









1 Department of Veterinary Anatomy and Animal Behavior, College of Veterinary Medicine and BK21 FOUR Program, Chonnam National University, 61186 Gwangju, Republic of Korea
2 Department of Basic Veterinary Sciences, College of Veterinary Medicine, University of the Philippines Los Baños, 4031 Los Baños, Philippines
Academic Editors: Ulises Gomez-Pinedo and Rafael Franco
Abstract
Background: The central nervous system (CNS) is enriched in lipids; despite this, studies exploring the functional roles of lipids in the brain are still limited. Sterol regulatory element binding protein (SREBP) signaling is a transcriptomic pathway that predominantly participates in the maintenance of lipid homeostasis; however, its involvement in the CNS dysfunction is not well-established. In this study, we aimed to characterize and pinpoint specific genes of the SREBP pathway which may be implicated in neurodegenerative, neurological, and neuropsychiatric diseases. Methods: In silico bioinformatic analysis was performed using the open-source databases DisGeNET and MSigDB. Protein-protein interaction data were visualized and analyzed using STRING, after which GO (Gene Ontology) and KEGG (Kyoto Encyclopedia of Genes and Genomes) enrichment analyses were conducted via DAVID (Database for Annotation, Visualization and Integrated Discovery). Results: Several common genes were identified between the SREBP pathway and CNS disorders. In GO enrichment analysis, the most enriched biological processes included lipid, cholesterol, and steroid biosynthetic processes; the most enriched molecular functions were transcription factor-related; and the most enriched subcellular compartments revealed that the genes involved in CNS disorders were mainly associated with the enzyme complexes of acetyl-CoA carboxylase (ACC) and fatty acid synthase (FASN). In KEGG enrichment analysis, the most enriched pathway was the AMP-activated protein kinase (AMPK) signaling pathway, and the top-ranked genes significantly enriched under this pathway were ACACA, ACACB, FASN, HMGCR, MTOR, PPARGC1A, PRKAA1, SCD, SIRT1, and SREBF1. Conclusions: The findings of this study strengthen the evidence linking the involvement of lipid homeostasis in CNS functions. We suggest herein the roles of downstream ACC and FASN enzymes and upstream AMPK signaling in the SREBP pathway as mechanisms underlying neurodegenerative, neurological, and neuropsychiatric CNS disorders.
Keywords
- AMPK
- Lipids
- Neurodegenerative
- Neurological
- Neuropsychiatric
- SREBP
Lipids serve various functions in animals. They act as sources of energy and are components of cellular membranes as well as being substrates of compounds participating in diverse biological activities, including steroid hormones, vitamins, bile acids, and eicosanoids [1, 2]. Both dietary and endogenous synthetic pathways provide the lipids we require [3, 4]. Fatty acid (FA) and cholesterol synthesis can occur in every cell; however, these processes are particularly essential in the liver and adipose tissues, which are organs that specialize in lipid export and storage [5, 6]. A class of transcription factors known as sterol regulatory element binding proteins (SREBPs) regulates both cholesterol and FA biosynthesis. SREBPs trigger a series of enzymes essential for endogenous cholesterol, FA, triglyceride (TG), and phospholipid production. SREBPs are thus thought to be key regulators of cholesterogenesis and lipogenesis [3].
SREBPs are transcription factors with a basic-helix-loop-helix-leucine zipper
(bHLH-LZ) structure; they are synthesized as 1150 amino acid (aa) inactive
precursors attached to the endoplasmic reticulum (ER) membrane [7]. Each SREBP
precursor is divided into three domains: (a) a 480-aa NH
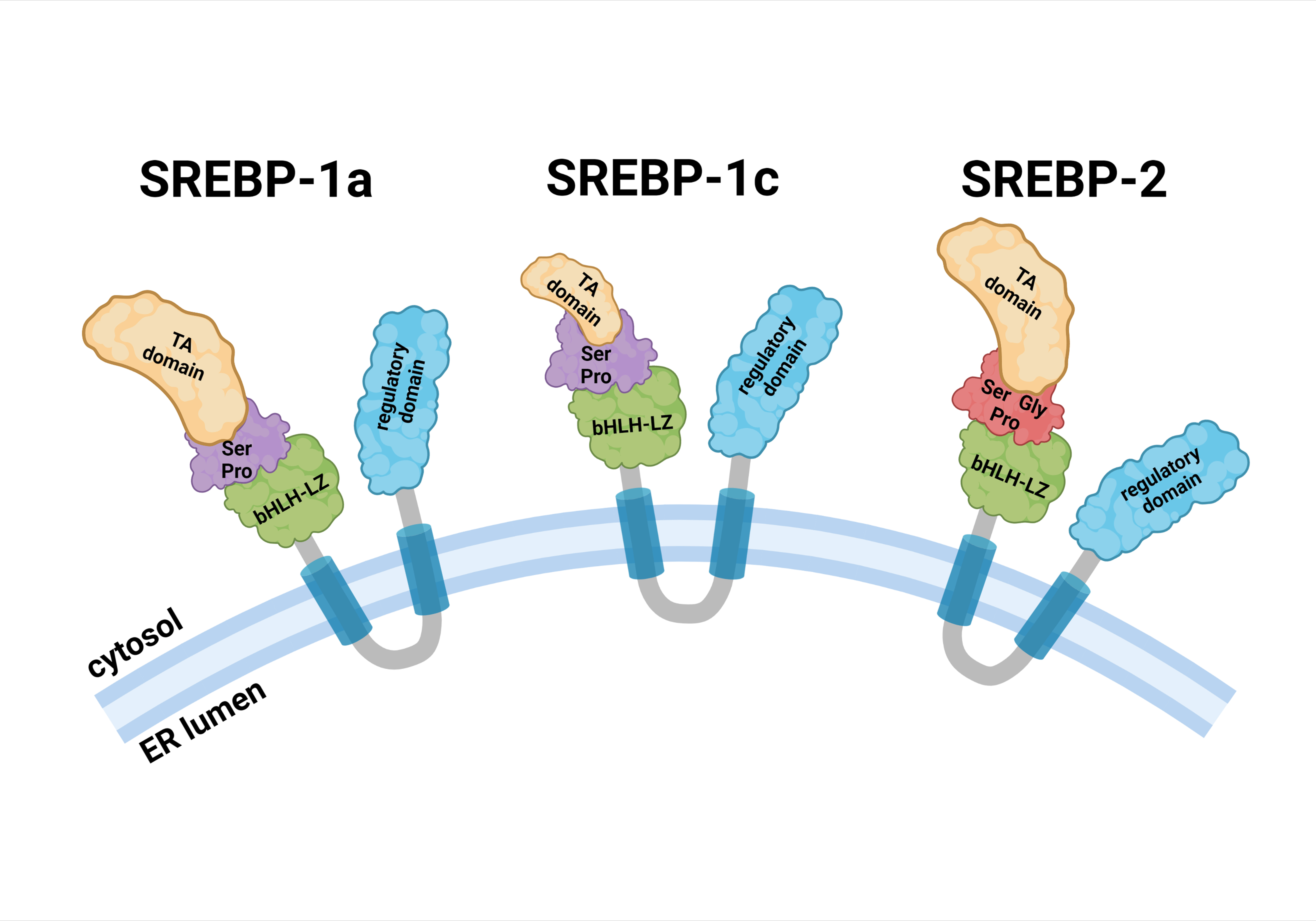 Fig. 1.
Fig. 1.Schematic structure of SREBP precursors. Each SREBP precursor is divided into three domains: (1) a 480-aa NH2-terminal domain containing the transactivation domain, a serine and proline-rich region, and the bHLH-LZ region for DNA binding and dimerization; (2) two hydrophobic transmembrane spanning segments, interrupted by a 30-aa short loop that projects into the ER lumen; and (3) a 590-aa COOH-terminal containing the regulatory domain. Abbreviations: bHLH-LZ, basic helix-loop-helix leucine zipper; ER, endoplasmic reticulum; Gly, glycine; Pro, proline; Ser, serine; SREBP, sterol regulatory element binding protein; TA, transactivation.
SREBP family members have been identified in numerous mammalian species:
SREBP-1a and 1c, which are generated from a single gene (sterol regulatory
element binding transcription factor 1; SREBF-1) on human chromosome
17p11.2 [12], and SREBP-2, which is produced from a different gene
(SREBF-2) on human chromosome 22q13 [13]. SREBP-1 and SREBP-2 proteins
have 47% similarity. The first exons of SREBP-1a and 1c transcripts differ owing
to different transcription start locations (exon 1a and exon 1c, respectively),
whereas the remaining exons are shared by both isoforms [8, 14]. Moreover,
alternative 3
Even though lipids are highly enriched in the brain [18, 19, 20, 21, 22], there is little research identifying and examining the role of lipids in brain structure and function. This matter has been acknowledged by some neuroscientists. For example, Montesinos, Guardia-Laguarta and Area-Gomez [18] have proposed numerous reasons for this obvious gap in knowledge: (1) lipids are not coded in the genome, and hence are not subject to the core dogma of biology, but rather to the rules of biophysics; (2) lipids do not possess intrinsic catalytic activity, and as a result, they have long been thought to be inert substances with little biological value or only a reflection of food and environmental adaptations; and (3) lipids cannot be studied as separate molecules. To maintain homeostasis and retain the form and function of cellular membranes, fluctuations in the proportion of one lipid typically trigger a cascade of changes in other lipid species. Thus, a large amount of the information carried by lipids must be investigated holistically. Collectively, these and other factors have driven lipid biology out of the ‘mainstream’ of neuroscience research.
Nevertheless, lipid research in neuroscience has been increasing, indicating that neural lipids are finally receiving the attention that they deserve [23]. This could be due to the emergence of novel technologies such as mass spectrometry [24, 25] and atomic-force microscopy [26]. These methods allow for the detailed screening of complex changes in lipid composition as well as the characterization of the topographical distribution of lipid species in the brain, down to the level of individual neuronal cells [18].
Lipids have an established set of roles that are equivalent with those of neurotransmitters, neuropeptides, and growth factors [27]. Specifically, lipids play key roles in structural activities (i.e., membrane compartmentalization) [28] and physiological functions (i.e., energy production, gene expression, neural communication, neurogenesis, synaptic transmission, as well as signal transduction and regulation) [29]. Other roles include the processing of complex behaviors and participation in neocortex development [19, 30, 31]. In neurons, synapses and neuromuscular junctions are specific sites wherein lipid dynamics regulate cellular activities, such as signal transduction and transmembrane gradients [30]. For example, changes in cholesterol and sphingolipids are required to produce membrane curvature for synaptic vesicle release and fusion, ion channel modulation, and protein activation signaling [32]. In glial cells, lipids are important components of oligodendrocytes, where they function in myelin biogenesis, axon-glia communication, and long-term maintenance of myelin [33]. Lipids in astrocytes play important roles in energy generation, membrane fluidity, and cell-to-cell signaling [34]. In microglia, increased lipid metabolism is vital in supporting protective cellular functions, such as phagocytosis [35] and neuroinflammatory sensitization of central pain pathways [36].
Central nervous system (CNS) disorders, including neurodegenerative, neurological, and neuropsychiatric disorders greatly impact quality of life and present an economic burden on society. These disorders are of major clinical importance because their definitive pathoetiology and therapeutics remain largely elusive. Alterations in lipid metabolism in the CNS have recently been implicated in the development of neurodegenerative diseases, mental disorders, and brain injury. Several review papers have described and detailed the crucial role of lipids in tissue physiology and cell signaling in CNS-related diseases [21, 28, 37, 38, 39, 40, 41]. Adibhatla and Hatcher [21, 22] have identified CNS injuries that implicate aberrations in lipid systems. Neurological disorders affected by lipid signaling include bipolar disorder and schizophrenia, and neurodegenerative disorders involving deregulated lipid metabolism include Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), multiple sclerosis (MS), and amyotrophic lateral sclerosis (ALS). CNS injuries involving altered lipid metabolism include stroke, traumatic brain injury, and spinal cord injury (SCI) [22].
Lipid-related mechanisms involved in CNS disorders include alterations in
cholesterol and lipid homeostasis (AD [42, 43]), decreased docosahexaenoic acid
levels (MS, epilepsy, and stroke [44]), excessive accumulation of cholesterol and
sphingolipids (increased activity of phospholipases A2 and generation of lipid
mediators (AD [45, 46], PD [45], MS [45], schizophrenia [47], and stroke [48]),
increased lipid peroxidation (AD [49], ALS [50]), inhibition of SREBP-1
(schizophrenia [51, 52, 53, 54]), lipid raft interaction with amyloid precursor protein
(AD [55]), polyunsaturated FAs promoting the aggregation of
SREBP signaling is a vital pathway responsible for maintaining transcriptional activities that promote lipid synthesis and homeostasis. Even though ample evidence of the associations between lipid deregulation and CNS disorders exists, studies exploring the possible involvement of the SREBP pathway in the development or occurrence of common CNS disorders are not well-established. Instead, most of the studies exploring this pathway have remained focused on disorders of lipid-rich tissues and lipid-regulating organs, such as adipose tissues and the liver, respectively. This warrants a need to discover specific lipid-processing pathways involved in CNS disorders. Thus, we sought to fill this gap in knowledge by conducting in silico bioinformatic analyses between the SREBP signaling pathway and CNS disorder-related genes and proteins. We accessed several open-source bioinformatic programs and open-source databases for gene data sets to discover overlaps in protein-protein interactions, gene ontology, and biological functions between CNS disorder-related genes and components of the SREBP signaling pathway.
Fig. 2 shows the schematic diagram of the in silico analysis performed in the study.
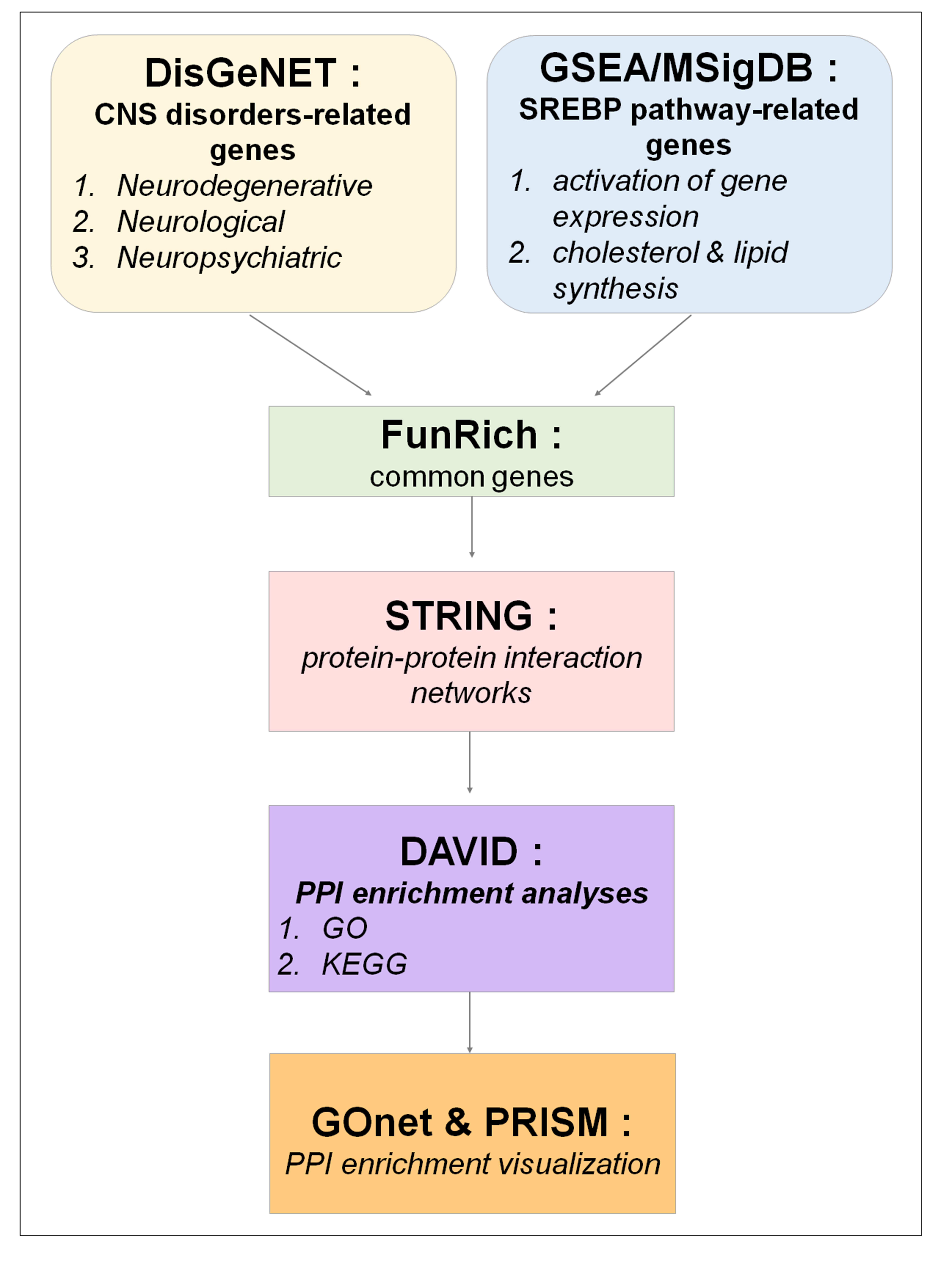 Fig. 2.
Fig. 2.Schematic diagram of the in silico analysis performed in this study. The methodology used in the study are outlined in flow-chart form. Genes related to CNS disorders were obtained from DisGeNET, and SREBP pathway-related genes were collected from the MSigDB feature of GSEA. Overlapping genes between datasets were identified using the FunRich program. The resulting gene sets were uploaded into STRING to obtain protein-protein interactions. Gene set enrichment analyses were conducted using DAVID for GO functional enrichment and KEGG pathway enrichment analyses. Abbreviations: CNS, central nervous system; DAVID, Database for Annotation, Visualization and Integrated Discovery; FunRich, Functional Enrichment analysis tool; GO, Gene Ontology; GSEA, Gene Set Enrichment Analysis; KEGG, Kyoto Encyclopedia of Genes and Genomes; MSigDB, Molecular Signatures Database; PPI, protein-protein interaction; STRING, Search Tool for Retrieval of Interacting Genes.
Related genes of CNS disorders previously associated with lipid deregulation were exported from the DisGeNET database [57]. DisGeNET is a publicly available collection of genes and variants associated with human diseases. Gene set enrichment analysis (GSEA v4.1.0, Broad Institute, Cambridge, MA, USA) is a computational method that statistically analyzes significant differences in gene sets under two biological states. The Molecular Signatures Database (MSigDB v7.4, Broad Institute, Cambridge, MA, USA) [58] is a collection of annotated gene sets utilized by the GSEA software. Genes involved in the SREBP signaling pathway were obtained from the MSigDB collection for GSEA [59]. Common genes between CNS disorder-related set and the SREBP pathway were retrieved using Functional Enrichment analysis tool (FunRich v3.1.4, La Trobe University, VIC, Australia) [60]. FunRich is an open-source program for analyzing gene and protein functional enrichment and interaction networks.
Associations between overlapping gene sets were revealed by constructing a
protein-protein interaction (PPI) network using the Search Tool for Retrieval of
Interacting Genes (STRING) database [61, 62]. STRING is a well-known database
that gathers, integrates, and assesses all available PPI data from various
sources, as well as computational predictions. Its purpose is to create a massive
network that is comprehensive and objective, with both direct (physical) and
indirect (functional) linkages. The combined STRING score represents the
estimated confidence of data support presented by seven networks, including three
genomic context prediction channels (gene neighborhoods, cooccurrence, and
fusions), as well as co-expression, experiments, databases, and text-mining
channels. Owing to its extensive and up-to-date data, user-friendly website, and
uniform scoring method, STRING is regarded as a useful and beneficial tool for
static PPI analysis. All seven STRING channels were utilized in this study.
PPI networks were generated with Homo sapiens as the species of
interest, medium confidence of
To characterize and explore the potential biological functions of CNS disorder-associated genes of the SREBP pathway, the Database for Annotation, Visualization and Integrated Discovery (DAVID) [63, 64] was utilized for Gene Ontology (GO) [65, 66] function and Kyoto Encyclopedia of Genes and Genomes (KEGG) [67] pathway enrichment analyses. The GO database is a bioinformatics tool that provides specific definition describing gene products in terms of biological process, molecular function, and cellular component. KEGG is a database that contains information on genomes, biological pathways, diseases, medications, and chemical compounds. The functional enrichment analysis results were visualized using GOnet [68] and GraphPad Prism (GraphPad Prism version 8.0.0 for Windows; GraphPad Software, San Diego, CA, USA; www.graphpad.com).
Gene sets associated with 11 CNS disorders previously reported to be related with lipid deregulatory mechanisms were obtained from the DisGeNET database. The gene sets were divided into three categories, namely (1) neurodegenerative disorder (NDDs): AD, HD, MS, and PD; (2) neurological disorder (NLDs): epilepsy and ischemic stroke; and (3) neuropsychiatric disorder (NPDs): anxiety, bipolar disorder, mental depression, post-traumatic stress disorder (PTSD), and schizophrenia. A total of 17,626 genes were identified, of which 8253, 2374, and 6999 were associated with NDDs, NLDs, and NPDs, respectively.
Two SREBP pathway gene sets were identified from the MSigDB dataset: the SREBP activation of gene expression (SAGE) pathway, which generated 42 genes, and the SREBP cholesterol and lipid homeostasis (SCLH) pathway, which generated 19 genes. The complete lists of gene sets per disorder and pathway are provided in Supplementary Tables 1 and 2. After importing the data into FunRich, an analysis of overlapping genes between CNS disorder-related genes and the SAGE pathway was conducted, yielding 25 genes (NDDs: 19, NLDs: 10, NPDs: 11) (Fig. 3A, left panel); the analysis of overlapping genes with the SCLH pathway generated 15 genes (NDDs: 14, NLDs: 8, NPDs: 11) (Fig. 3B, left panel).
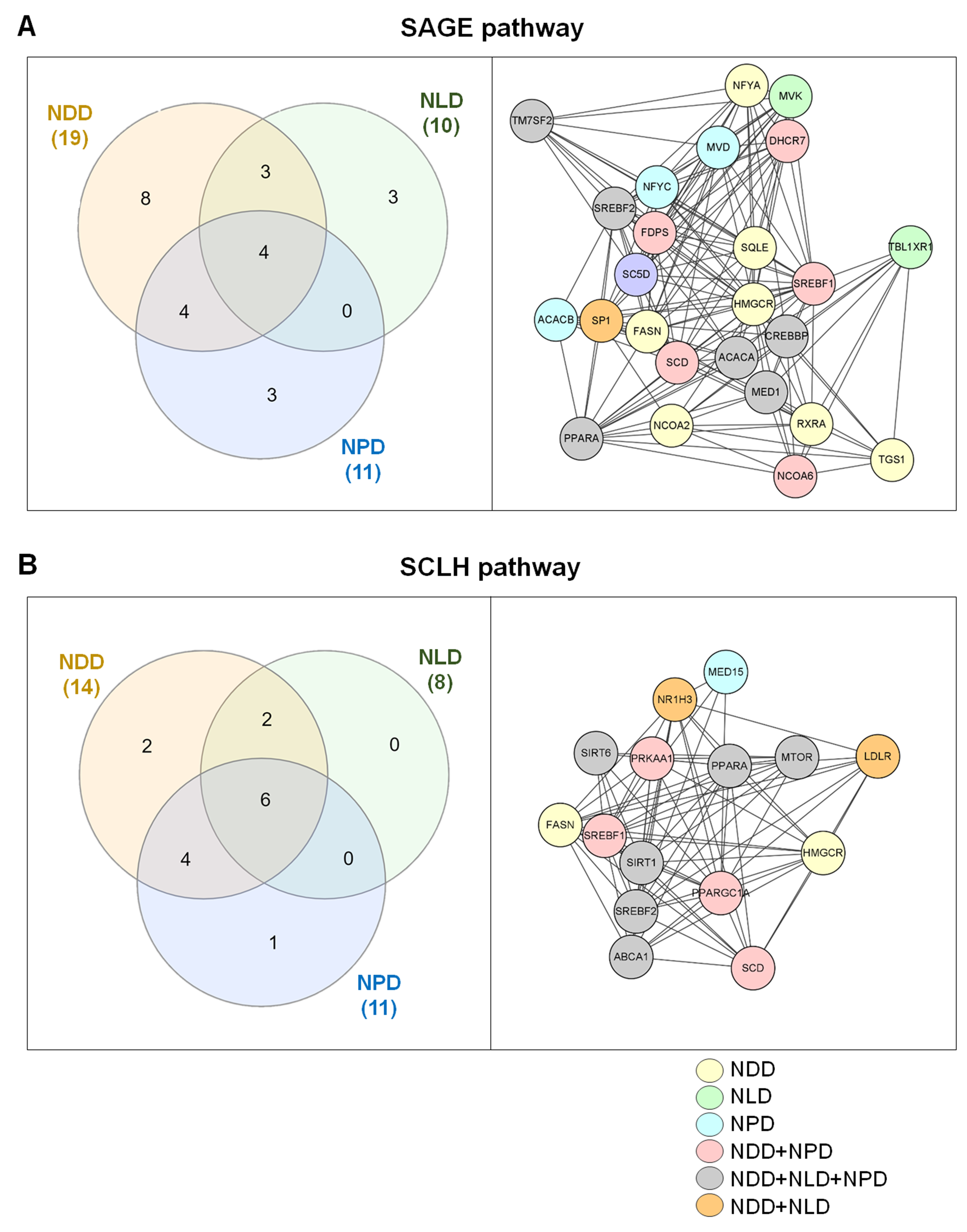 Fig. 3.
Fig. 3.Venn diagram and PPI networks of overlapping genes between the
SREBP pathways and CNS disorders. Venn diagrams show the number of
overlapping genes between two pathways related to SREBP (the SAGE and SCLH
pathways) and common CNS disorders, (organized into NDDs, NLDs, and NPDs). A
total of 25 and 15 genes were found to be in common between CNS disorders and the
SAGE (A, left panel) and SCLH (B, left panel) pathways, respectively. PPI network
constructs generated via STRING revealed genes that are specific for NDDs (yellow
nodes), NLDs (green nodes), or NPDs (blue nodes) only; shared between two CNS
disorder categories (pink or orange nodes); or shared among all (grey nodes) CNS
disorder categories (A and B, right panels). PPI networks were generated with
Homo sapiens as the species of interest, medium confidence of
In Table 1, a detailed list of SREBP pathway-related genes associated with CNS disorders has been provided. In Table 2, the percentages of common genes (‘FunRich recognized’ in SupplementaryTable 1) between SREBP pathways and CNS-related disorders have been presented. Among NDDs, PD (0.54%) exhibited the highest percentage of SREBP pathway-related genes in SAGE, while HD (0.78%) exhibited the highest percentage in SCLH pathways. Among NLDs, epilepsy (0.54%) presented the highest percentage of SAGE pathway-related genes, and ischemic stroke (0.79%) exhibited the highest percentage of common SCLH pathway genes. Among NPDs, mental depression displayed the highest percentage of common genes for both the SAGE (0.52%) and SCLH (0.59%) pathways.
| SREBP pathway | Activation of gene expression | Cholesterol & lipid homeostasis |
| NDD | ||
| AD | ACACA, CREBBP, FASN, FDPS, HMGCR, NCOA6, PPARA, RXRA, SCD, SP1, SQLE, SREBF2, TM7SF2 | ABCA1, FASN, HMGCR, LDLR, MTOR, NR1H3, PPARA, PPARGC1A, PRKAA1, SCD, SIRT1, SIRT6, SREBF2 |
| HD | CREBBP, RXRA, SP1, TM7SF2 | ABCA1, MTOR, NR1H3, PPARGC1A, PRKAA1, SIRT1, SIRT6 |
| MS | DHCR7, HMGCR, NCOA2, NFYA, PPARA, SP1 | ABCA1, HMGCR, LDLR, MTOR, NR1H3, PPARA, PPARGC1A, SIRT1 |
| PD | ACACA, FASN, MED1, PPARA, RXRA, SCD, SP1, SREBF1, TGS1, TM7SF2 | ABCA1, FASN, LDLR, MTOR, PPARA, PPARGC1A, PRKAA1, SCD, SIRT1, SREBF1 |
| NLD | ||
| Epilepsy | CREBBP, MED1, PPARA, SC5D, SP1, TBL1XR1 | ABCA1, MTOR, PPARA, SIRT1 |
| Ischemic stroke | ACACA, MVK, PPARA, SREBF2, TM7SF2 | ABCA1, LDLR, MTOR, NR1H3, PPARA, SIRT1, SIRT6, SREBF2 |
| NPD | ||
| Anxiety | ACACA, FDPS, MVD | PPARGC1A, SIRT1, SIRT6 |
| Bipolar disorder | ACACA, NFYC, SCD, SREBF2 | PPARGC1A, SCD, SIRT1, SREBF2 |
| Mental depression | ACACA, ACACB, FDPS, MVD, PPARA, SCD, SREBF1 | ABCA1, MTOR, PPARA, PPARGC1A, SCD, SIRT1, SIRT6, SREBF1 |
| PTSD | PPARA | PPARA, SIRT1 |
| Schizophrenia | ACACA, CREBBP, NCOA6, PPARA, SCD, SP1, SREBF1, SREBF2 | ABCA1, MED15, MTOR, PPARA, PPARGC1A, PRKAA1, SCD, SIRT1, SREBF1, SREBF2 |
| ABCA1, ATP binding cassette subfamily A member 1; ACACA, acetyl-CoA carboxylase alpha; ACACB, acetyl-CoA carboxylase beta; AD, Alzheimer’s disease; CREBBP, CREB binding protein; CNS, central nervous system; DHCR7, 7-dehydrocholesterol reductase; FASN, fatty acid synthase; FDPS, farnesyl diphosphate synthase; HD, Huntington’s disease; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; LDLR, low density lipoprotein receptor; MED1, mevalonate diphosphate decarboxylase; MED15, mediator complex subunit 15; MS, multiple sclerosis; MTOR, mechanistic target of rapamycin kinase; MVD, mevalonate diphosphate decarboxylase; MVK, mevalonate kinase; NCOA2, nuclear receptor coactivator 2; NCOA6, nuclear receptor coactivator 6; NDDs, neurodegenerative diseases; NFYA, nuclear transcription factor Y subunit alpha; NFYC, nuclear transcription factor Y subunit gamma; NLDs, neurological diseases; NPDs, neuropsychiatric disease; NR1H3, nuclear receptor subfamily 1 group H member 3; PD, Parkinson’s disease; PPARA, peroxisome proliferator-activated receptor alpha; PPARGC1A, PPARG coactivator 1 alpha; PRKAA1, protein kinase AMP-activated catalytic subunit alpha 1; PTSD, post-traumatic stress disorder; RXRA, retinoid X receptor alpha; SC5D, sterol-C5-desaturase; SCD, stearoyl-CoA desaturase; SIRT1, sirtuin 1; SIRT6, sirtuin 6; SP1, Sp1 transcription factor; SQLE, squalene epoxidase; SREBF1, sterol regulatory element binding transcription factor 1; SREBF2, sterol regulatory element binding transcription factor 2; SREBP, sterol regulatory binding protein; TBL1XR1, TBL1X receptor 1; TGS1, trimethylguanosine synthase 1; TM7SF2, transmembrane 7 superfamily member 2. | ||
| SREBP pathway | Activation of gene expression | Cholesterol & lipid homeostasis |
| NDD | ||
| AD | 13 out of 3007 (0.43%) | 13 out of 3007 (0.43%) |
| HD | 4 out of 896 (0.45%) | 7 out of 896 (0.78%) |
| MS | 6 out of 1574 (0.38%) | 8 out of 1574 (0.51%) |
| PD | 10 out of 1846 (0.54%) | 10 out of 1846 (0.54%) |
| NLD | ||
| Epilepsy | 6 out of 1111 (0.54%) | 4 out of 1111 (0.36%) |
| Ischemic stroke | 5 out of 1019 (0.49%) | 8 out of 1019 (0.79%) |
| NPD | ||
| Anxiety | 3 out of 962 (0.31%) | 3 out of 962 (0.31%) |
| Bipolar disorder | 4 out of 1057 (0.38%) | 4 out of 1057 (0.38%) |
| Mental depression | 7 out of 1345 (0.52%) | 8 out of 1345 (0.59%) |
| PTSD | 1 out of 381 (0.26%) | 2 out of 381 (0.52%) |
| Schizophrenia | 8 out of 2524 (0.32%) | 10 out of 2524 (0.40%) |
| AD, Alzheimer’s disease; CNS, central nervous system; HD, Huntington’s disease; MS, multiple sclerosis; NDD, neurodegenerative disease; NLD, neurological disease; NPD, neuropsychiatric disease; PD, Parkinson’s disease; PTSD, post-traumatic stress disorder; SREBP, sterol regulatory binding protein. | ||
Gene product interactions between both SREB pathways and CNS disorders were
constructed using STRING (Fig. 3, right panels). The resulting SAGE
pathway-related CNS disorder genes PPI network had 25 nodes with 159 edges
(vs. 8 expected edges), a clustering coefficient of 0.798, an enrichment
p-value of
Potential common mechanisms between SREBP pathway genes associated with CNS
disorders were examined using GO and KEGG analyses. In the SAGE pathway, GO
enrichment analysis resulted in 81 terms under the biological process category,
11 terms under the cellular component category, and 28 terms under the molecular
function category. SAGE pathway-related GO enrichment analyses were mapped
graphically using GOnet (Fig. 4). Only biological processes with a
p-value threshold of
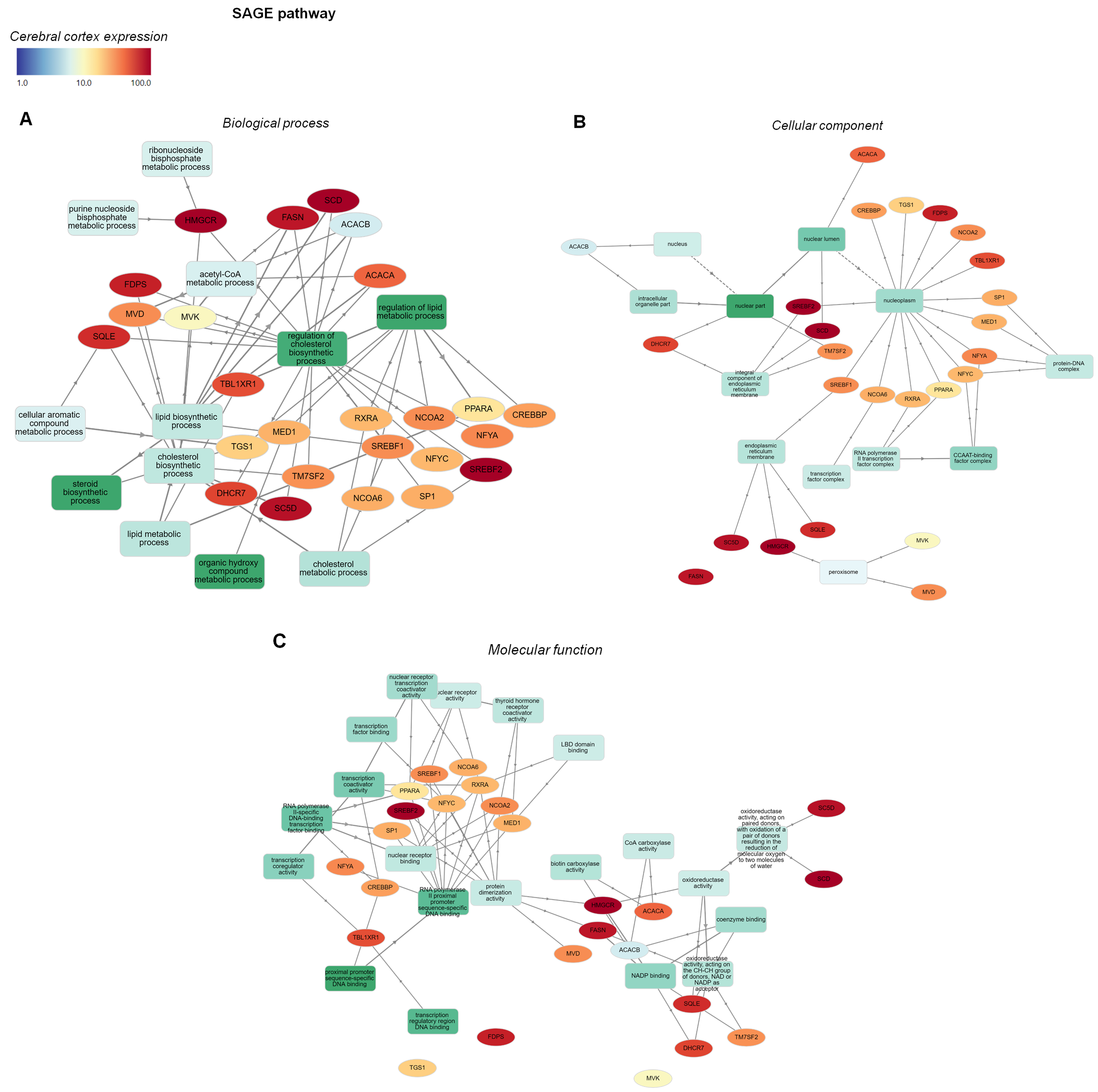 Fig. 4.
Fig. 4.GO functional enrichment analysis network of SAGE pathway genes
overlapping with CNS disorder genes. GO enrichment analyses of SAGE pathway
genes were mapped graphically using GOnet. Only biological processes (A) with a
p-value threshold of
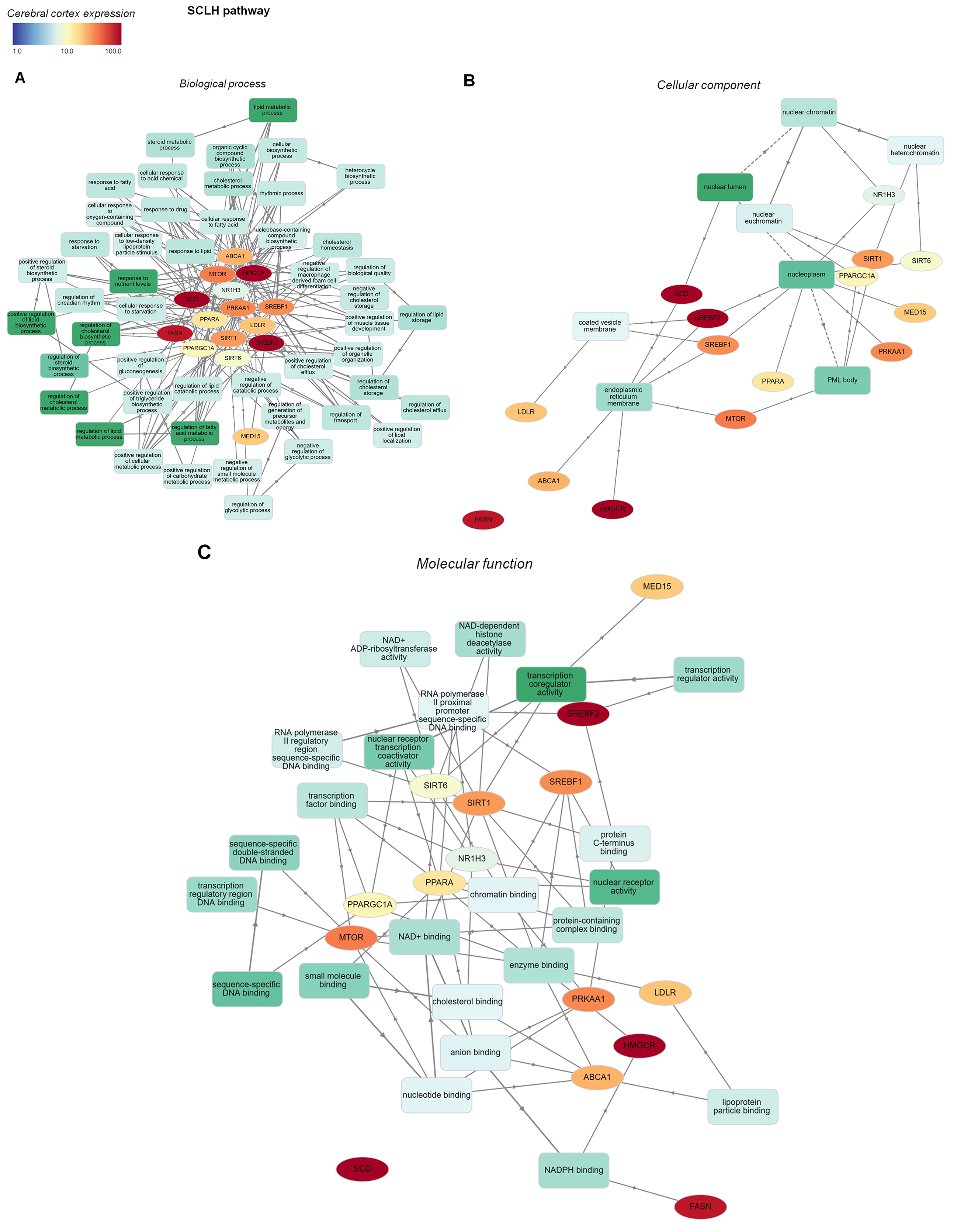 Fig. 5.
Fig. 5.GO functional enrichment analysis network of SCLH pathway genes
overlapping with CNS disorder genes. GO enrichment analyses of SCLH
pathway-related genes were mapped graphically using GOnet. Only biological
processes (A) with a p-value threshold of
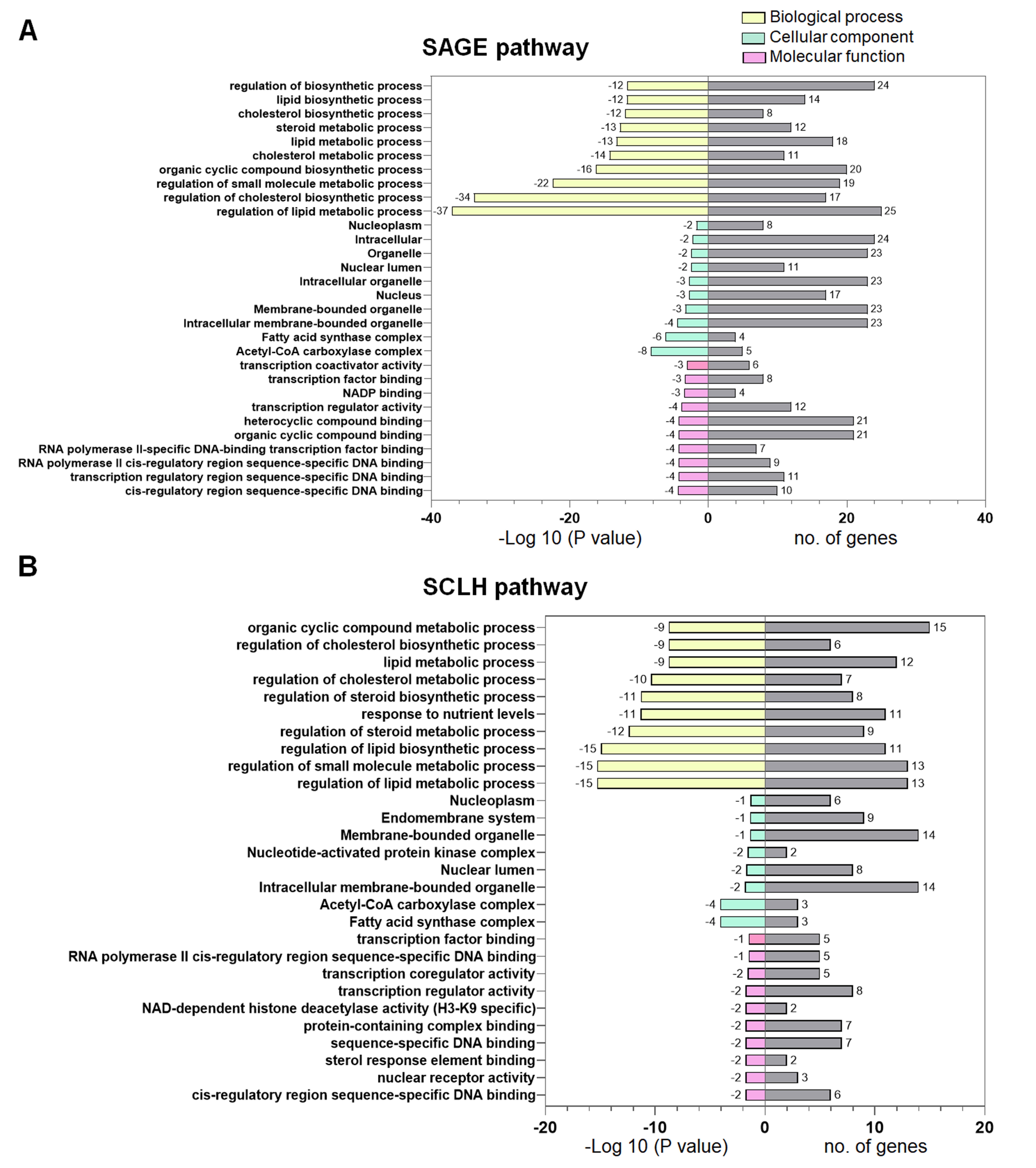 Fig. 6.
Fig. 6.Top biological functions of common genes between the SREBP pathways and CNS disorders, as analyzed by GO. Top enriched GO functional processes presented in bar graph format, with their corresponding p-values and corresponding number of genes associated for the SAGE (A) and SCLH (B) pathways. Yellow bars represent enriched biological processes, green bars represent enriched cellular components, and pink bars represent enriched molecular functions. Abbreviations: Acetyl-CoA, acetyl coenzyme A; NAD, nicotinamide adenine dinucleotide; NADP, nicotinamide adenine dinucleotide phosphate; SAGE, SREBP activation of gene expression; SCLH, SREBP cholesterol & lipid homeostasis pathway; SREBP, sterol regulatory element binding protein.
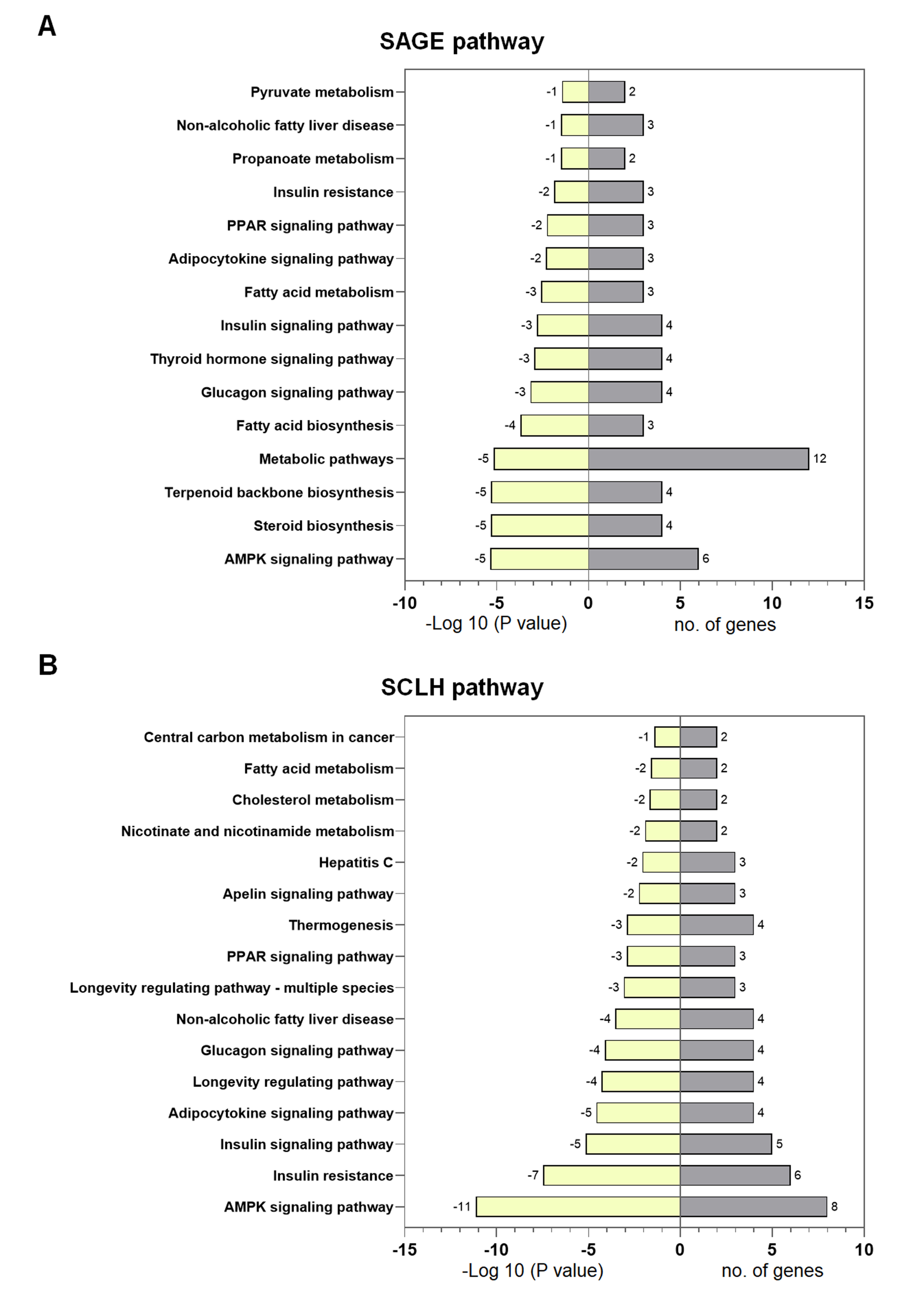 Fig. 7.
Fig. 7.Top enriched biological pathways between the SREBP pathways and CNS disorders, as analyzed by KEGG. Top enriched KEGG pathways presented in bar graph format, with their corresponding p-values and corresponding number of genes associated with the SAGE (A) and SCLH (B) pathways. Abbreviations: AMPK, AMP-activated protein kinase; PPAR, peroxisome proliferator-activated receptor; SAGE, SREBP activation of gene expression; SCLH, SREBP cholesterol & lipid homeostasis pathway; SREBP, sterol regulatory element binding protein.
In this study, using in silico bioinformatic analyses, we present evidence for the involvement of lipid deregulation in the signaling of most common CNS disorders. This is indicated by the presence of overlapping genes and biological pathways between common CNS disorders (NDDs, NLDs, and NPDs) and two pathways (SAGE and SCLH) of SREBP signaling.
The SAGE pathway is primarily involved in regulating the expression of genes encoding enzymes that are responsible for lipid homeostasis and control endogenous cholesterol, FA, and triacylglycerol levels, and phospholipid synthesis. There are three isoforms of SREBP: SREBP-1a, SREBP-1c, and SREBP-2. All isoforms have varying roles in lipid synthesis. SREBP-1c is primarily associated in FA synthesis and insulin-induced glucose metabolism (especially in lipogenesis), whereas SREBP-2 is implicated in cholesterol synthesis. In contrast, SREBP-1a seems to participate in both FA and cholesterol synthesis. SREBP transcription factors are generated from inactive precursors attached to the membranes of the ER. The precursor goes through a two-step cleavage process after activation to release the NH (2)-terminal active domain in the nucleus. The amount of cellular sterol influences SREBP processing. When sterol levels fall, the precursor is cleaved, allowing cholesterogenic genes to be activated and cholesterol homeostasis to be maintained. This sterol-sensitive mechanism appears to represent a primary point of regulation for the SREBP-1a and SREBP-2 isoforms, but not for SREBP-1c. Furthermore, insulin appears to be the primary regulator of SREBP-1c at the transcriptional level. The distinct regulatory and activation features of each SREBP isoform make it easier to coordinate lipid metabolism control [3, 14, 69].
The SCLH pathway, on the contrary, is a more specific subset of the SAGE pathway. It focuses on the cholesterol- and lipid-regulating activities of the SREBF2 isoform in conjunction with miR-33, a short noncoding RNA found inside the genes that code SREBPs. This microRNA inhibits the expression of the adenosine triphosphate-binding cassette transporter A1, a protein that controls the synthesis of high-density lipoprotein (“good” cholesterol) and aids in the removal of low-density lipoprotein (“bad” cholesterol) from the bloodstream [70]. Moreover, miR-33 regulates lipid homeostasis through the reduction of FA breakdown. This is accomplished through inhibition of the translation of numerous transcripts encoding proteins involved in FA oxidation, such as carnitine palmitoyltransferase 1A (CPT1A), hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit beta (HADHB), and carnitine O-octanoyltransferase (CROT) [71]. Despite sharing some common genes with the SAGE pathway, most of the genes involved in the SCLH pathway are unique. Thus, a separate analysis was deemed necessary for each pathway. Predictably, despite the separate analyses, functional enrichment results still elicited major congruencies in the results, with commonalities in the GO and KEGG functional enrichment analyses.
As expected, almost all the top enriched GO biological processes for both the SAGE and SCLH pathway-related genes included the regulation of lipid, cholesterol, and steroid metabolic and biosynthetic processes, as this is the well-established purpose of the SREBP pathway. The GO molecular functions also naturally involved transcription-related functions, including DNA, organic cyclic compound, and transcription factor binding, as SREBPs are, in fact, transcription factors. The top enriched GO subcellular compartments essentially mirrored the transcription-related molecular functions, with most of the enriched terms reflecting the nuclear compartment and its subparts, where transcription occurs. Moreover, the enzyme complexes for ACC and FASN were also among the top subcellular compartments enriched in SAGE pathway genes. Not surprisingly, KEGG analysis revealed AMPK signaling as the most significantly enriched pathway for both SAGE and SCLH pathway-related genes.
ACC contributes significantly to the overall control of energy metabolism. ACC catalyzes the formation of malonyl-CoA, an essential substrate for FA synthesis in lipogenic tissues and a key regulatory molecule in muscle, brain, and other tissues. The specific activity of ACC is also rapidly modulated, being increased in response to insulin and decreased following exposure of cells to catabolic hormones or environmental stress. The acute control of ACC activity results from integrated changes in substrate supply, allosteric ligands, phosphorylation of multiple serine residues, and interactions with other proteins [72].
In CNS disorders, both the activation and inhibition of ACC play paradoxical
roles in the action mechanisms of therapeutic candidates. In AD, some drug
candidates primarily exert their therapeutic effects by inhibiting ACC. In animal
models of AD, inhibition of ACC1 reduced cognitive decline and markers of aging
via preservation of mitochondrial homeostasis in terms of increased levels of
acetyl-CoA [73]. Further, reductions of beta-secretase activity and amyloid beta
levels are induced by ACC inhibition in neuronal cells via the action of lipid
homeostasis modulators, such as the pluripotent peptide leptin [74]. In
Parkinson’s disease, ACC1 is recruited and colocalized with a neurotoxic species
of
In humans and birds, FASN plays a key role in de novo lipogenesis. FASN catalyzes all chemical steps in the conversion of acetyl-CoA and malonyl-CoA to palmitate using its seven active sites. Mitochondrial glycerol-3-phosphate acyltransferase (GPAT) catalyzes the first committed step in glycerophospholipid biosynthesis by acylating glycerol-3-phosphate with fatty acyl-CoA to produce 1-acyl-glycerol-3-phosphate (lysophosphatidic acid), which is acylated to create triacylglycerol for storage. Allosteric effectors or covalent modification are not known to regulate FASN activity. FASN concentration, however, is extremely sensitive to nutritional, hormonal, and developmental status; when animals are subjected to different nutritional and hormonal manipulations, the concentration or activity of FASN in lipogenic tissues, such as liver and adipose tissues, changes dramatically [82, 83, 84].
In NDDs, FASN activity has been implicated as a target for therapeutics. A FAS
inhibitor successfully alleviated cognitive loss, modulated lipid metabolism, and
reduced inflammation and lipid peroxidation in the brains of transgenic AD mouse
models [85]. In genetic models of PD, such as flies, mouse cells, patient-derived
fibroblasts, and induced pluripotent stem cell-derived dopaminergic neurons,
partial genetic and pharmacological inhibition of FASN decreases toxicity
generated by PINK1 deficiency [86]. This is corroborated by studies reporting
that the level of
The top enriched pathway for both SAGE and SCLH genes related with CNS disorders is the AMPK signaling pathway. This is not surprising because the top enriched subcellular compartments were ACC and FASN, enzymes that are both known to be regulated by AMPK as its downstream targets. Both ACC and FASN are mainly inhibited by the phosphorylation of AMPK. ACC inhibition by AMPK is generally coupled with thrombus formation [88], hypolipogenic pathways in the liver [89], and the insulin-sensitizing effect of metformin action [90]. FASN inhibition by AMPK is also implicated in cancer [91, 92] and tumor suppression [93].
AMPK pathway genes associated with the SAGE pathway included HMGCR, FASN, ACACB, SREBF1, SCD, and ACACA, whereas those related to the SCLH pathway comprised SIRT1, PPARGC1A, HMGCR, FASN, PRKAA1, SREBF1, MTOR, and SCD. AMPK is a serine/threonine kinase found in several tissues, and it serves as a major energy sensor. It is a heterotrimeric complex made up of a catalytic component and regulatory subunits [94]. The AMPK signaling pathway boosts ATP-producing pathways whilst inhibiting ATP-consuming pathways. Several factors, including low glucose, hypoxia, ischemia, and heat shock, can activate the kinase [95].
The modulation of lipid metabolism is one of the most well-known functions of AMPK; it promotes FA oxidation whilst inhibiting FA synthesis. AMPK phosphorylation inhibits ACC synthesis and lowers malonyl-CoA levels. Malonyl-CoA is an FA elongation and de novo synthesis substrate [96]. AMPK is also a carnitine palmitoyl transferase I inhibitor, which is essential for the transfer of primed cytosolic FAs into the mitochondrion for degradative beta-oxidation [97]. This kinase has long been a therapeutic target for metabolic disorders and malignancies. In particular, the involvement of aberrant AMPK pathway activity in CNS illnesses and diseases is well documented, with particular focus on the clinical repercussions of the pathway and its role as a therapeutic target [98, 99, 100].
Several review papers have elucidated the connection of the AMPK pathway with
NDDs [98, 101, 102]. In AD, the AMPK pathway predominantly confers deleterious
roles in the late stages of the disease [102]. Excitotoxicity, as well as
metabolic and oxidative stress, are all hallmarks of AD [103, 104]. Mitochondrial
failure eventually results in the generation of reactive oxygen species and a
rise in the AMP/ATP ratio, which are correlated to oxidative and metabolic
stressors, respectively [105]. These two events activate AMPK, which reduces
protein synthesis and contributes to memory loss by causing synaptic loss and
deficiency of long-term potentiation [106, 107]. AMPK is also involved in tau and
amyloid protein modulation. AMPK phosphorylates tau protein, modifying
microtubule assembly and, as a result, vesicle, and mitochondria axonal transit
[108]. In addition, AMPK is involved in the generation and degradation of
amyloid-
Collectively, the role of AMPK in NDDs may be viewed as a double-edged sword [102]. During the early stages of NDDs, the activation of AMPK may be beneficial, as it may help restore energy homeostasis and eliminate protein aggregates that can be harmful to neurons [99]. Nevertheless, chronic AMPK activation is detrimental to neurons in late-stage diseases. Moreover, AMPK over-activation may cause neurodegeneration via various signaling mechanisms. Reduction of protein synthesis may lead to synaptic loss and decreased synaptic plasticity, eventually leading to neurodegeneration. Finally, increased autophagosome synthesis coupled with deregulated lysosomal clearance (known to occur in these disorders) leads to an increase in hazardous proteins aggregates and mitochondrial defects [100, 118, 119].
In NLDs such as epilepsy and ischemic stroke, most evidence points to beneficial
effects of AMPK pathway activation, leading to neuroprotection. In epilepsy, the
action mechanism of several anti-epileptic drug candidates has been attributed to
the activation of the AMPK pathway. The neuroprotective effects of AMPK
phosphorylation via amelioration of oxidative stress through reduction of
reactive oxygen species and malondialdehyde, as well as strengthening of
glutathione peroxidase and superoxide dismutase, were associated with the
epilepsy-attenuating properties of
The involvement of the AMPK pathway in NPDs is also well-reported. AMPK
signaling plays a dual role in NPDs, in which both beneficial and deleterious
involvements have been documented. In anxiety, decreased levels of phosphorylated
AMPK are associated with anxiety, decreasing synaptic protein levels in the
cortex of mice subjected to unpredictable chronic mild stress. This suggests that
AMPK inactivation may be a mechanism by which unpredictable chronic mild stress
induces anxiety [128]. In bipolar disorder, AMPK upregulation is associated with
the manic phase of the disorder, which is characterized by increased
mitochondrial respiration and ATP production [129]. In mental depression,
antidepressant drugs exert their therapeutic effects via increased
phosphorylation of AMPK. In a mouse model of inflammatory bowel disease, liver
hydrolysate prevented depressive-like behavior by enhancing hippocampal
neurogenesis through the AMPK/brain-derived neurotrophic factor (BDNF) pathway
and anti-neuroinflammation in the hippocampus [130]. Moreover, the ability of
metformin to produce antidepressant effects is credited to the upregulation of
BDNF via activation of AMPK and cAMP-response element binding protein [131]. In
PTSD, increased AMPK activity was associated with the ability of
electroacupuncture to improve hippocampal neurogenesis and ameliorate
anxiety-like behavior in an enhanced single prolonged stress model of PTSD in
rats [132]. In schizophrenia, common differentially methylated genes were found
to be significantly involved in the AMPK signaling pathways [133]. AMPK
expression was also reported to be altered in the cortical excitatory neurons of
patients with mutations in the disrupted schizophrenia 1 (DISC1) gene
[134]. The deletion of 10 genes in human 1q21.1, a genetic risk of schizophrenia,
also involves the
In summary, we identified commonly expressed genes, biological processes, molecular functions, subcellular compartments, and biological pathways between the SREBP pathways and common CNS disorders. GO biological process and molecular function analyses confirmed the lipid regulatory action of the genes, as well as their transcription-related functions, all of which agree with the primary characteristic and main mandate of the SREBP pathways. Interestingly, most of the common genes were part of the subcellular compartments of either or both ACC and FASN enzymes, which play a key role in the regulation of energy metabolism and possibly serve as downstream targets of AMPK. Unsurprisingly, the top enriched pathway of the overlapping genes was the AMPK signaling pathway. We identified ACACA, ACACB, FASN, HMGCR, MTOR, PPARGC1A, PRKAA1, SCD, SIRT1, and SREBF1 as the top genes significantly enriched under this pathway. Both animal models and clinical studies consistently showed the role of AMPK signaling in mediating the involvement of the SREBP pathway in the lipid deregulation-related mechanisms underlying NDDs, NLDs, and NPDs. However, the relevance of gene-environment interaction is another noteworthy factor that may affect disease outcome, and thus cannot be discounted.
Further studies involving a more systematic literature review of the specific roles of each implicated common gene may shed more light in the treatment and diagnostic procedures currently available for CNS disorders. Likewise, gain- or loss-of-function experiments involving the same genes may provide an insight to which new models can be generated, replicating the lipid deregulated pathways of CNS disorders. Finally, investigation of other pathways related to lipid metabolism other than the SREBP signaling pathway may also uncover novel mechanisms underlying CNS disorders.
The elucidation of the importance of lipids in neuroscience is evolving with the advent of techniques allowing for a more detailed analysis of the types and roles of lipids in the nervous system. Several studies have already suggested the involvement of lipid homeostasis in the mechanism of CNS disorders. SREBP signaling is a transcriptomic pathway regulating lipid homeostasis, frequently implicated with organs that store or process large amounts of lipids, such as the liver and adipose tissue. The brain, which predominantly consists of lipids, is also affected by SREBP signaling. This is evidenced by the commonality between the genetic components of the pathway and the genes involved in NDDs, NLDs, and NPDs. In silico bioinformatic analysis revealed that the enzymes ACC and FASN, as well as the AMPK signaling pathway, are indisputably involved in the mechanism of disease progression and/or resolution in the CNS (Fig. 8). Lastly, lipid dysregulation-related animal models of NDDs, NLDs, and NPDs may present a new avenue by which research discovering novel, safe, and effective therapeutics may be propelled.
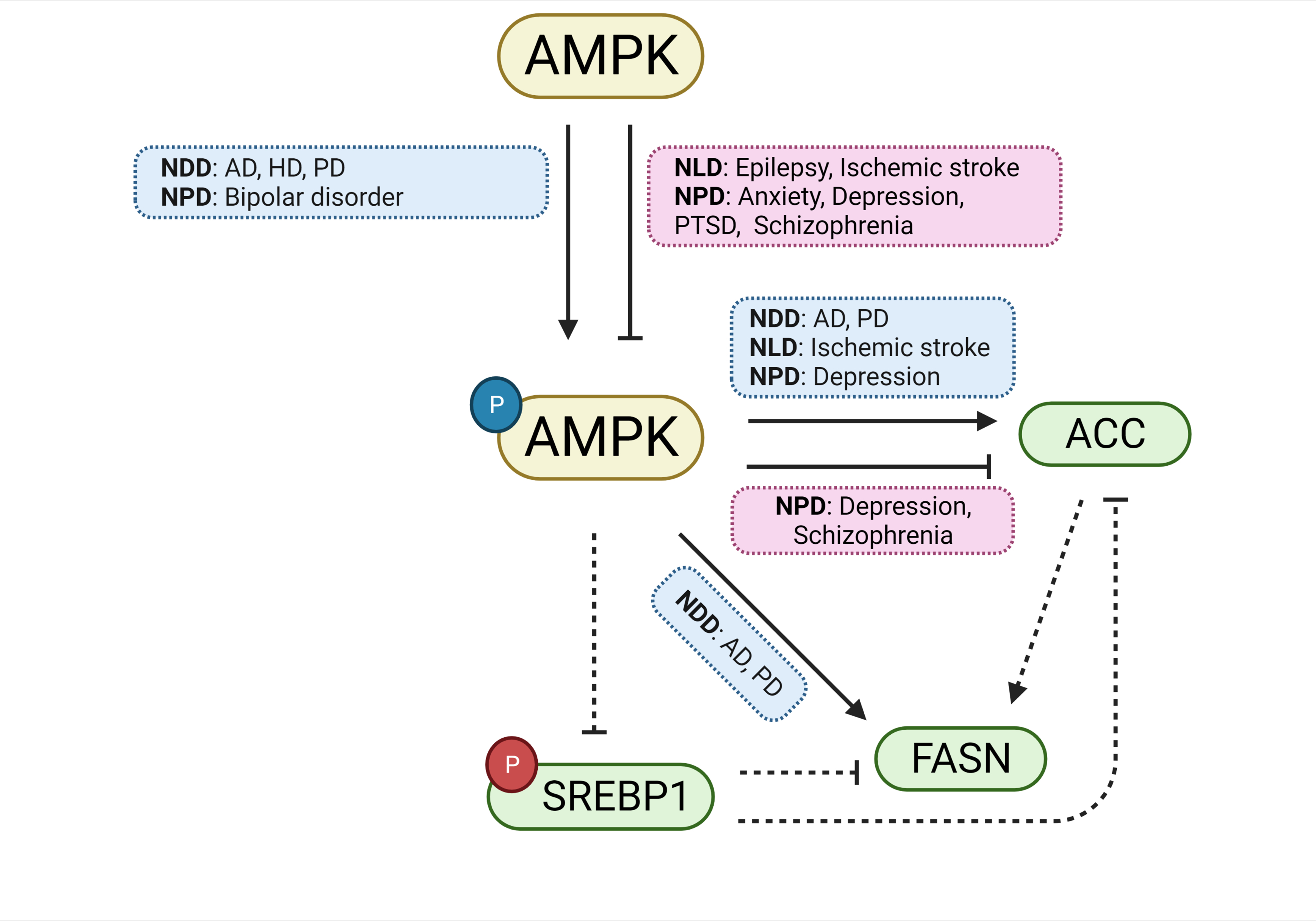 Fig. 8.
Fig. 8.ACC, FASN, and AMPK are involved in the genetic overlap between SREBP and CNS disorders. A schematic diagram summarizing the pathways governing ACC, FASN, and AMPK, and their involvement in CNS disorders. Both the activation and inhibition of AMPK are involved in the mechanisms underlying several CNS disorders. Likewise, both the activation and inhibition of the ACC enzyme are involved in CNS disorders. For FASN, only its activation is implicated in CNS disorders. Boxes in blue indicate involvement with CNS disorders during activation, whereas red boxes indicate involvement with disorders during inhibition. Inhibitory phosphorylations are shown in red and activating phosphorylations are shown in blue. Solid lines are evidenced by studies included in the current study; broken lines represent possible pathways not directly implicated by the reports included in the current study. Abbreviations: ACC, acetyl-CoA carboxylase; AD, Alzheimer’s disease; AMPK, AMP-activated protein kinase; FASN, fatty acid synthase; HD, Huntington’s disease; NDDs, neurodegenerative diseases; NLDs, neurological diseases; NPDs, neuropsychiatric diseases PD, Parkinson’s disease; PTSD, post-traumatic stress disorder; SREBP, sterol regulatory element binding protein.
ABCA1, ATP binding cassette subfamily A member 1; ACACA, acetyl-CoA carboxylase alpha; ACACB, acetyl-CoA carboxylase beta; ACC, acetyl-CoA carboxylase; Acetyl-CoA, acetyl coenzyme A; AD, Alzheimer’s disease; AMPK, AMP-activated protein kinase; bHLH-LZ, basic helix-loop-helix leucine zipper; CNS, central nervous system; CREBBP, CREB binding protein; DAVID, Database for Annotation, Visualization and Integrated Discovery; DHCR7, 7-dehydrocholesterol reductase; ER, endoplasmic reticulum; FASN, fatty acid synthase; FDPS, farnesyl diphosphate synthase; FunRich, Functional Enrichment analysis tool; Gly, glycine; GO, Gene Ontology; GSEA, Gene Set Enrichment Analysis; HD, Huntington’s disease; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; KEGG, Kyoto Encyclopedia of Genes and Genomes; LDLR, low density lipoprotein receptor; MED1, mevalonate diphosphate decarboxylase; MED15, mediator complex subunit 15; MTOR, mechanistic target of rapamycin kinase; MVD, mevalonate diphosphate decarboxylase; MVK, mevalonate kinase; NADP, nicotinamide adenine dinucleotide phosphate; NCOA2, nuclear receptor coactivator 2; NCOA6, nuclear receptor coactivator 6; NDDs, neurodegenerative diseases; NFYA, nuclear transcription factor Y subunit alpha; NFYC, nuclear transcription factor Y subunit gamma; NLDs, neurological diseases; NPDs, neuropsychiatric diseases; NR1H3, nuclear receptor subfamily 1 group H member 3; MSigDB, Molecular Signatures Database; PD, Parkinson’s disease; PPARA, peroxisome proliferator-activated receptor alpha; PPARGC1A, PPARG coactivator 1 alpha; PPI, protein-protein interaction; PRKAA1, protein kinase AMP-activated catalytic subunit alpha 1; Pro, proline; PTSD, post-traumatic stress disorder; RXRA, retinoid X receptor alpha; SAGE, SREBP activation of gene expression; SCLH, SREBP cholesterol & lipid homeostasis pathway; SC5D, sterol-C5-desaturase; SCD, stearoyl-CoA desaturase; Ser, serine; SIRT1, sirtuin 1; SIRT6, sirtuin 6; SP1, Sp1 transcription factor; SQLE, squalene epoxidase; SREBF1, sterol regulatory element binding transcription factor 1; SREBF2, sterol regulatory element binding transcription factor 2; SREBP, sterol regulatory element binding protein; STRING, Search Tool for Retrieval of Interacting Genes; TA, transactivation; TBL1XR1, TBL1X receptor 1; TGS1, trimethylguanosine synthase 1; TM7SF2, transmembrane 7 superfamily member 2.
MJA and CM conceived and designed the experiments; MJA designed the methodology; MJA did the formal analysis; MJA wrote the original draft; CM edited and reviewed the draft; CM supervised the study, CM was responsible for funding acquisition.
Not applicable.
Not applicable.
This work was supported by a grant from the National Research Foundation (NRF) of Korea funded by the Korean Government (NRF-2020R1A4A1019395).
The authors declare no conflict of interest. CM is serving as one of the Editorial Board members of this journal. We declare that CM had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to UGP and RF.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/j.jin2103095.