

 , Evgeny Borisov 2, Andrey Grechko 3, Mikhail Popov 4, Vasily Sukhorukov 2, Alexander Orekhov 2,*
, Evgeny Borisov 2, Andrey Grechko 3, Mikhail Popov 4, Vasily Sukhorukov 2, Alexander Orekhov 2,*1 Laboratory of Angiopathology, Institute of General Pathology and Pathophysiology, 125315 Moscow, Russia
2 Petrovsky Scientific Center of Surgery, 119991 Moscow, Russia
3 Federal Research and Clinical Center of Intensive Care Medicine and Rehabilitology, 109240 Moscow, Russia
4 Department of Cardiac Surgery, Moscow Regional Research and Clinical Institute (MONIKI), 129110 Moscow, Russia
Abstract
The fight against neurodegenerative diseases is one of the key direction of modern medicine. Unfortunately, the difficulties in understanding the factors underlying the development of neurodegeneration hinder the development of breakthrough therapeutics that can stop or at least greatly slow down the progression of these diseases. In this review, it is considered the disruption of mitochondrial transport as one of the pathogenesis factors contributing to neurodegeneration using the examples of Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, and Huntington’s disease. Here, the mechanism of mitochondrial transport under normal conditions and the mechanisms of disturbances for the indicated diseases will be considered.
Keywords
- neurodegenerative diseases
- Alzheimer's disease
- Parkinson's disease
- mitochondrial transport
- mitochondria
Neurodegenerative diseases are a heavy burden for the global health system and
pose a serious threat to health, especially for the elderly. Clinically,
neurodegenerative diseases can manifest themselves both in the form of impaired
motor functions and in the form of dementia. The main feature that unites this
group of diseases is the progressive death of neurons, which determines the
chronic nature of the diseases and the absence of effective methods of treatment,
and also distinguishes neurodegenerative diseases from static neuronal damage
caused by exposure to toxic compounds [1]. In addition to clinical
manifestations, neurodegeneration also differs depending on the brain regions
(frontotemporal lobes, extrapyramidal system of the brain, spinal nerve pathways
of the spinal cord) where pathological processes occur [1]. However, the main
classification of neurodegenerative diseases is related to the difference in
abnormal structural modifications and the accumulation of the certain proteins in
neurons, which are identified as the main causes of the disease progression.
According to these anomalies, neurodegenerative diseases are classified into
tauopathies (Alzheimer’s disease), alpha-synnucleopathies (Parkinson’s disease),
amyloidosis, Fused in sarcoma gene (FUS/FET) proteinopathies, prion diseases,
neuroserpinopathies, ferritinopathies, trinucleotide repeat expansion disorders
and TAR DNA-binding protein 43 (TDP-43) proteinopathies [1]. The most common neurodegenerative diseases are
Alzheimer’s disease (AD) and Parkinson’s disease (PD). The clinical manifestation
of AD is characterized by developing dementia, which is based on a
neurodegenerative process, which is determined by two characteristic properties:
the accumulation of plaques formed from amyloid-beta, which is a breakdown
product of the amyloid precursor protein, and the formation of neurofibrillary
tangles formed from hyperphosphorylated modifications of the tau protein,
associated with microtubules [2]. The clinical manifestation of PD is associated
with impaired motor functions, the main symptoms of such disorders are rigidity,
bradykinesia, rest tremor and postural instability. The pathogenesis of PD is
based on the degeneration of dopaminergic neurons of the substantia nigra, which
is determined by the accumulation of Lewy bodies, consisting of fibrillar
formations containing
Due to the dependence of local ATP concentrations on the subcellular location of
mitochondria, the regulation of their distribution is an extremely important
mechanism responsible for the bioenergetic state of the cell. In neurons,
mitochondria are in a balance of movement and stationary state: new mitochondria
move from the neuron body along the axon towards synaptic endings to ensure
high-energy processes, such as the recycling of synaptic vesicles; old
mitochondria move in the opposite direction to the neuron body, where they are
degraded by mitophagy [9]. Mitochondrial transport occurs as a result of the
movement of motor proteins that carry out their activity mainly along
microtubules. Kinesin motor proteins promote mitochondrial transport in the
anterograde direction, i.e., from the neuron body to synapses, while dynein motor
proteins ensure retrograde transport, which runs in the opposite direction [10].
Motor proteins bind to mitochondria as a result of interaction with receptor and
adapter proteins. Mammalian receptor proteins are the Miro1 and Miro2 proteins,
which are localized on the outer mitochondrial membrane [11]. Adapter proteins
act as anchors between the receptor and motor proteins. These proteins are known
as Milton proteins or Trak1 and Trak2 [12]. Together, the complex of these
proteins ensures the movement of mitochondria along microtubules and,
accordingly, their movement into various cellular compartments. The regulation of
mitochondrial transport is of great importance for the vital activity of neurons.
The implementation of the transport of mitochondria within the axon is a complex
process that requires maintaining a balance between movement and stopping of
mitochondria in the process of their movement. One of the signals to stop
mitochondrial movement in the axon is an excess level of calcium, which is
necessary for concentrating mitochondria in areas that require the most energy
supply. The Miro protein is able to bind calcium, which provides a determination
of the calcium concentration in the environment of mitochondria. There are
several hypotheses for the stopping of the mitochondrial movement. According to
the first of them, excess calcium triggers the detachment of the complex of
motor-adapter and receptor proteins from microtubules [13]. According to another
hypothesis, a high concentration of calcium leads to the dissociation of the
motor protein from the Milton/Miro complex, which causes detachment of
mitochondria from microtubules [14]. According to the third hypothesis,
mitochondrial arrest is mediated by the activity of the syntaphilin protein,
which anchors mitochondria to microtubules [15]. In addition, the energy state of
the cell directly regulates the transport of mitochondria. Thus, it is quite
obvious that the lack of ATP and the state of hypoxia favor the anterograde
movement of mitochondria. This regulation is based on the activation of the 5’
Adenosine monophosphate-activated protein kinase (AMPK) or Hypoxia-inducible
factor 1-alpha (HIF-1
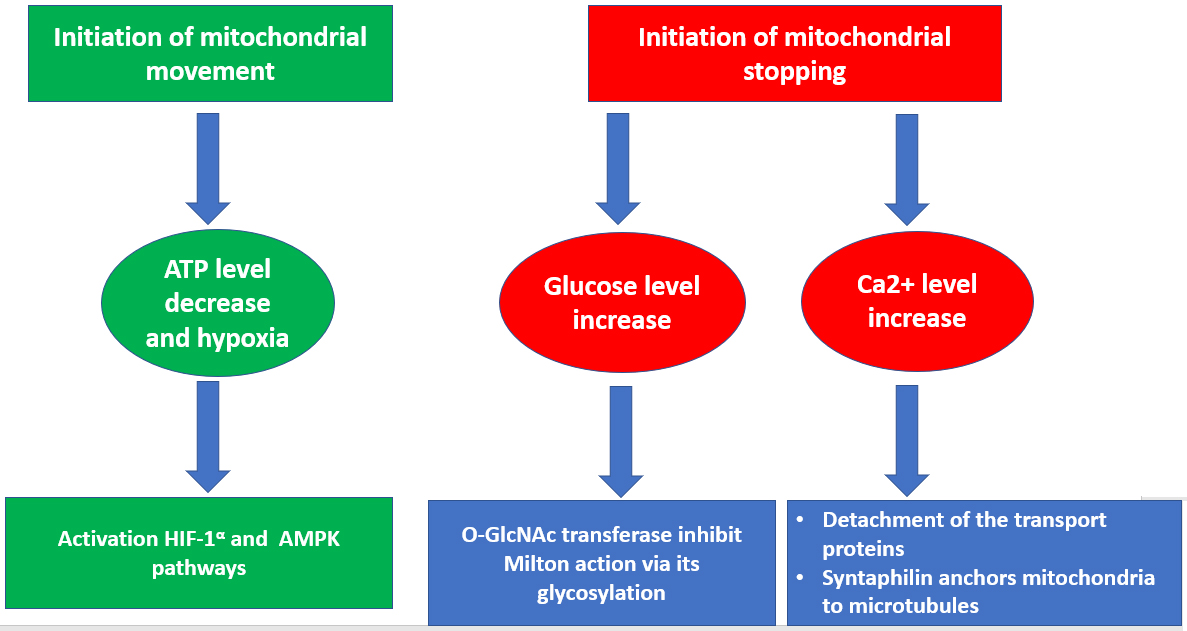 Fig. 1.
Fig. 1.General scheme of regulation of mitochondrial transport.
Modulation of mitochondrial transport can occur in response to a number of stimuluses, including changes in the concentration of calcium ions and metabolites, in the energy state of the cell as well as the development of hypoxia. In response to these stimuluses, either inhibition or activation of motor proteins of mitochondrial transport occurs, which in turn either enhances or weakens the movement of mitochondria.
It is noted that in AD, the occurrence of abnormalities in mitochondrial
transport is a closely related event in therelation to the formation of
aggregates of the toxic proteins and is directly related to the synaptic
dysfunction and a decrease in the integrity of axons [18]. The exact mechanisms
of the development of mitochondrial transport disorders in AD in neurons are not
fully known, however, there is a clear relationship between inhibition of
mitochondrial movement and such pathological processes associated with AD, such
as an imbalance in the division and fusion of mitochondria, an increase in the
concentration of A
As noted above, due to the high energy requirements of neurons for the
transmission of a nerve impulse, mitochondria are actively recruited to cellular
compartments which are remoted from the neuron body, such as axonal and dendritic
endings. The target neurons that are affected in PD, called dopaminergic neurons,
have long and relatively thin axons and minimal or no myelination. This
characteristic makes this population of neurons more sensitive to possible
changes in mitochondrial transport due to increased vulnerability to the supply
of ATP for the nerve impulse conduction due to the weak axon myelination and the
long path that mitochondria have to travel to get from the neuron body to the
axon endings [25]. Thus, it has been shown that dopaminergic neurons, unlike
other types of neurons, have a smaller number of mitochondria, which also have
smaller sizes, and, moreover, mitochondrial transport is much slower [26]. There
is evidence that proteins whose mutant forms are genetic risk factors for the
development of PD, such as Leucine rich repeat kinase 2 (LRRK2), Parkin,
PTEN-induced kinase 1 (PINK1), and
ALS is the most common neurodegenerative disease affecting motor neurons, including both upper and lower motor neurons. As a result of the dysfunction of motor neurons, muscle denervation occurs, which ultimately leads to respiratory failure, paralysis and death, which occurs within five years from the onset of the disease [2]. To date, there are no effective ways to completely cure ALS: there are only two drugs approved by the Food and Drug Administration (FDA) for use in the treatment of ALS, but they only slightly increase life expectancy [2]. This circumstance makes the search for new therapeutic targets in the pathogenesis of ALS a potentially promising direction. It is noted that one of the most frequent pathological events that occur in ALS are mitochondrial abnormalities. These abnormalities include disturbances in mitochondrial dynamics such as mitochondrial fission, fusion, and transport. The resulting disturbances in these processes lead to an increase in the number of dysfunctional mitochondria and a defective distribution of mitochondria in a neuron [32]. Since motor neurons are CNS cells with a highly elongated axoplasm, the disruption of mitochondrial function at sites that require more energy, such as muscle-bound synapses, can lead to the detrimental consequences, resulting in muscle denervation. Some evidence indicates a correlation between defects in the axonal transport and the development of the motor neuron degeneration in ALS [33]. Impaired axonal mitochondrial transport has been shown in mouse models of ALS [34, 35]. Based on the results of these studies, it was concluded that impaired transport of mitochondria in motor neurons was a process preceding degeneration. To date, the reasons underlying the development of impaired mitochondrial transport in motor neurons in ALS are not yet sufficiently understood. One of the mechanisms in this case is a direct effect on the motor proteins. Thus, in studies [36, 37], it was hypothesized that the mutant form of the antioxidant enzyme superoxide dismutase type 1 (SOD1) interacted with the complex of motor proteins dynein and dynactin, which led to the formation of aggregates and blocking of axonal transport. According to another hypothesis, mutant forms of proteins in ALS, including SOD1, form aggregates that damage mitochondria, which causes a disruption of energy metabolism and, accordingly, the production of ATP, which is also necessary for mitochondrial transport [38]. At the same time, it was shown that ATP concentrations affected the initiation of the mitochondrial movement [39]. This hypothesis directly links mitochondrial dysfunction to impaired mitochondrial dynamics in ALS. Mitochondrial transport over long distances is based on the interaction of the transport proteins with microtubules, while the movement of mitochondria over short distances is carried out with the help of the actin filaments [40]. At the same time, the actin cytoskeleton is a highly vulnerable cellular component in the diseases associated with the dysfunction of motor neurons [41]. The mutant form of the profilin 1 protein associated with the development of ALS causes a disruption in the structure of the actin cytoskeleton in motor neurons, with a decrease in the level of fibrillar actin [42, 43]. At the same time, it has been shown that the induced cytoskeletal disorders have an effect on mitochondria, which may be associated with the inhibition of their transport [42].
HD has a clearly identified etiology associated with a mutation of the gene
encoding the huntingtin protein, which has an unknown function in the norm
condition. This mutation is a multiple expansion of cytosine, adenine, and
guanine (CAG) repeats in the first exon of the gene. HD is typical for the
middle-aged people, its frequent symptoms are uncontrolled movements, the
development of dementia and the rapid onset of death after the onset of the
disease. The pathogenesis of HD is characterized by degeneration of neurons,
which are predominantly represented by striatal neurons; however, in the later
stages of the disease, neurodegeneration also affects other areas of the brain
[44]. At the same time, it has been shown in animal models that degeneration of
striatal neurons can be caused by the introduction of compounds toxic to
mitochondria, which is a prerequisite for the hypothesis of a significant role of
mitochondrial dysfunction in the development of HD [45]. Mitochondrial
dysfunction in HD was confirmed on the basis of the activity analysis for the
number of enzymes of the tricarboxylic acid cycle and oxidative phosphorylation,
which was carried out from the brain tissues of deceased patients with HD [46].
According to the results of the analysis, a decrease in the concentration of
detectable enzymes in the striatum of the brain was found. At the same time, it
was confirmed that the mutant huntingtin protein directly had a negative effect
on the metabolism of neurons. It is known from its properties that it is able to
penetrate into the cell nucleus, where it can interact with transcription
factors, thus acting as a regulator of gene transcription. In particular, it was
found that the mutant form of huntingtin is able to change the transcriptional
activity of p53 and PGC-1
As indicated above, mitochondria are highly dynamic organelles, and all processes of mitochondrial dynamics must function properly not only separately, but also in interconnection with each other, which makes it possible to effectively satisfy the energy needs of cells. Neurons obtain theestablished system of distribution, quality control and quantity of mitochondria because they have a high vulnerability associated with disturbances in the processes of mitochondrial dynamics. The fusion of mitochondria is necessary for the exchange of molecular components between the connecting mitochondria, which allows mitochondria with morphological or functional disorders to restore their function as the result of the receiving components from healthy mitochondria. The fission of mitochondria allows the damaged mitochondria to retain functioning components in one of the daughter mitochondria, and to concentrate dysfunctional elements in the other, which will undergo mitophagy, which, thus, contributes to the preservation of the functional components of mitochondria. The subtle interaction between these processes and the high dependence of neurons on their conduction means that even seemingly minor disturbances can lead to the development of the serious neurodegenerative disorders.
Since the size of the mitochondria directly affects its mobility, the disturbances that occur during mitochondrial division also affect mitochondrial transport. One of the key regulatory proteins of mitochondrial division is Dynamin-related protein (DRP1). In a culture of hippocampal neurons, it was shown that induced disturbances in DRP1-mediated mitochondrial division led to abnormal mitochondrial distribution associated with an increase in the number of mitochondria in the neuron body and a decrease in their number in dendritic endings [58]. This indicates that the deficiency of division leads to a decrease in the efficiency of mitochondrial transport to the distal regions of neurons. In turn, dysfunction of the mitochondrial transport protein Miro1, in addition to inhibition of mitochondrial transport, causes enhanced mitochondrial fission, and its overexpression has the opposite effect [59].
Fusion initiation requires the convergence of neighboring mitochondria, so mitochondrial transport is essential for mitochondrial fusion to occur. In turn, the fusion of mitochondria also affects their movement. It has been shown that in neurons with a mutation of the gene encoding the main regulatory fusion protein Mitofusin 2 (MFN2), the overall mitochondrial mobility decreases, which leads to disruption of both retrograde and anterograde mitochondrial transport [60]. In a study [61], it was demonstrated that the interaction of MFN2 with the Miro-Milton protein complex was required for the axonal transport of mitochondria.
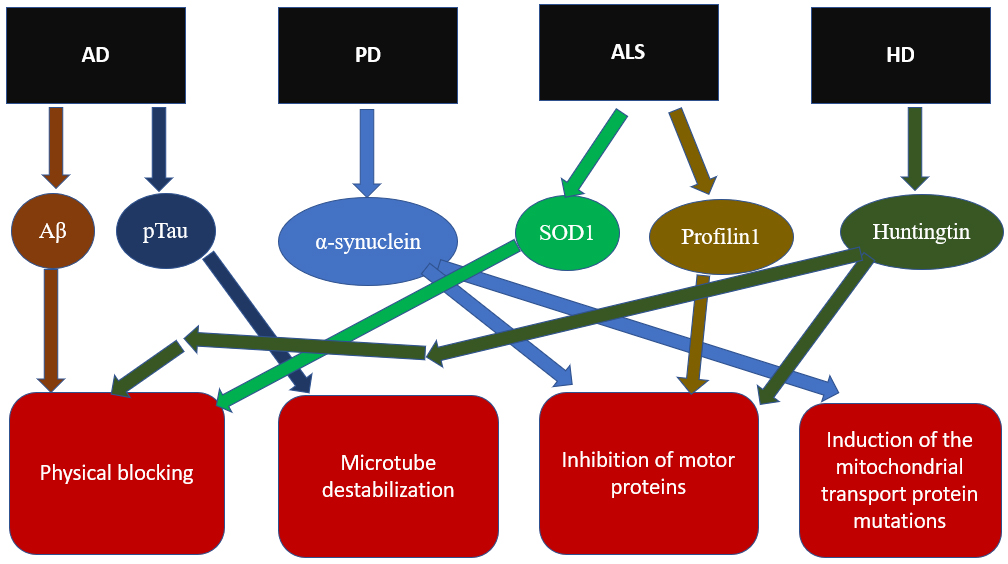
A direct relationship between the processes of mitophagy and mitochondrial transport has not yet been fully established. At the same time, the mitophagy regulatory enzyme PINK1 is known to be able to bind to the Miro-Milton complex [62]. Also, in a study on a culture of neurons, it was shown that at the early stage of mitophagy, degradation of proteins of the outer mitochondrial membrane developed, among which the transport proteins Miro1 and Miro2 were also present [63]. This circumstance shows that in the course of mitophagy, suppression of mitochondrial transport occurs. As a result, the movement of mitochondria stops due to the detachment of the motor proteins from mitochondria. The roles of the pathological proteins of the described neurodegenerative diseases in the disruption of mitochondrial transport are shown in Fig. 2.
 Fig. 2.
Fig. 2.Mechanisms of impaired mitochondrial transport in the neurodegenerative diseases.
Despite differences in the type of affected neurons, the variants of initiating
pathological factors, and other features that characterize neurodegenerative
diseases, there are some common patterns that are characteristic of progressive
neuronal damage. In this review, we described the most well-known and common
examples of neurodegenerative diseases, and for each of them data have been shown
which indicating the occurrence of the mitochondrial transport disorders in
neurons, which characterize this pathology as a rule rather than an exception in
the pathogenesis of neurodegenerative disorders. Despite the differences, there
are also common signs in the mitochondrial transport disorders for the different
diseases. First of all, defects in mitochondrial transport are most often
initiated by the main characterized mutant proteins associated with the
progression of the disease under consideration: A
| Disease | Inhibitor | Inhibition mechanisms |
| AD | A |
Possible physical blocking of transport during the formation of A |
| AD | pTau | Glycogen synthase kinase 3 phosphorylates Tau at the AT8 site, leading to Tau fibril formation and microtubule destabilization |
| PD | (1) Impaired binding of kinesin to microtubules | |
| (2) Possible induction of mutations in the genes of mitochondrial transport proteins | ||
| ALS | SOD1 | (1) Formation of protein aggregates blocking mitochondrial transport |
| (2) It is possible that aggregates damage mitochondria, which leads to their dysfunction and a lack of ATP for transport | ||
| ALS | Profilin1 | Disruption of actin cytoskeleton structure |
| HD | Huntingtin | (1) Formation of protein aggregates blocking mitochondrial transport |
| (2) N-terminal peptide fragments block the binding of mitochondria to motor proteins |
AD, Alzheimer’s disease; PD, Parkinson’s disease; ALS, amyotrophic lateral sclerosis; HD, Huntington’s disease; SOD1, superoxide dismutase type 1; ATP, adenosine triphosphate.
As noted in the previous section, mitochondrial transport is not an isolated
process of mitochondrial dynamics, but works in close relationship with
mitochondrial fission and fusion, as well as mitophagy. Accordingly, disorders
that directly affect mitochondrial transport in neurons also potentially lead to
changes in mitochondrial fusion and fission, and vice versa. These indirect
mechanisms of influence may be important for understanding the pathogenesis of
neurodegenerative diseases, but require further study. Also of great importance
is the accumulation of a large amount of data on impaired axonal movement of
mitochondria during the course of the disease in patients with neurodegenerative
disorders, since it is still not completely known at what stages of the disease
these disorders occur and how they are associated with the clinical
manifestations of diseases. The other step in this direction is the study of the
modulation of internal apoptosis regulated by the mitochondrial proteins. In
connection with the disruption of mitochondrial function, the regulation of
internal apoptosis is also disturbed, which leads to the spread of
neurodegeneration. To a certain extent, this is facilitated by the main
pathological proteins of neurogenerative diseases: A
The greater vulnerability of neurons to mitochondrial function identifies mitochondria as one of the key targets of neurodegenerative diseases. At the same time, an important component of the work of mitochondria, especially in neurons, is the correct implementation of mitochondrial transport, which contributes to the required distribution of mitochondria in neuron compartments depending on energy needs. Impaired mitochondrial transport is one of the important events in the pathogenesis of neurodegenerative diseases. At the same time, mutant proteins, which are pathological factors of these diseases, play a key role in the initiation of this disorder. The main mechanisms of inhibition of mitochondrial axonal movement are physical blocking of transport caused by the formation of protein aggregates and competitive binding of mutant proteins to microtubule motor proteins. The development of drugs that inhibit the spread of pathological protein complexes in neurons or their binding to microtubules may be a promising direction for the therapy of neurodegenerative diseases because of the restoration of the energy state of neurons and further decline in neurodegeneration.
AO, EB and VS designed the review plan and wrote the manuscript. AB, AG and MP aquized, analyzed and interpreted the data and wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by the Russian Science Foundation (Grant # 23-25-00237).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.