






 , Miguel Ángel Pozo 1,2,6, Luis García-García 1,2,3
, Miguel Ángel Pozo 1,2,6, Luis García-García 1,2,31 Unidad de Cartografía Cerebral, Instituto Pluridisciplinar, Universidad Complutense de Madrid, 28040 Madrid, Spain
2 Unidad de Cartografía Cerebral, Instituto de Investigación Sanitaria, Hospital Clínico San Carlos (IdISSC), 28040 Madrid, Spain
3 Departamento de Farmacología, Farmacognosia y Botánica, Facultad de Farmacia, Universidad Complutense de Madrid, 28040 Madrid, Spain
4 Bioimagen Complutense (BIOIMAC), Universidad Complutense de Madrid, 28040 Madrid, Spain
5 Servicio de Medicina Nuclear, Instituto de Investigación Sanitaria, Hospital Clínico San Carlos (IdISSC), 28040 Madrid, Spain
6 Departamento de Fisiología, Facultad de Medicina, Universidad Complutense de Madrid, 28040 Madrid, Spain
Abstract
Background: Epilepsy is one of the most common neurologic diseases, and
around 30% of all epilepsies, particularly the temporal lobe epilepsy (TLE), are
highly refractory to current pharmacological treatments. Abnormal synchronic
neuronal activity, brain glucose metabolism alterations, neurodegeneration and
neuroinflammation are features of epilepsy. Further, neuroinflammation has been
shown to contribute to dysregulation of neuronal excitability and the progression
of epileptogenesis. Flufenamic acid (FLU), a non-steroidal anti-inflammatory
drug, is also characterized by its wide properties as a dose-dependent ion
channel modulator. In this context, in vitro studies have shown that it
abolishes seizure-like events in neocortical slices stimulated with a
gamma-aminobutyric acid A (GABA
Keywords
- epilepsy
- flufenamic acid
- neuroinflammation
- PET
- epileptogenesis
- FDG
Epilepsy is one of the most common and disabling neurological disorders exerting a considerable suffering on more than 50 million afflicted patients worldwide according to the World Health Organization (https://www.who.int/news-room/fact-sheets/detail/epilepsy). Temporal lobe epilepsy (TLE) is one of the most predominant forms of focal epilepsy [1] being highly refractory to the currently available pharmacological treatments [2]. Research focused on finding safe and effective therapeutical drugs is still a need [1, 2]. Nowadays, common pathways relating epilepsy and seizures to neuroinflammation have been reported [3, 4, 5]. In fact, neuroinflammation is considered both, consequence and cause of various forms of epilepsy, playing an important role in the etiopathophysiology of seizures [6, 7, 8, 9]. In the last two decades, mounting data, even though yet inconclusive, suggest potential therapeutic effects of a non-steroidal anti-inflammatory drugs (NSAIDs) in epilepsy [10] pointing towards this pharmacologic group as a new potential therapeutic tool to tackle epilepsy by means of managing the neuroinflammatory response.
Flufenamic acid (FLU), 2-[3-(trifluoromethyl) phenyl]aminobenzoic acid, is a NSAID included with mefenamic, meclofenamic and niflumic acids in the fenamate group. FLU is a non-selective inhibitor of cyclooxygenases 1 and 2 (COX-1, COX-2), thereby reducing the synthesis of prostaglandins, prostacyclins, and thromboxanes [11]. FLU, in a regimen of 600 mg/day for 3 months, has currently limited clinical applications for the management of dysmenorrhea [12] as well as mild to moderate pain associated with joint and musculoskeletal disorders [13]. Nevertheless, multiple experimental evidences indicate that FLU is a broad spectrum, dose-dependent ion channel complex modulator, including gap junction channels, chloride channels, with a preference for non-selective cation channels, including certain subtypes of potassium and calcium channels [11, 14, 15, 16, 17]. Given the role of neuroinflammation and channel activity modulation as contributors in the etiology of neurological and neurodegenerative diseases, the repurposing of fenamate NSAIDs as potential therapeutic tools has been considered. In this context, its potential therapeutic role in neurological and neurodegenerative diseases has been considered given the role of neuroinflammation and channel activity modulation as contributors in the etiology of these pathological entities [18, 19].
The use of animal models of seizures and epileptogenesis constitute a valuable experimental tool to study the potential antiseizure, antiepileptic, neuroprotective and anti-inflammatory effect of new pharmacological agents. The lithium-pilocarpine model of status epilepticus (SE) and epileptogenesis in rodents is considered as an improved evolution from the original pilocarpine model of SE [20, 21, 22] given that it is characterized by a lower mortality rate while able to generate in a reproducible manner consistent and prolonged seizures [23]. The lithium-pilocarpine model is a promising model for studying SE resembles many, although not all, the behavioral, electrographic, proteomic and neuropathological features of human TLE [24, 25, 26].
The epileptogenic process in the lithium-pilocarpine model is characterized by a
rapid occurrence of the SE, followed by a latent silent period characterized by
the absence of spontaneous seizures. This silent stage is accompanied by: (i) a
generalized hypometabolism measured by 2-deoxy-2-[
In vivo studies, using different animal models of seizures and epileptogenesis have reported inconsistent results regarding the potential antiseizure or anticonvulsant activity of non-selective NSAIDs as well as COX-2 selective inhibitors [10, 32, 33, 34]. Nevertheless, and as far as we know, no animal studies have been conducted to evaluate the effects of FLU in the pilocarpine or lithium-pilocarpine rat models. Thus, in the current study, we have sought to explore the effects of a single dose of FLU when administered prior to pilocarpine on the behavioral and neurobiological damage associated with the SE that are characteristically observed during the early phase of the latent period.
Adult Sprague-Dawley male rats (Charles Rivers, Spain) were housed in pairs in
standard cages on a ventilated rack (Tecniplast, Buguggiate, Italy). The animal
room was under controlled conditions of temperature (22
The lithium-pilocarpine model was used to trigger the SE as previously reported
[28, 29, 30]. Briefly, lithium chloride (3 mEq/kg (
The LD50 for FLU in rats when administered i.p. is 185 mg/kg (https://cdn.caymanchem.com/cdn/msds/21447m.pdf). In addition, studies in rats have shown that FLU administered i.p at a dose of 66 mg/kg did not affect movement, feeding and drinking behavior, alertness, and sustained attention to the environment, while reaching body fluids concentrations able to block TRPM2 channels [40]. Based on this information, and to evaluate the eventual effects of FLU, a single intermediate dose of this NSAID (100 mg/kg in DMSO, i.p; Sigma–Aldrich, St. Louis, MO, USA) was chosen to be administered 2 h before methyl-scopolamine. The respective control groups were injected with DMSO as vehicle (1 mL/kg). Thus, initially the experiment consisted of 4 experimental groups with 32 rats distributed as follows: (i) vehicle + saline (VEH + SAL, n = 6), FLU + saline (FLU + SAL, n = 6), vehicle + pilocarpine (VEH + PILO, n = 8) and FLU + pilocarpine (FLU + PILO, n = 12) (Fig. 1A). The number of rats included in the groups subjected to SE was higher having in view the risk of mortality associated to the procedure.
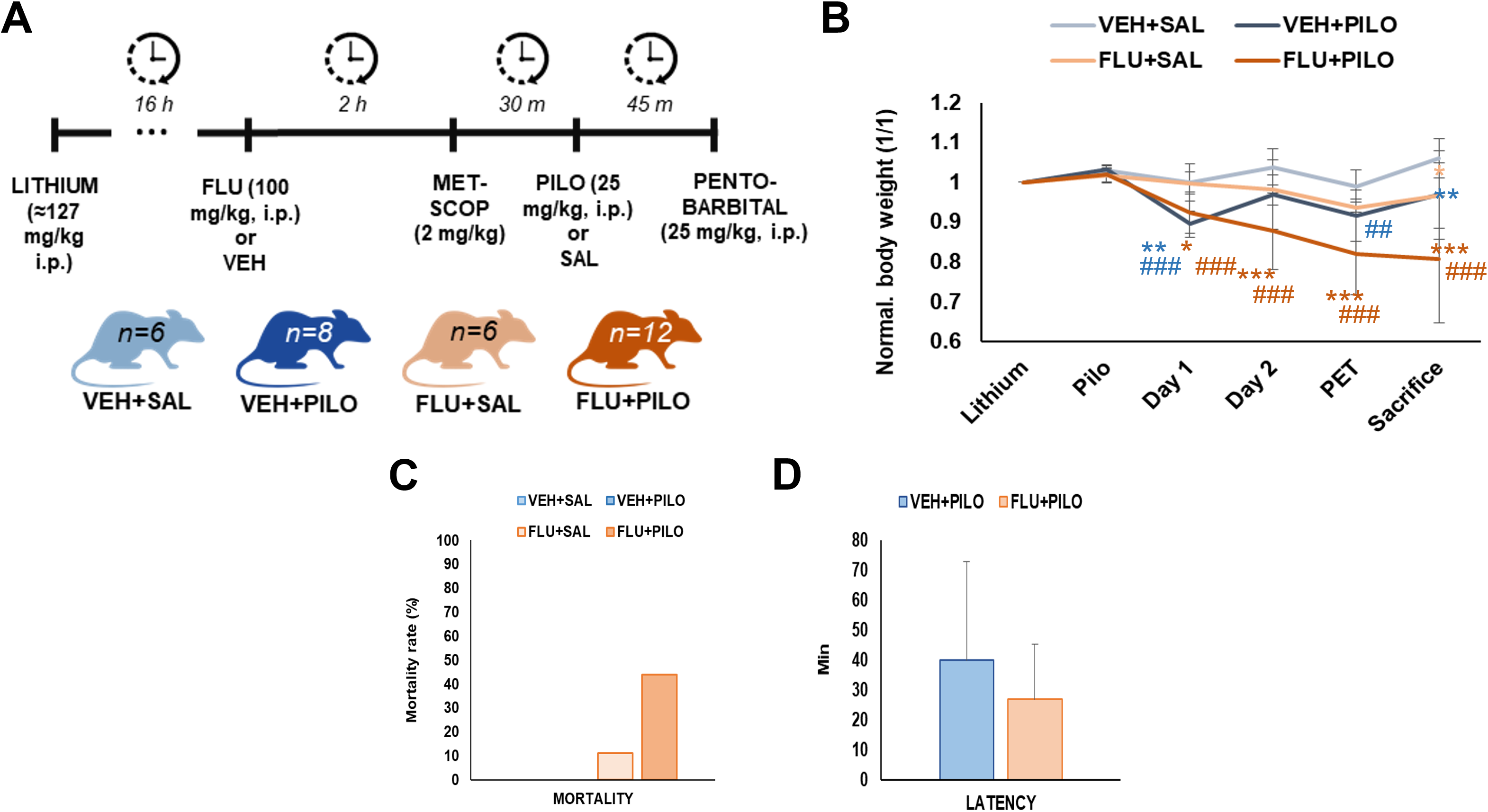 Fig. 1.
Fig. 1.Descriptive data. (A) Schematic representation of seizure model
and drug treatment methodology with the resultant experimental groups. (B)
Graphical representation of animals BW change relative to the initial BW. Data is
expressed as mean
Three days after the SE, during the latent period, brain glucose metabolism was
evaluated by in vivo [
After acquisitions, PET images were reconstructed by maximum likelihood expectation maximization algorithm applying decay, random and scatter corrections (Albira Reconstructor, Bruker, Ettlingen, Germany). CT scans were co-registered to a magnetic resonance imaging (MRI) rat brain with pre-defined volumes of interest (VOIs). Each CT mathematical transformation was applied to co-register PET images to the MRI template to obtain tracer uptake values in the different brain areas (kBq/mL; PMOD 4.1, PMOD Technologies Ltd., Zurich, Switzerland). The regional glucose metabolic activity was evaluated in brain areas known to be involved in epileptogenesis by calculating the standard uptake value (SUV), in order to correct the individual weight differences, by applying the following equation: [Tracer uptake (kBq/mL) * BW (g)) / (Tracer dose administered (kBq))].
Animals were sacrificed by decapitation the day after the PET scan. Brains were
collected and stored at –80 °C. Thirty-
Nissl staining was performed to obtain a qualitative histological representation of hippocampal integrity. Sections containing the anterior hippocampus were fixed in formaldehyde (4% in phosphate buffer saline, 30 min) and then transferred to a solution of 0.5% of cressyl violet in 0.1% acetic acid for 1 h. Slices were washed in distilled water, dehydrated in ethanol graded solutions (70%, 95% and 100%), cleared in xylene and finally covered with DPX mounting medium (Merck Sigma-Aldrich, Darmstadt, Germany). The images were collected with a digital camera (DFC425 camera, Leica, Wetzlar, Germany) coupled to a microscope (Leitz Laborlux S, Leica, Wetzlar, Germany).
Fluoro-Jade C labeling was used as a marker of hippocampal neurodegeneration [41]. Briefly, brain slices were fixed in 4% formaldehyde, washed in phosphate buffer, and then rinsed in basic alcohol, 100% ethanol and distilled water. Afterwards, slides were rinsed in 0.06% potassium permanganate, 0.1% acetic acid solution containing 0.0001% Fluoro-Jade C (Merck Millipore, Darmstadt, Germany), distilled water and xylene, and mounted with DPX.
GFAP immunofluorescence was evaluated as a marker of reactive astrogliosis in the hippocampus. The sections were fixed in 4% formaldehyde, rinsed in Tris-buffered saline (TBS pH 7.6, 30 minutes) and blocked in 3% bovine serum albumin (BSA), 0.1% triton X-100 in TBS one hour. For GFAP immunofluorescence, slices were incubated with conjugated anti-GFAP-Cy3 antibody (1:500 1% BSA/TBS; Sigma–Aldrich, St. Louis, MO, USA) at 4 °C overnight and cleared in 0.1% Tween 20 and mounted with Mowiol.
NeuN immunofluorescence was evaluated as a biomarker of neuronal integrity. To this end, slides were incubated at 4 °C overnight with a primary antibody (mouse anti-NeuN 1:200, ref. MAB377, Millipore, Darmstadt, Germany), washed in TBS-0.1% tween 20 and, incubated with the anti-mouse secondary antibody (1:140, 1% BSA/TBS, ref. T5393, Sigma–Aldrich, St. Louis, MO, USA) conjugated with tetramethylrhodamine isothiocyanate (TRITC) for 2 h at room temperature. Then, slides were washed again in TBS-0.1% tween 20 and covered with Mowiol.
Images from Fluoro-Jade C, GFAP and NeuN were acquired using a Leitz Laborlux S microscope (Leica Biosystems, Wetzlar, Germany). The filters used were as follows: FITC filter for Fluoro-Jade C and TRITC filter for GFAP and NeuN. In order to carry out the quantification, the different hippocampal subareas (CA1, CA3 and hilus) of each brain slice were manually delimited by an operator blinded to the animal’s identification and the mean fluorescence signal value of those regions were obtained. The average value for each animal was obtained from 4 different brain slices containing dorsal hippocampus. Both delimitation and quantification steps were performed with ImageJ software (NIH, available at the following website: https://imagej.nih.gov/ij/).
Data are shown as mean
Results were analyzed as cumulative BW changes relative of the initial BW. When
groups were compared to their main control (VEH + SAL), VEH + PILO animals only
showed significant differences in BW at days 1 and 4 after SE (p = 0.003
and p = 0.01) whether FLU + PILO was different until the sacrifice of
animals (p = 0.04, p = 0.01, p = 0.005 and p
Pilocarpine administration triggered SE after 39.9
Unexpectedly, there was no mortality associated with the SE in the VEH + PILO group. By contrast, FLU pre-treatment arkedly increased mortality (z = 2.513; p = 0.012). Thus, from the 12 initial animals in the FLU + PILO group, 8 rats died (66.6%). The temporal mortality occurrence was as follows: 1 rat died 1h after the SE, 2 rats died 24 h after the SE and 5 rats 48 h after the SE. Surprisingly, 2 rats out of 6 also died in the FLU + SAL group (Fig. 1C). Thus, the following results were obtained from data collected from the surviving rats. The final number in the 4 experimental groups were: (i) VEH + SAL, n = 6; (ii) FLU + SAL, n = 4; (iii) VEH + PILO, n = 8 and (iv) FLU + PILO, n = 4.
Comparing VEH + PILO vs VEH + SAL (Fig. 2), pilocarpine-induced SE resulted in
significant brain glucose hypometabolism, measured as percentage of [
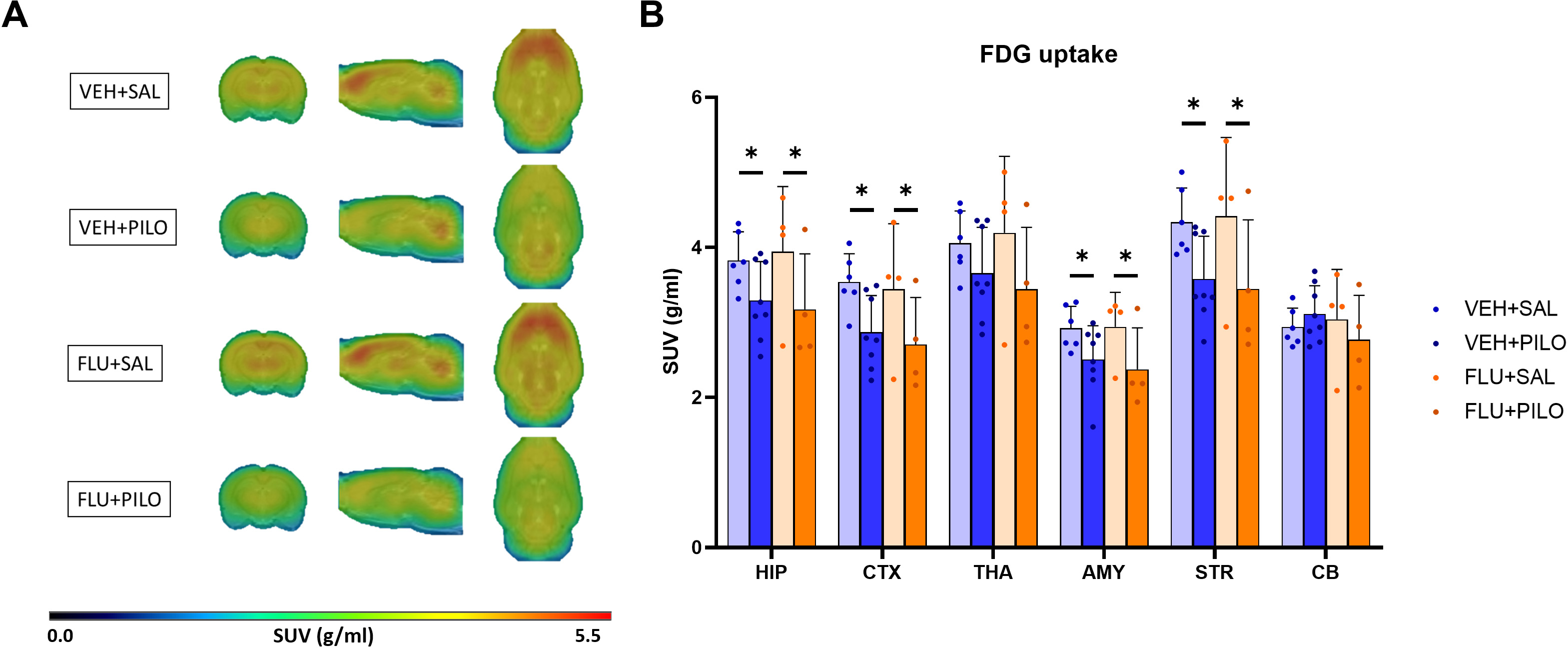 Fig. 2.
Fig. 2.SE induced by pilocarpine decreased [
The Fig. 3 depicts representative images obtained from the Nissl staining in brain sections containing the anterior hippocampus. The images are shown as qualitative data. As can be observed, pilocarpine reduced the intensity of the staining in all the hippocampal subregions, being more marked in the CA1. Regarding FLU, pre-administration of this NSAID had no apparent effects in FLU + SAL or in FLU + PILO in comparison to their vehicle control groups.
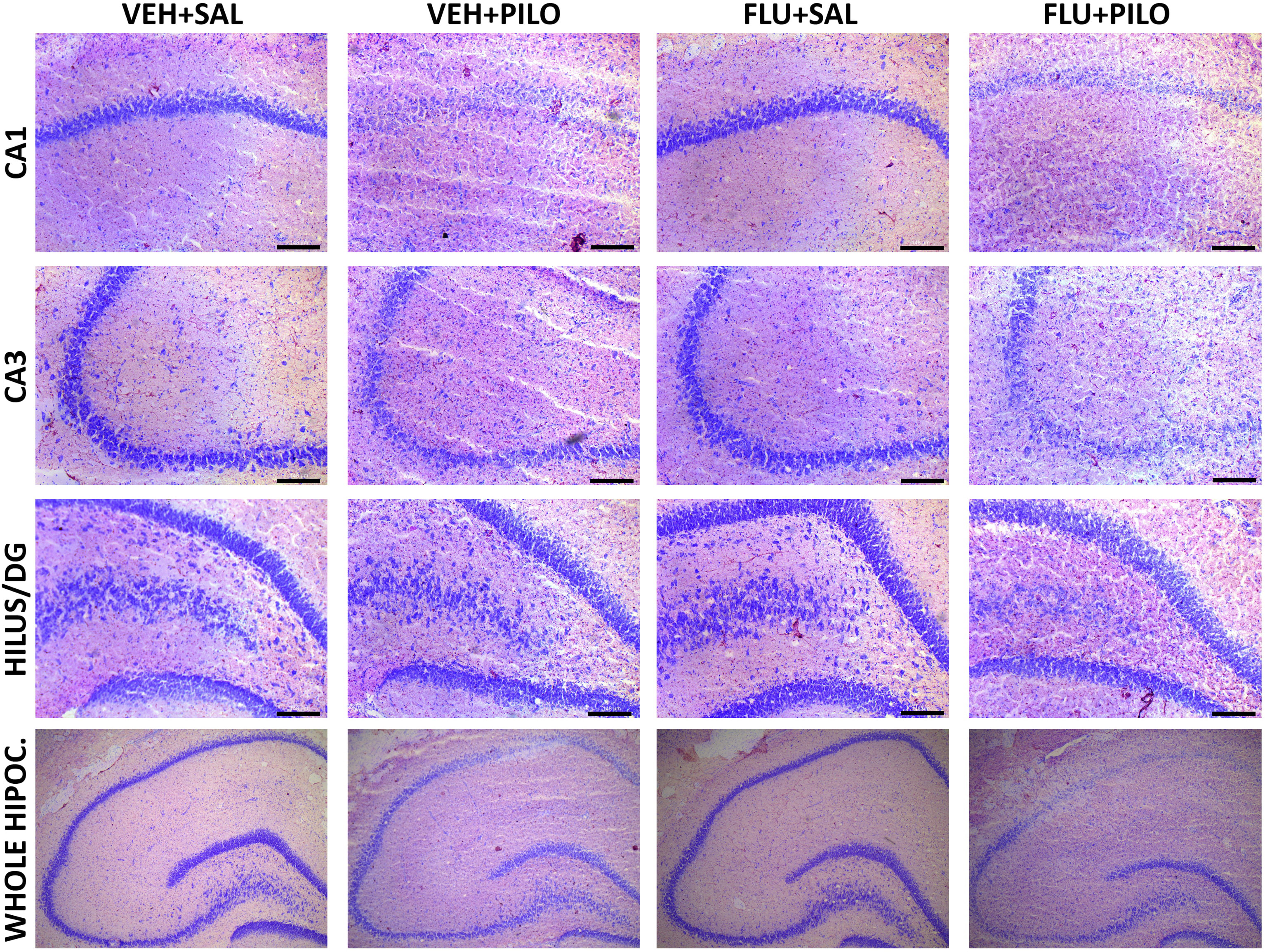 Fig. 3.
Fig. 3.Neurohistochemical images of cresyl violet (Nissl staining) show a severe degeneration of CA1 and CA3 regions in pilocarpine-insulted groups. As shown, this disruption of the hippocampal integrity was not ameliorated by previous administration of 100 mg/kg FLU.
Results from Fluoro-Jade C fluorescence intensity in the different hippocampal
areas were calculated in the VEH + PILO and FLU + PILO as percentage of their
respective control groups (refer to section 2.9). Pilocarpine treatment increased
hippocampal neurodegeneration. Comparing VEH + PILO vs VEH + SAL, the increase in
Fluoro-Jade C fluorescence were as follows: CA1: 179.1%, p
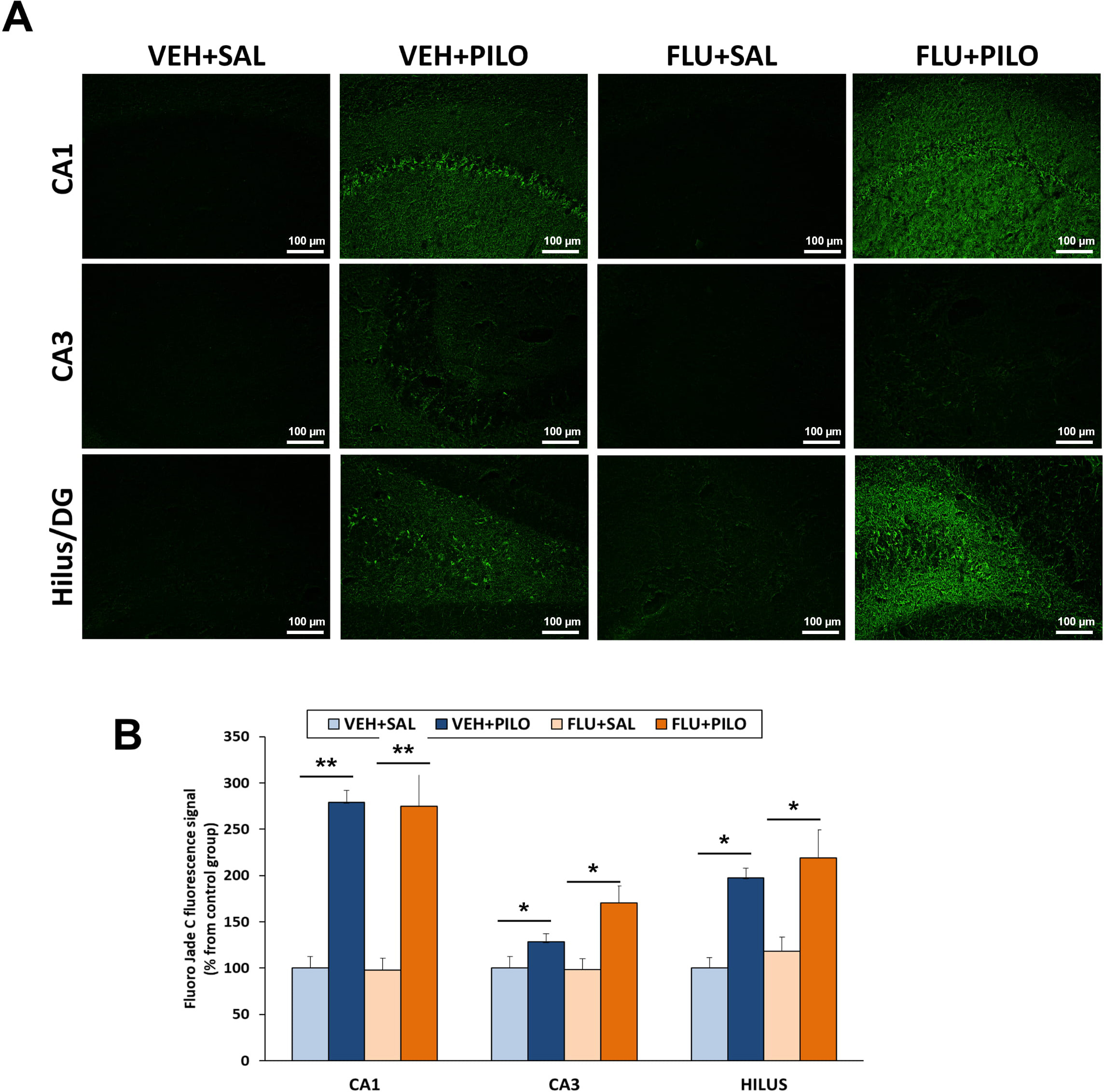 Fig. 4.
Fig. 4.Hippocampal neurodegeneration (assessed by Fluoro-Jade C
staining) triggered by pilocarpine injection was not reduced by FLU
administration. (A) Representative images from Fluoro-Jade C staining. (B) Plot
of fluorescence intensity values of Fluoro-Jade C (mean
As shown in Fig. 5, pilocarpine treatment, independently from administration of
FLU, significantly reduced NeuN immunofluorescence intensity in the hippocampal
CA1 subregion (p = 0.013). Neither pilocarpine nor FLU significantly
altered NeuN signaling in the hippocampal CA3 and hilus subregions. Nonetheless,
there was a trend (p
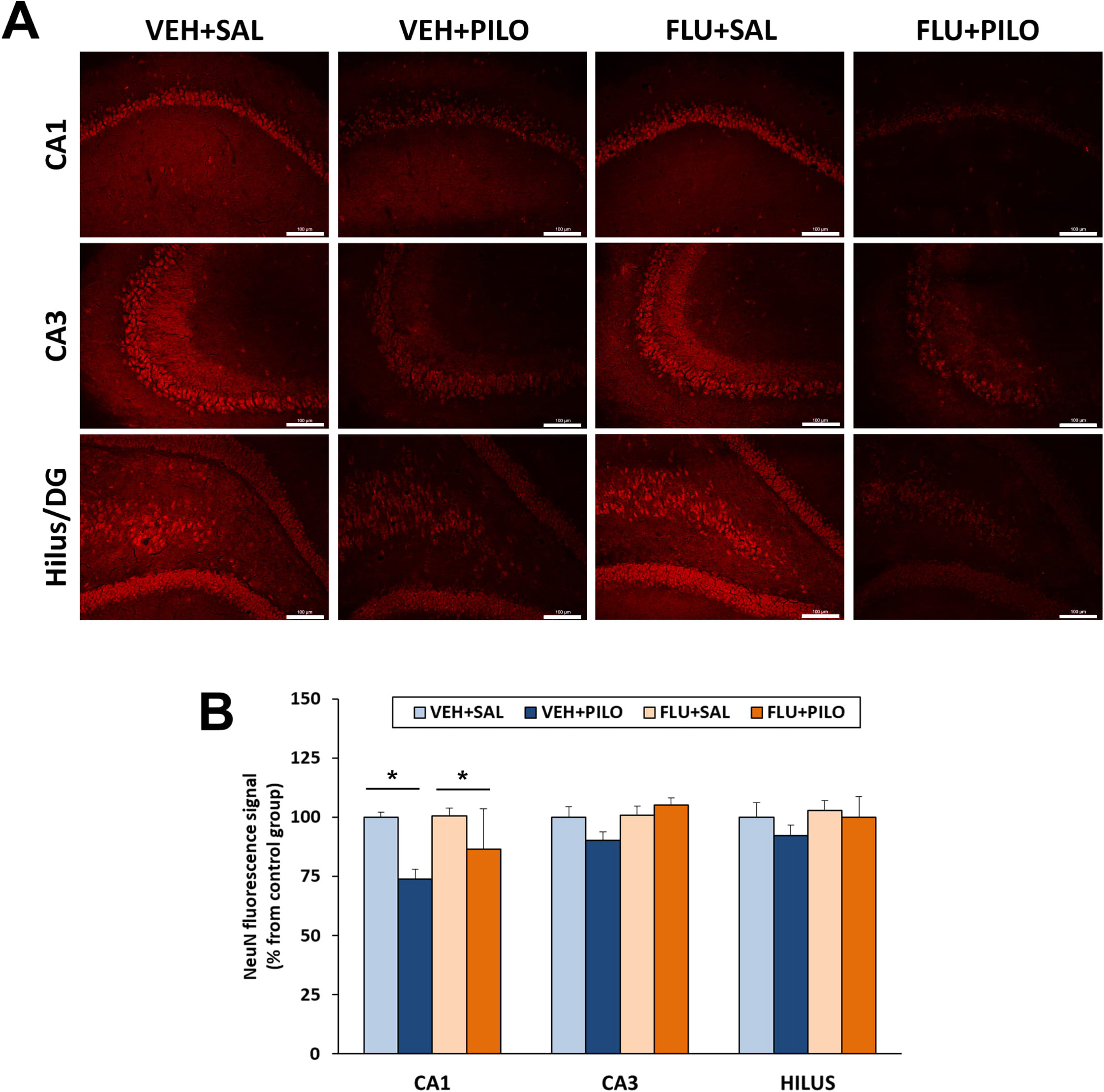 Fig. 5.
Fig. 5.FLU pretreatment (100 mg/kg) was ineffective at rescuing CA1
neuronal loss (evaluated by NeuN immunofluorescence) in rats injected with
pilocarpine. (A) Representative images of hippocampal subregions showing
immunofluorescence NeuN signal. (B) Plot of NeuN fluorescence signal intensity
(mean
GFAP immunofluorescence intensity was measured as percentage vs their respective
control groups. Pilocarpine treatment resulted in reactive astrogliosis in the 3
hippocampal subregions analyzed, but FLU by itself did not have significant
effects. Thus, comparing VEH + PILO vs VEH + SAL, the increments in GFAP
intensity were as follows: CA1: 63.6%, p
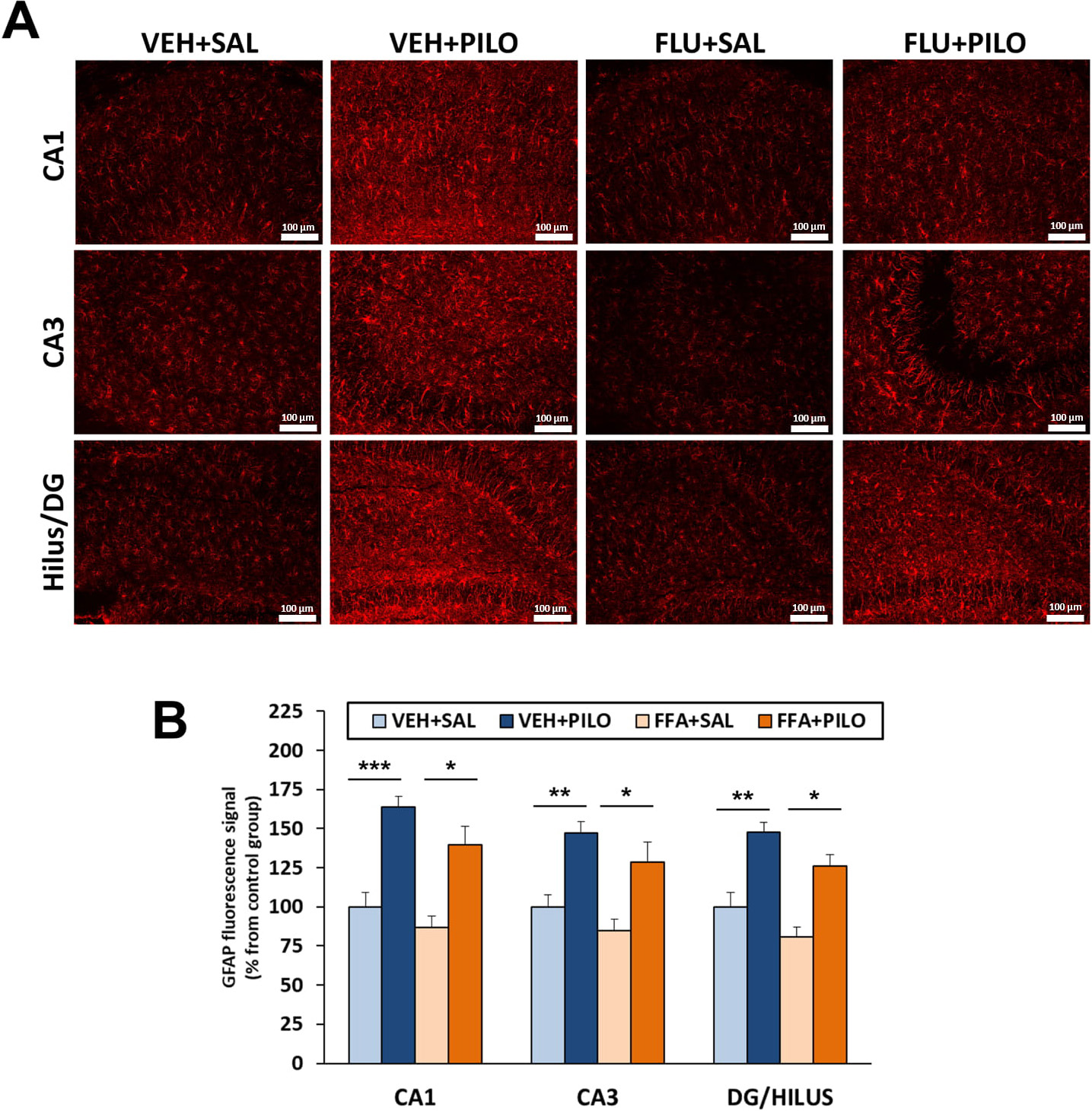 Fig. 6.
Fig. 6.SE triggered by pilocarpine injection induced a marked astrocyte
activation (evaluated by GFAP immunofluorescence), which was not significantly
modified by a previous administration of FLU. (A) Representative images of
hippocampal subregions showing immunofluorecent GFAP signal. (B) Plot of GFAP
fluorescence signal intensity (mean
In the current study we have investigated the eventual effects of administering a single high dose of FLU previous to the pilocarpine-induced SE on latency time, mortality rate, BW changes, brain glucose metabolism and several neurohistochemical signs of hippocampal damage typically associated with the SE that consistently manifest on the latent period in this model.
Since brain inflammation is promoted in epileptogenesis [6, 7, 8], anti-inflammatory drugs have been proposed as therapeutical agents with beneficial effects against the deleterious outcomes of convulsions and epilepsy [42]. Moreover, accumulation of prostaglandins, endogenous inflammatory mediators, and final products of the arachidonic acid cascade has also been reported in experimental epilepsy [43], having a pro-epileptogenic role [44]. Nevertheless, and despite numerous animal studies, no agreement has been reached about the actual anti-seizure or neuroprotective effects of different types of NSAIDs assayed up till now [10, 32, 33, 34].
The results herein presented do not support that FLU pre-treatment prevents or alleviates the severity of SE or would have neuroprotective or anti-inflammatory effects, since it did not prevent the occurrence of SE or the damage triggered by the lithium-pilocarpine model. On the contrary, FLU significantly increased the mortality rate associated with the severity of this chemical model of epileptogenesis and slightly impaired the ability of the rats to maintain their BW.
BW change is widely accepted as a marker of the overall wellbeing of the animals. As expected, pilocarpine-induced SE resulted in a significantly acute BW loss. However, the animals were able to partially recover from the acute effects of SE inducing BW loss, even though the overall consequence of SE was a significant BW loss. In rats that did not undergo SE, FLU pre-treatment resulted in a slightly lower BW gain compared with the rats from the VEH-SAL. Furthermore, the group FLU + PILO consistently lost weight during the experiment with no signs of recovery. It is likely that the transient effect of FLU acutely impairing the ability of the rats to defend their BW reflects a potential toxic effect of FLU. Studies in albino rats have shown that chronic treatment with FLU (with doses ranging from 4 to 29 mg/kg, doses lower than the one used acutely in this study) reduced food intake and weight gain [45]. Other studies in male rats have shown that FLU (doses ranging from 1–9 mg/kg/day) dose-dependently improves BW gain in the model of adjuvant-induced arthritis [46], showing that the intense damage induced by SE does not only relay on unchaining an inflammatory response.
Regarding the potential anticonvulsant effect of FLU, in vitro studies
have shown that FLU reversibly abolishes seizure-like events in neocortical
slices stimulated with a GABA
The lithium-pilocarpine model is characterized by a high rate of mortality [29]. Even so, it has been shown that there is a high variability in sensitivity to pilocarpine resulting in a great unpredictability in mortality depending on factors such as strains, sub-strains, age, commercial providers, and the time of purchase of animals [48, 49]. Surprisingly, in the current study, no rats died in the VEH + PILO group but FLU significantly increased mortality rate. In line with our results, other studies have shown an increase both in mortality rate (and seizure susceptibility) after NSAIDs administration in the pilocarpine rodent model [34, 50]. The high mortality rate found in animals pretreated with FLU in the current study may be attributed both the relatively high dose administered as well as to the complex (including opposite) effects on multiple ion channels of this particular NSAID [11].
Finally, the histological studies further confirmed that FLU had no beneficial effects in this rather aggressive model. It is known that the pilocarpine and lithium-pilocarpine models compromise the hippocampal structure as it courses with neurodegeneration and neuroinflammatory processes that result in a large neuronal loss [28, 29, 30, 51]. In fact, other authors have shown that Tenidap, a COX-2 selective NSAID, administered after the SE induced by pilocarpine, had neuroprotective effects in the hippocampal CA3 area [52, 53]. However, in this study, no beneficial effect of FLU in preventing neuronal death or astrogliosis was observed, although it did not worsen these processes. However, it is necessary to mention again the high mortality ratios in the FLU + PILO group that could be masking deleterious effects. Further studies in vivo, with different FLU regimes and/or animal models, might be needed to either unmask or discard the potential antiepileptogenic or neuroprotective effects of this anti-inflammatory drug.
In conclusion, the administration of a single high dose of FLU (100 mg/kg, i.p.)
before SE induced by lithium-pilocarpine did not prevent or ameliorated the
occurrence of SE. Besides, it did not show beneficial effects neither on brain
glucose metabolism as assessed by [
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
LGG, MD and MAP designed the research study. LGG, ÁS, MD and RFR conducted the experiments. LGG, FG, RFR, ÁS, NHM and PB analyzed the data and performed the statistical analysis. LGG, FG, NHM and PB wrote the manuscript. MAP was the principal investigator (PI) responsible of funding acquisition and project administration. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The project in which was included this study was approved by the Animal Research Ethical Committee of the Universidad Complutense de Madrid and the Autonomous Community of Madrid (PROEX: 238/15), and it was carried out in accordance with regulations of the European Union (2010/63/UE) and Spain (RD53/2013) regarding animal welfare.
Not applicable.
This work was financially supported by the Spanish Ministerio de Ciencia e Innovación (Retos PID2019-106968RB-100). PB is a Miguel Servet researcher from ISCIII (CP21/00020).
Pablo Bascuñana Almarcha is serving as one of the Guest editors of this journal. We declare that Pablo Bascuñana Almarcha had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Gernot Riedel. The other authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.