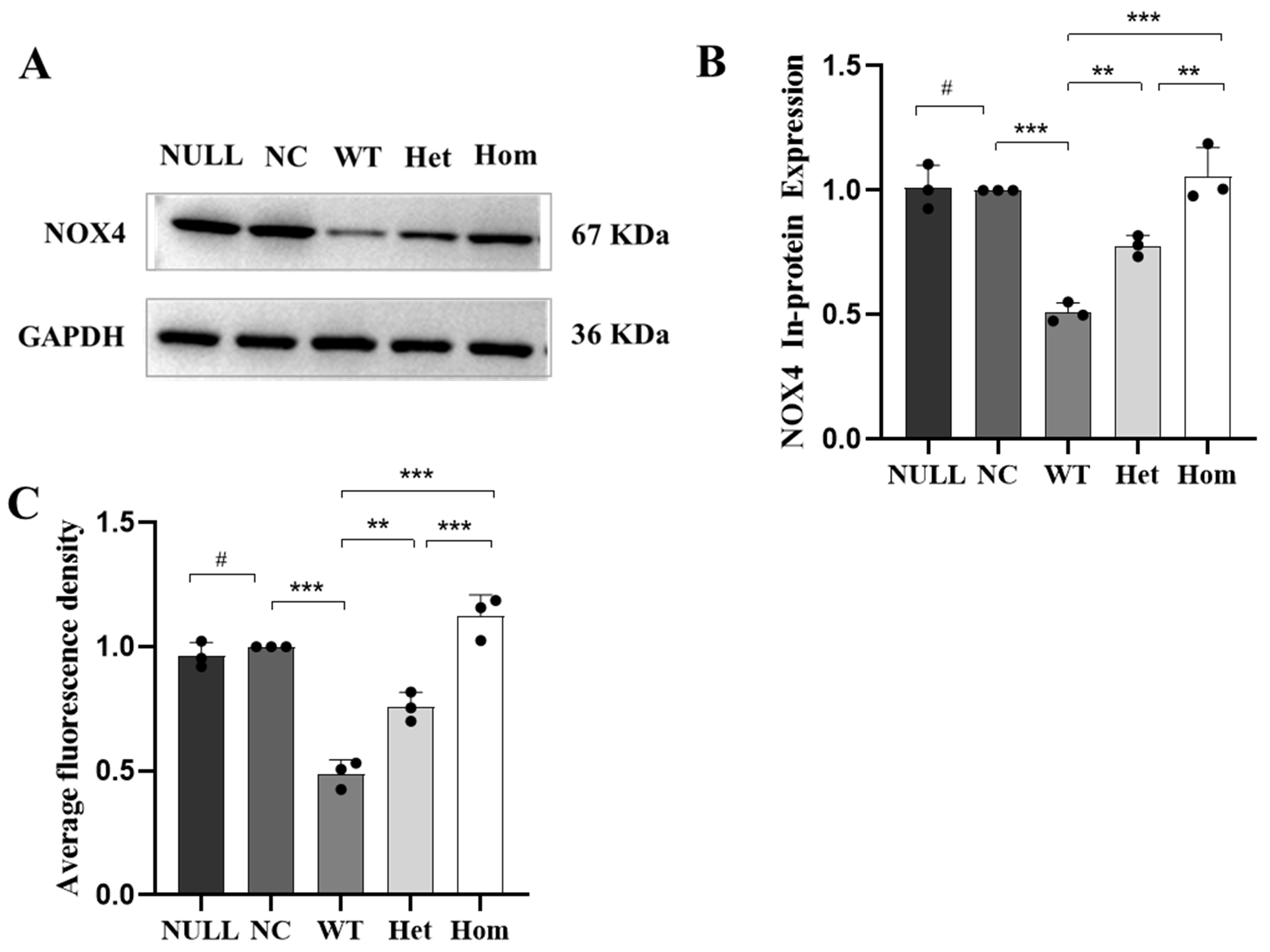 Fig. 5.
Fig. 5.
Evaluation of
oxidative stress levels. (A) Western blot analysis of NOX4 protein expression in
bEnd.3 cells. (B) Expression level of NOX4 protein in the cell groups. (C) bEnd.3
cell fluorescence intensity as determined by the DCFH-DA probe. #p
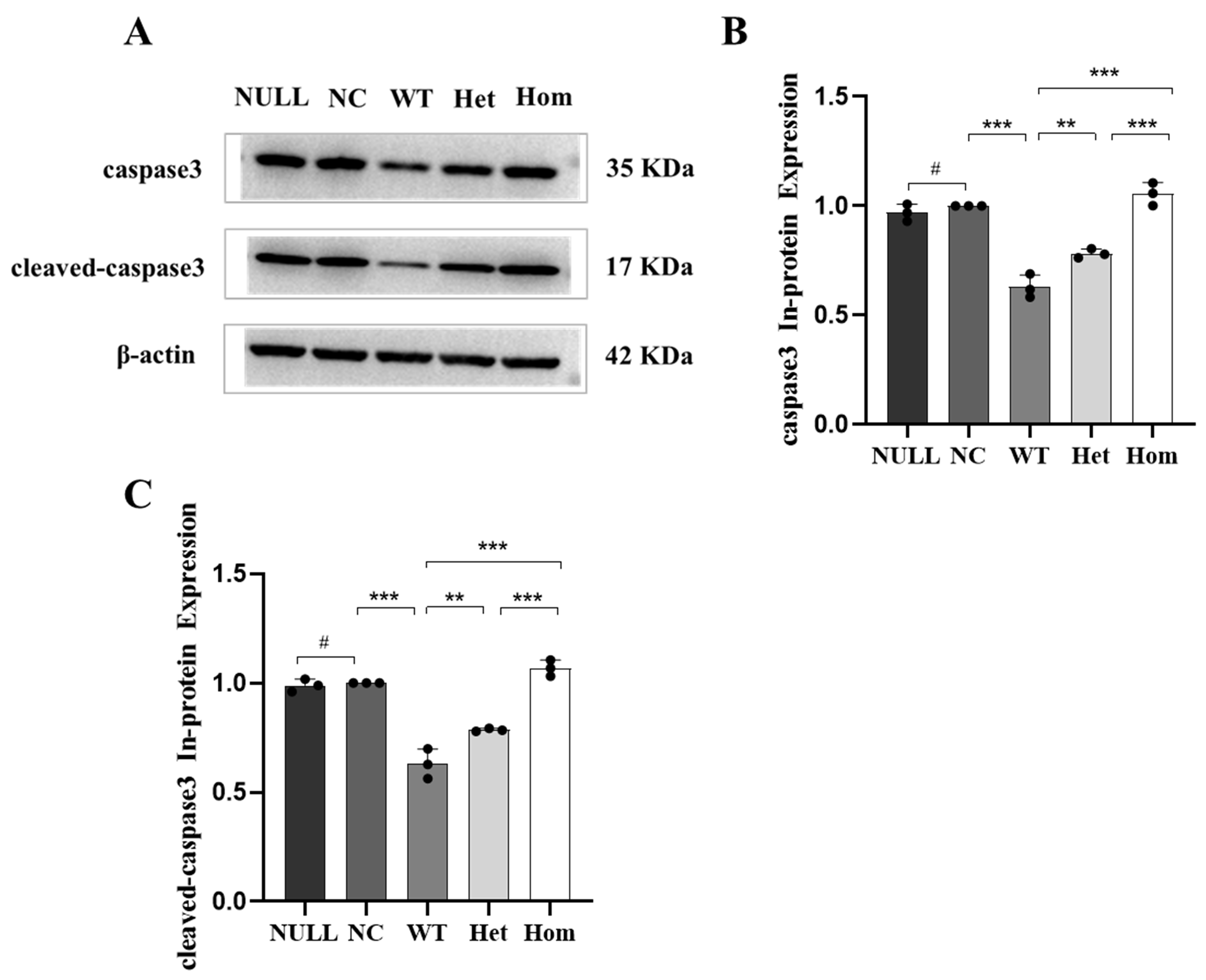
Western blot analysis was used to evaluate the expression of intracellular
caspase3 and cleaved-caspase3 protein in each cell group (Fig. 6A–C). No
significant difference in caspase3 and cleaved-caspase3 expression was apparent
between NULL and NC cells (p
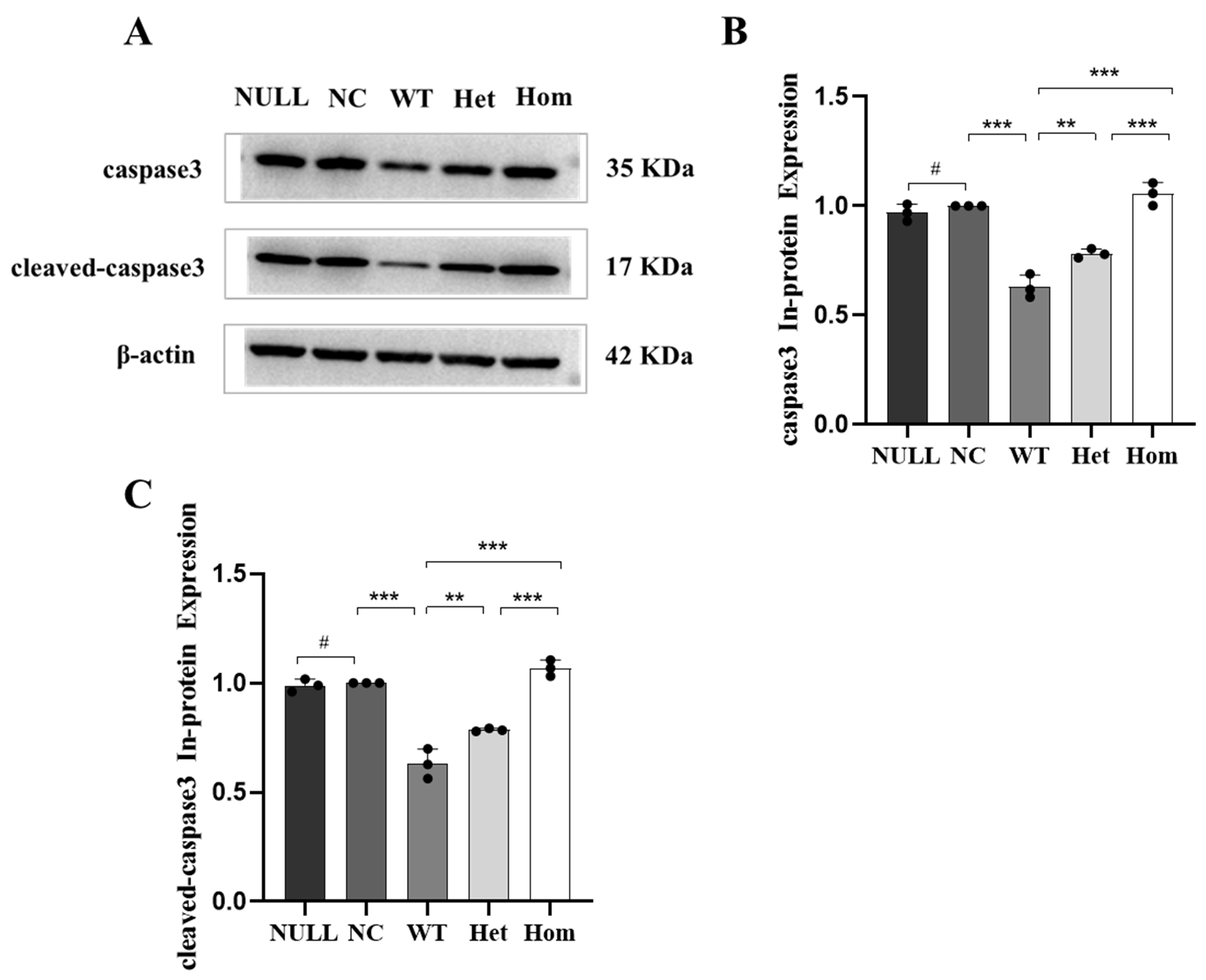 Fig. 6.
Fig. 6.
Expression of the apoptosis-related proteins caspase3
and cleaved-caspase3. (A) Western blot analysis of caspase3 and cleaved-caspase3
protein expression in bEnd.3 cells. (B,C) Relative protein expression level of
caspase3 and cleaved-caspase3 in each cell group. #p
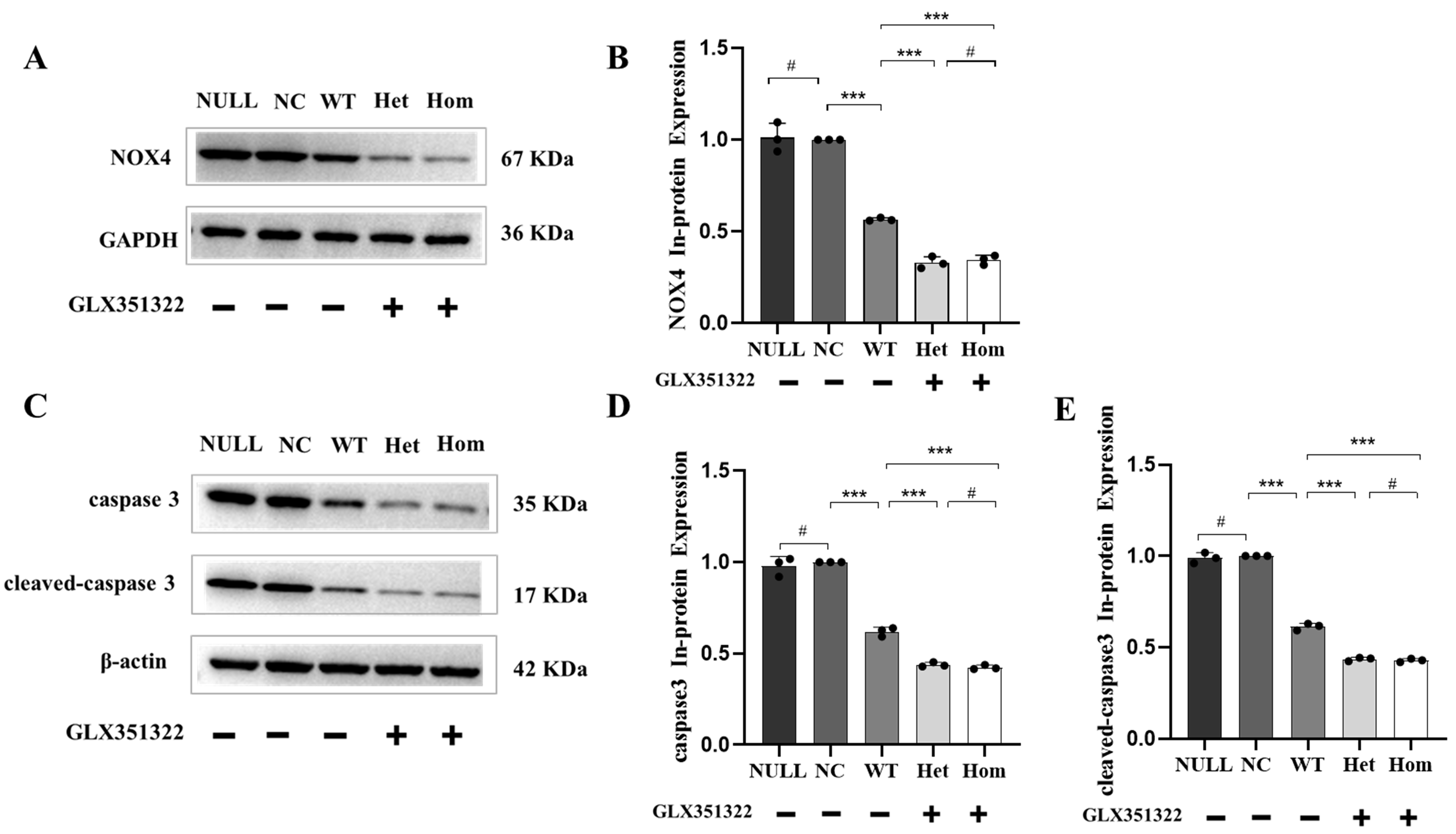
Loss-of-function experiments using the NOX4 inhibitor GLX351322 (MedChemExpress,
Monmouth Junction, NJ, USA) were performed in bEnd.3 cells with HtrA1
gene mutation (Fig. 7A–E). GLX351322 at 5 µM was added to the Het and Hom
groups for 24 h to downregulate NOX4 expression. NOX4 expression was lower in the
Het and Hom groups compared to WT (p
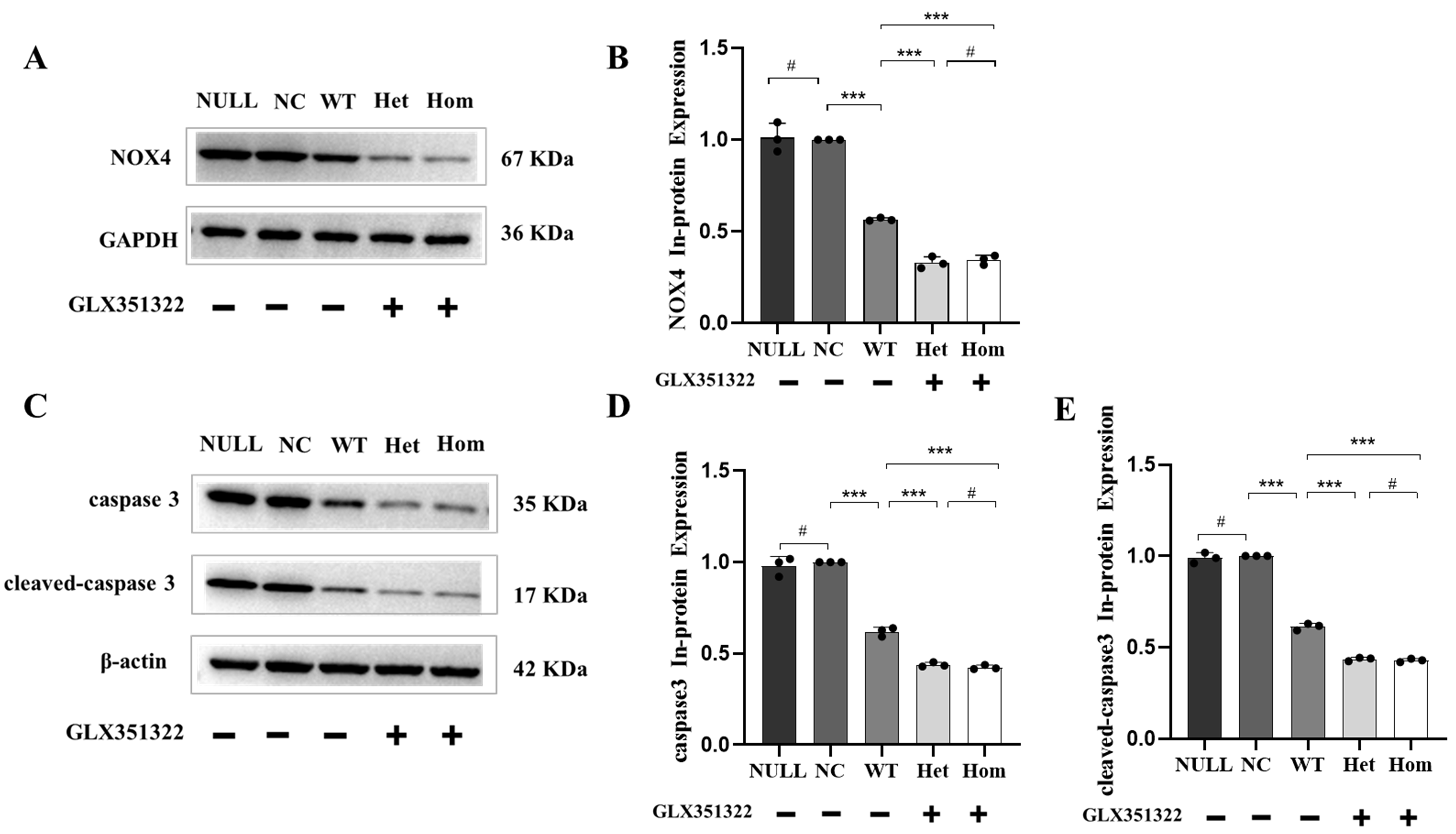 Fig. 7.
Fig. 7.
Evaluation of NOX4, caspase3 and cleaved-caspase3 expression
after addition of the NOX4 inhibitor GLX351322. (A,C) Western blot analysis of
NOX4, caspase3 and cleaved-caspase3 protein expression in bEnd.3 cells. (B,D,E)
Expression levels of NOX4, caspase3 and cleaved-caspase3 protein in the cell
groups. #p
This research found that lentivirus-mediated missense mutation in HtrA1 leads to
activation of the TGF-
An autopsy study of patients with HTRA1-associated cerebral small-vessel disease by Japanese researchers found that vascular changes were the main pathological manifestations of this disease [22]. In the present study, bEnd.3 cells were transfected with an HtrA1 overexpression lentiviral vector. Differences in HtrA1 mRNA and protein expression were found between the Hom and Het groups, indicating successful construction of the model. Homozygous and heterozygous mutations in HtrA1 show different clinical phenotypes [23, 24]. Heterozygous HtrA1 carriers have a later age of onset, fewer severe lesions, and fewer concomitant extra-neurological symptoms (e.g., baldness, spinal disorders) compared to patients with CARASIL [6, 25, 26].
In the current study, the expression of TGF-
Previous studies have shown that HtrA1 has pro-apoptotic effects in cancer cell lines [28, 29]. However, in a wound healing model it was found that HtrA1 promotes fibroblast survival and exerts anti-apoptotic effects [30]. HtrA1 is involved in estrogen-induced oxidative stress and is a member of the oxidative stress family of proteases [31, 32]. In the present study, expression of the oxidative stress-related indicators NOX4 and ROS, and of the apoptosis-related proteins caspase3 and cleaved-caspase3, were higher in the Hom and Het groups relative to the WT group. Based on our results, we conclude that changes in HtrA1 protein expression in bEnd.3 cells can alter the expression of apoptosis-related proteins and of oxidative stress-related proteins.
After inhibiting NOX4 expression in the Het and Hom groups, we found that caspase3 and cleaved-caspase3 were expressed at significantly lower levels in these cells. We therefore infer that homozygous and heterozygous missense mutations in HtrA1 result in increased expression of caspase3 and cleaved-caspase3 in bEnd.3 cells compared to controls, which may be mediated through the oxidative stress signaling pathway. Oxidative stress is closely linked to endothelial cell apoptosis, and NOX4 is the most important ROS-generating enzyme in endothelial cells [33, 34]. NOX4-dependent accumulation of ROS is a major cause of apoptosis in vascular endothelial cells [35]. NOX4 induces apoptosis in brain endothelial cells during inflammation-induced oxidative stress [36]. Furthermore, NOX4 knockdown significantly reduces ROS production, attenuates caspase3 activity, and can reduce the expression of Bcl-2 family members [37]. Therefore, the biological role of HtrA1 in different cell types is often contradictory (either protective or deleterious), depending on the cell type and its environment.
Lentivirus-mediated missense mutation of HtrA1 leads to activation of the
TGF-
The datasets during the study are available from the corresponding author upon reasonable request.
SX and CL designed the study. SS, HL, JL, and QR conducted experiments. SS drafted the manuscript. SS, HL, JL, and QR analyzed the data. SX and CL revised the manuscript accordingly. All authors read and approved the final manuscript. All authors contributed to editorial changes in the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The Ethics Committee of the First Hospital of Shanxi Medical University waived the requirement for approval of the experiment, as the vector construction were not conducted by the laboratory.
We would like to thank Editage (https://www.editage.com) for English language editing.
This research was funded by grants from Doctoral Fund of the First Hospital of Shanxi Medical University (YB161706, BS03201631, SD2215); Shanxi Applied Basic Research Program (20210302124404, 202303021221222, 202403021212232). Shanxi Scientific and Technologial Innovation Programs of Higher Education Institutions (2023L105).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/j.jin2311201.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.