







1 Department of Neonatology and Pediatric Intensive Care, University Medicine Greifswald, 17475 Greifswald, Germany
2 Department of Neurology, University Medicine Greifswald, 17475 Greifswald, Germany
3 Department of Congenital Heart Disease and Pediatric Cardiology, German Heart Center Munich, Technical University of Munich, 80636 Munich, Germany
†These authors contributed equally.
Abstract
Preterm birth causes neurological deficits. Previously, we demonstrated that fetal zone steroids reduce hyperoxia-mediated cell death in vitro. In immature oligodendrocytes (OLN-93 cells), dehydroepiandrosterone + 17
Keywords
- Fetal zone steroid
- Dehydroepiandrosterone
- 17
-estradiol - Preterm infant
- Estrogen receptor
- Androgen receptor
- Neuroendocrinology
- Hypertoxicity
Preterm infants are more vulnerable to central nervous system injury than term
neonates, typically white matter injuries [1]. Hyperoxia is one key factor in the
pathogenesis [2, 3]. The arterial oxygen partial pressure (PaO
The fetal zone is a compartment of the fetal adrenal cortex in humans and other higher primates. Dehydroepiandrosterone sulfate (DHEAS) is the main precursor from the fetal zone and is synthesized until the term in increasing amounts of up to 100-200 mg per day [10, 11]. In the placenta, DHEAS is used for biosynthesis of different estrogens, which has led to the term “feto-placental unit” [12]. Preterm birth disconnects this unit, which results in a 100-fold drop in plasma estrogen levels compared to intrauterine levels [13, 14]. Notably, preterm infants’ fetal zone produces persistently high quantities of DHEAS until the time of the termed birth [15, 16].
It was hypothesized that estrogen and progesterone’s substitution after preterm birth potentially protects the immature brain in case of neurological complications [17]. In a preliminary study, 83 premature infants were randomized to supplement E2 and progesterone to maintain intrauterine concentrations. When the infants were discharged home, no effects on survival or vulnerability for intracerebral hemorrhage were observed [18]. Five years later, no benefits were found [19], which underlines the need for a more detailed understanding of the role of fetal zone steroids (FZS), E2 and progesterone for the preterm brain.
In our recent work [20], we investigated the discrepancy between in
vitro models and findings of the preclinical studies of Trotter et al. [17, 18, 19].
By using three different types of immature glial cells (OLN-93, C6 and
PDGFR
In general, E2 and DHEA’s effect can be mediated by two classical estrogen
receptors (ERs), ER-
The amount of functional receptors expressed by a cell is crucial for its response to hormonal ligands and can change with development, physiological and pathological status [28]. The steroid hormone receptors can be regulated by the number of ligands (autoregulation - induction or suppression) [28]. Tissue dependent autoinduction and autorepression are described for ERs and ARs. In cross-regulation, the binding of one hormone to its receptor modulates another receptor. For example, E2 can induce the progesterone receptor’s expression via cross-regulation [29]. We, therefore, investigated the effects of hyperoxia and steroid hormones on receptor density.
Different effects of hormones are mediated by different receptor type and
subsequent different involved signal transduction pathways which include ERK1/2,
Akt (Thr308 and Ser473) [30], AMPKa, S6RP, Bad, p70S6K, PRAS40 and GSK-3
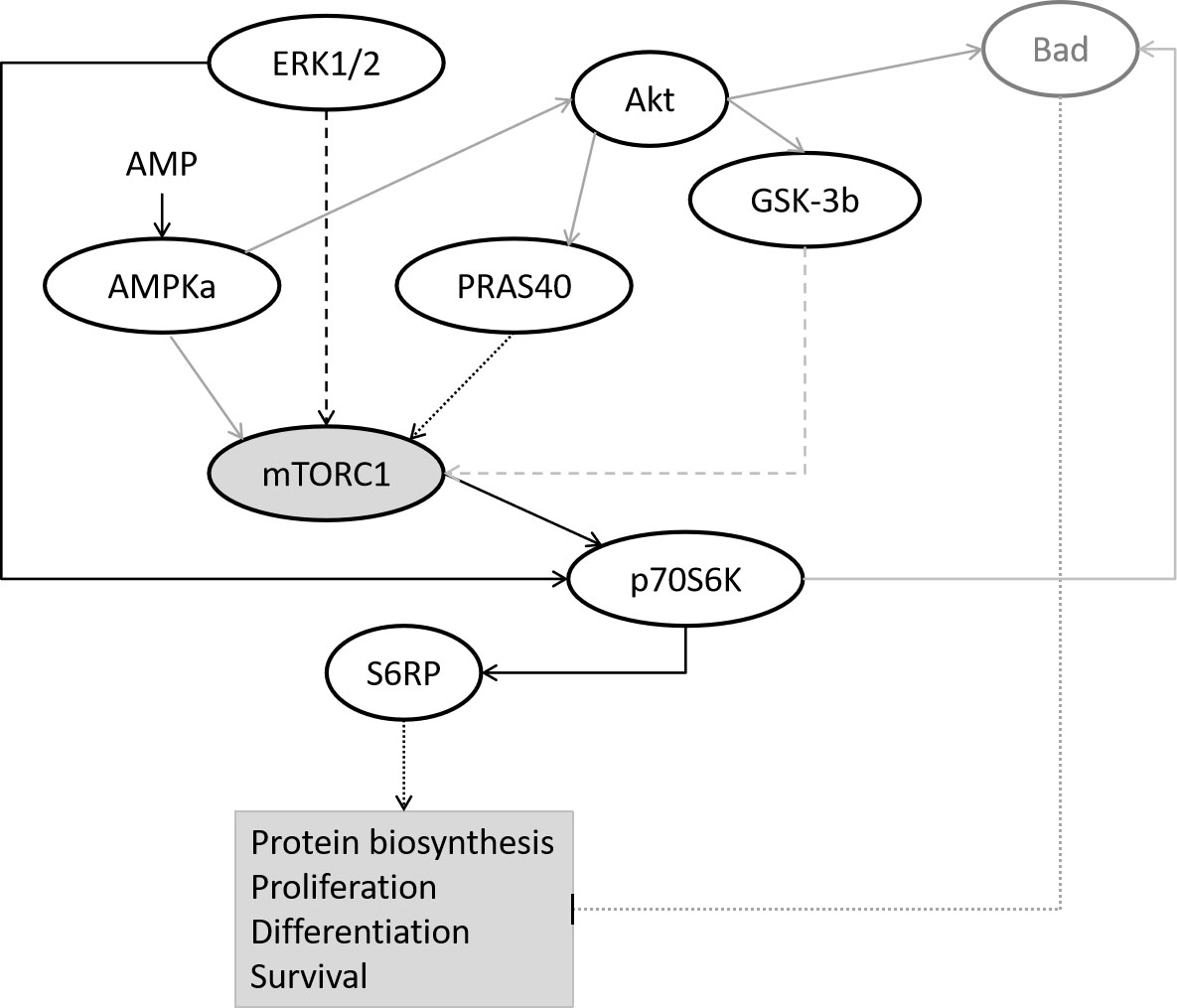 Fig. 1.
Fig. 1.Simplified overview of investigated signaling pathways modified from [31, 33]. Black line: dotted - activation, solid - activation by phosphorylation, dashed - intermediate steps. Grey line: dotted - inhibition, solid - deactivation of a protein complex by phosphorylation, dashed - intermediate steps.
Exploring the basics of E2 and DHEA influence on receptor density and signal transduction can help to develop effective neuroprotective treatments for preterm infants. Thus, we investigated the effects of E2 and DHEA on receptor density and signal transduction pathways in a hyperoxia cell culture model. Therefore, we chose two different immature glial cell types to take potential cell-type-specific outcomes into account: (i) the rat-derived immature oligodendroglial cell line OLN-93 and (ii) the rat-derived immature astroglial cell line C6.
The rat-derived oligodendrocyte progenitor cell line OLN-93 [34] was cultured as described previously [35].
The rat-derived immature astroglial cell line C6 [36, 37] was purchased from the American Type Culture Collection (ATTC) cultured as recommended.
Both cell lines were cultured to stable replication for up to ten passages after
thawing in a humidified atmosphere at 37
To investigate the causes of the protective effects, concentrations were
selected which exhibit a long-lasting protective effect. Only by this approach,
further experiments can be conducted to investigate the underlying mechanisms.
Therefore, different concentrations were used for the different substances, which
exhibited a long-lasting protective effect in earlier experiments. Therefore,
different concentrations were used for the different substances or cell
lines. Stock solutions (1000
After 48 h of recovery in culture medium, OLN-93 and C6 media were exchanged to
experimental media as previously described [20] containing steroids or DMSO.
After 2 h of preincubation, the medium was renewed, and the cells were exposed to
hyperoxia (humidified atmosphere at 37
C6 and OLN-93 cells were preincubated for 2 h with 1
A 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Promega) was used according to the manufacturer’s instructions to measure the viability of C6 cells. In this assay, MTT, a yellow tetrazole, is reduced to purple formazan in living cells. The conversion was measured by a plate reader at OD 595 nm and OD 650 nm as a reference and subtracted from OD 595 nm. For higher comparability among the survival assays, the viability results were processed to cell death by the formula: % cell death = 100- (% viability -100). Results are given as % cell death compared to DMSO-treated cells under hyperoxia.
To investigate the influence of steroid treatment and hyperoxia on the
ER-
| Name of Antibody | Manufacturer, catalog #, and/or name of individual providing the antibody | Species raised in; monoclonal or polyclonal | Dilutionused |
| Rabbit Anti-Estrogen Receptor- |
Santa Cruz, #sc-543 | Rabbit, polyclonal | 1 : 200 |
| Rabbit Anti-Estrogen Receptor- |
Affinity BioReagent, #PA1-311 | Rabbit, polyclonal | 1 : 1,000 |
| Rabbit Anti G-Protein Coupled Receptor 30 | Abcam, #ab39742 | Rabbit, polyclonal | 1 : 250 |
| Mouse Anti-Androgen Receptor (Clone AR441) | Abcam, #ab9474 | Mouse, monoclonal | 1 : 200 |
| Rabbit Anti- |
Sigma-Aldrich, #A6022 | Rabbit, polyclonal | 1 : 5,000 |
| Peroxidase AffiniPure F(ab’)2 Fragment Goat Anti-Rabbit IgG (H + L) | Jackson ImmunoResearch, # 111-036-003 | Goat | 1 : 5,000 |
| Peroxidase AffiniPure F(ab’)2 Fragment Goat Anti-Mouse IgG (H + L) | Jackson ImmunoResearch, #115-036-146 | Goat | 1 : 5,000 |
C6 and OLN-93 cells were pretreated for 2 h with DMSO, E2, DHEA, or E2 and DHEA
for signaling array experiments. Cells were collected after 6 h and 24 h of
hyperoxia, according to the manufacturer’s instruction. As recommended by the
manufacturer, the lysis buffer was supplement with Halt Protease and Phosphatase
Inhibitor Single-Use Cocktail (100
According to the manufacturer’s instructions, cell lysates were added to the nitrocellulose-coated glass slides pre-coated with primary antibodies for 2 h. Afterward, the slides were incubated for 1 h with biotinylated antibodies, followed by a horseradish peroxidase-conjugated secondary antibody for 30 min. The slides were exposed to a substrate, and chemiluminescence was measured. Each array was processed with the “Protein Array Analyzer” macro in ImageJ (NIH) for background subtraction and grid measurement [38]. Normalization between arrays was calculated by scaling the individual intensities so that the mean intensities were the same across all compared arrays. The results show only proteins for which signal intensities were stronger than the background after 1 min of exposure. The signal intensities are given as the ratio to DMSO-treated cells, calculated for each experiment.
Statistical significance was assessed with GraphPad Prism 8.3.1 software
(GraphPad Software, LLC) by Two-way ANOVA or Mixed Model depending on missing
values. We corrected for multiple comparisons using Tukey’s post hoc test.
Matched values were factored in as appropriate. Friedman Test was used for data
of Fig. 2. The significance levels were denoted as *P
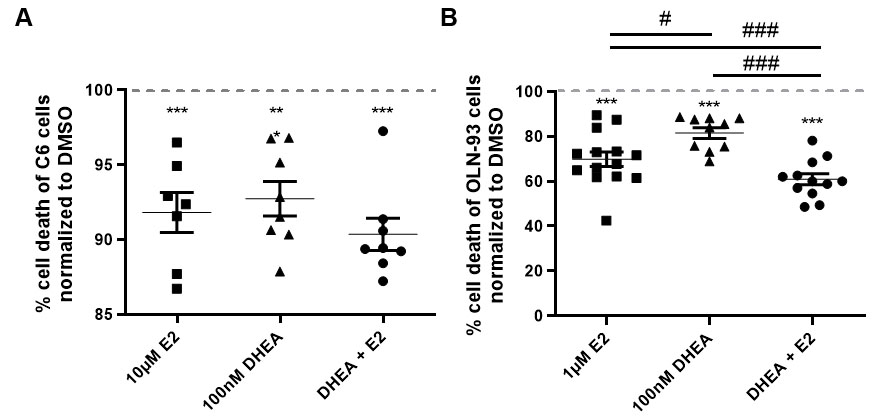 Fig. 2.
Fig. 2.Effects of E2 and DHEA co-treatment on hyperoxia-mediated cell
death. Cell survival was measured (A) in C6 cells by MTT assays and (B) in
OLN-93 cells by LDH assays after treatment with E2 alone (10 or 1
Under hyperoxia, the effect of treatment on cell survival with DHEA alone,
co-treatment with E2 or E2 alone was compared to DMSO control (set to 100%)
(Fig. 2A, B). While all hormone conditions reduced cell death significantly in
both cell types, co-treatment significantly increased cell survival compared to
DHEA or E2 single treatment in OLN-93 cells (P
To investigate the influence of steroid treatment and hyperoxia on the steroid
receptor availability, the expression of ER-
In C6 cells, neither steroid treatment nor hyperoxia led to significant
ER-
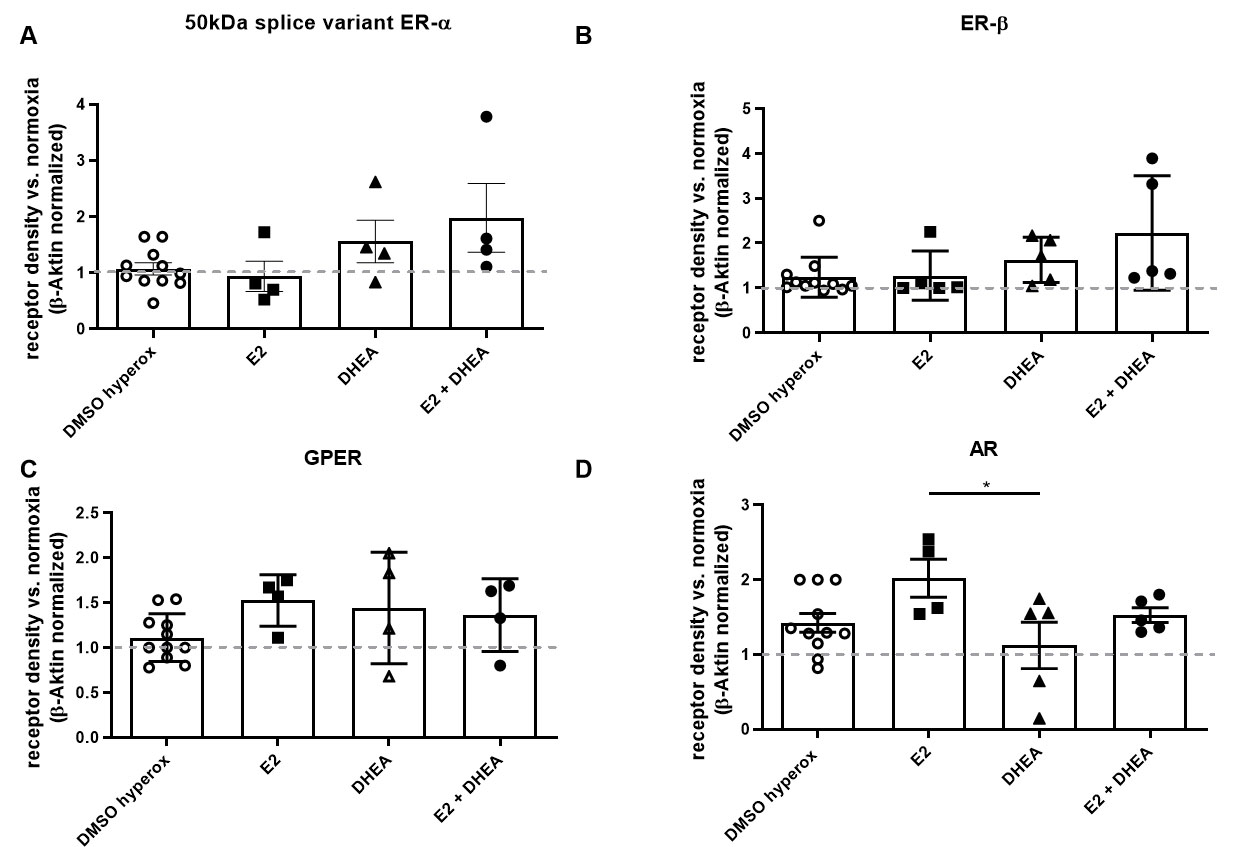 Fig. 3.
Fig. 3.Effects of steroid treatment and hyperoxia on receptor
expression in C6 cells. The expression of (A) 50 kDa splice variant of
ER-
In OLN-93 cells, treatment with DHEA plus E2 led to a significant increase of
ER-
There were no significant differences for GPER or AR observed under hyperoxia (Fig. 4C, D).
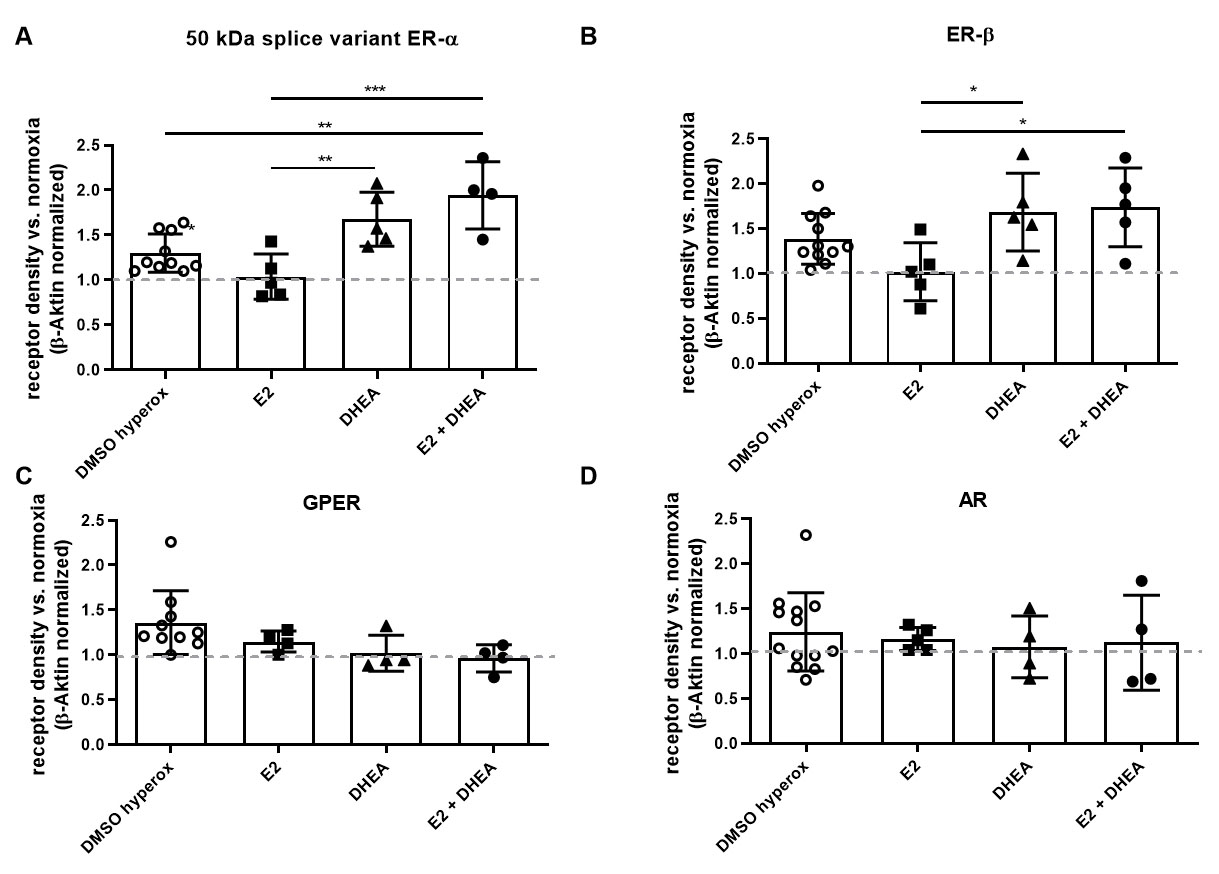 Fig. 4.
Fig. 4.Effects of steroid treatment and hyperoxia on receptor
expression in OLN-93 cells. The expression of (A) 50 kDa splice variant of
ER-
In C6 cells, ERK1/2 (Thr202/Tyr204), Akt (Thr308 and
Ser473), S6 ribosomal protein (Ser235/236; S6RP), Bad (Ser112),
ribosomal protein S6 kinase (Thr389; p70S6K), proline-rich Akt/PKB substrate 40
kDa (Thr246; PRAS40), and glycogen synthase kinase-3
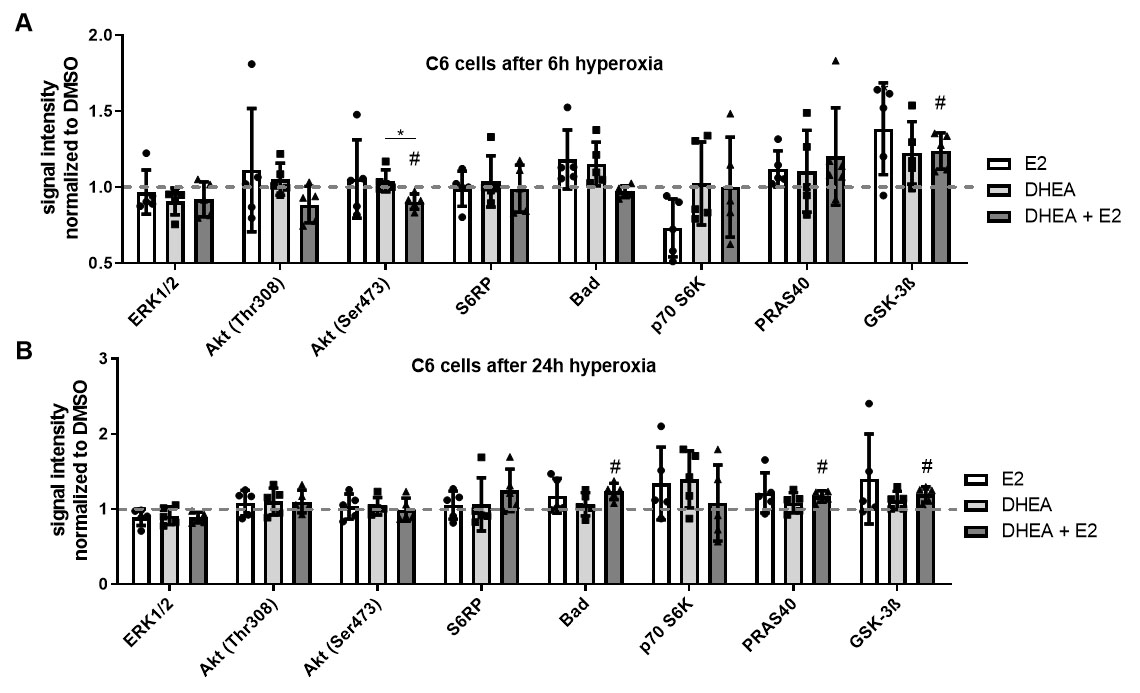 Fig. 5.
Fig. 5.Intracellular signaling among hyperoxia and treatments in C6
cells. To explore intracellular signaling, a signaling array was used. After 2 h
treatment with DMSO, E2, DHEA, or DHEA/E2 co-treatment, cells were exposed to 6 h
and 24 h of hyperoxia. C6 cells were treated with DMSO, 10
After 6 h of hyperoxia, Akt (Ser473) was significantly downregulated, whereas
GSK-3
After 24 h of hyperoxia, there were no significant differences in E2- and
DHEA-treated cells (Fig. 5B). Under DHEA + E2 co-treatment, Bad, PRAS40, and
GSK-3
In OLN-93 cells, the signaling mentioned above molecules and AMP-activated
protein kinase
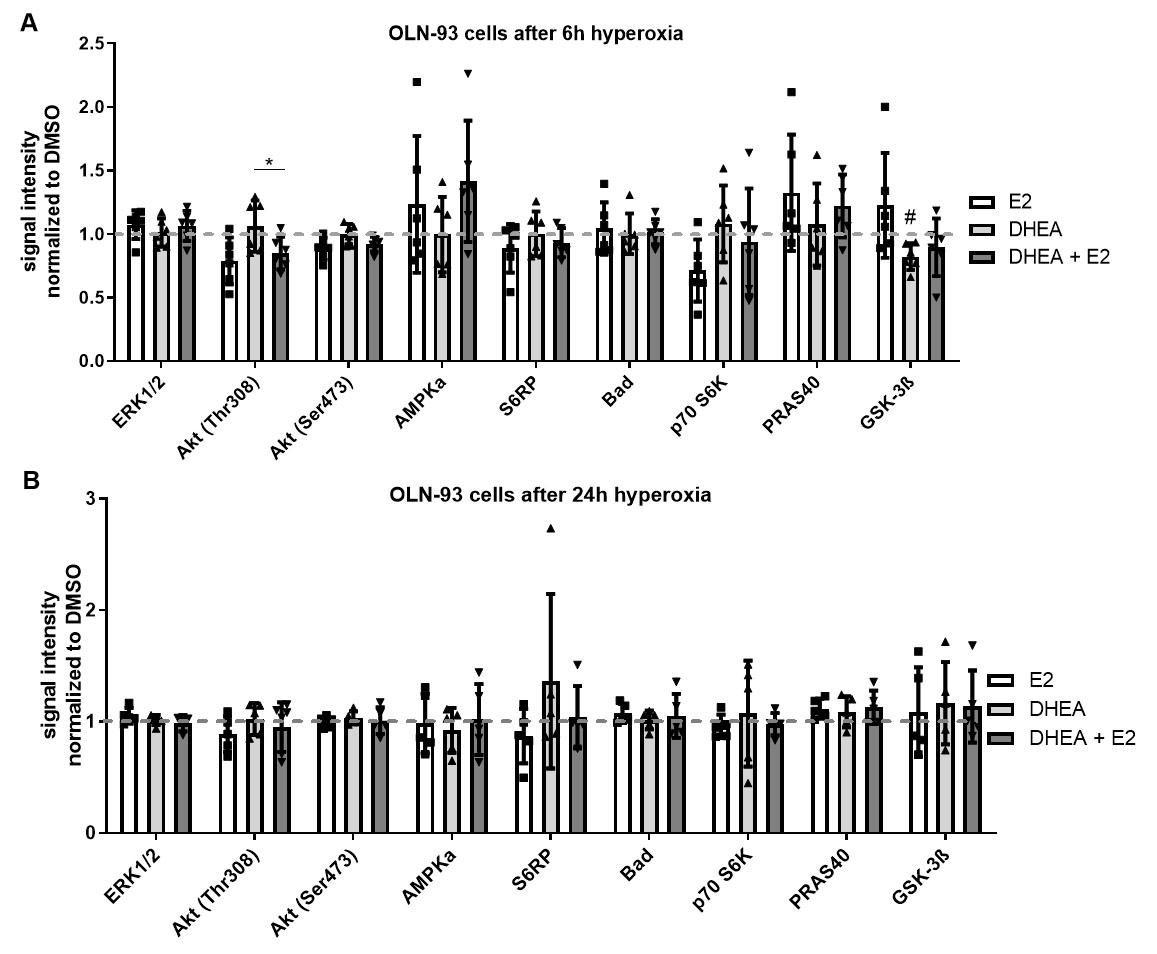 Fig. 6.
Fig. 6.Intracellular signaling among hyperoxia and treatments in OLN-93
cells. To explore intracellular signaling, a signaling array
was used. After 2 h treatment with DMSO, E2, DHEA, or DHEA/E2 co-treatment, cells
were exposed to 6 h and 24 h of hyperoxia. OLN-93 cells were treated with DMSO, 1
After 6 h of hyperoxia, there was a significant downregulation of GSK-3b under DHEA treatment (Fig. 6A). Of interest, there was a significant difference between DHEA single and its combination treatment with E2 in phosphorylation of Akt (Thr308). The activation of Akt in E2 and DHEA/E2 co-treated cells seems to be transient as in the C6 cells because there was a significant upregulation of the Akt pathway (Fig. 6A).
After 24 h of hyperoxia, no significant differences were observed (Fig. 6B).
During pregnancy, FZS is synthesized by the fetus and serve as precursors for estrogen synthesis in the placenta [12]. It was previously shown that FZS synthesis in preterm infants is persistently high until term [15, 16]. Our recent work showed that FZS activates classical ERs, which lead to protective properties against hyperoxia-induced cell death [20]. The experimental hyperoxia model was applied to mimic the oxygen supplementation used in neonatal intensive care in resuscitation and treatment of neonatal lung disease. Due to a dramatic rise in oxygen tissue tension, premature infants are subjected much earlier to relative hyperoxia than intrauterine conditions. However, in C6 cells under hyperoxia, co-treatment of DHEA and E2 showed no synergistic effects while both hormones competed for the same receptor [20]. In contrast, in OLN-93 cells under hyperoxia, co-treatment led to synergistic effects while E2 and DHEA mediated their effects via different receptors as published earlier by our group [20]. To further elucidate the circumstances of synergism, we now aimed to investigate the effects of DHEA and E2 alone and their combination on the receptor availability and the induced signaling pathways.
The effects of hyperoxia and steroid hormones on receptor density have been
investigated in only two studies to date. In one study, astrocytes were shown to
downregulate the amount of progesterone receptor by hyperoxia [39]. The other
study showed in cultured smooth muscle cells of the respiratory tract that
hyperoxia (with 50% O
Hyperoxia induced significant changes in receptor expression compared to DMSO control in OLN-93 cells. The effects on the C6 cells were the only minor. Since a similar trend in regulation could be observed, the adverse finding could be based on the limited number of experiments.
Since full-length ER-
In C6 cells, E2 seems to induce AR via cross-regulation under hyperoxia. The induction of AR via E2 has already been described for other tissues [44]. However, it remains to be clarified whether the effect of DHEA is an amplification of autoinduction and/or a cross-regulation since DHEA can bind to classical ERs and AR.
In OLN-93 cells, ER-
Receptor-mediated survival can be initiated via genomic and non-genomic pathways. The activation of signal transduction pathways can be temporary (transient activation) or persistent (permanent activation). Transient signals can be activated just a few minutes after adding factors and can disappear after minutes or hours [46, 47]. E2 and DHEA can induce non-genomic neuroprotective signaling pathways via ERK1/2 and Akt [8, 48, 49].
It was previously shown that PI3K/Akt/Bad signaling mediates protection of E2 against oxidative stress-induced apoptosis in C6 cells [50], and treatment with DHEA prevents cell death via PI3K/Akt/mTOR signaling in newborn neurons [49]. Even without sustained upregulation of Akt itself, we found downstream signaling proteins of the PI3K/Akt/Bad and mTOR pathways significantly upregulated in C6 cells after 24 h of hyperoxia in the co-treatment group. Of interest, it was previously reported that induction of cell death could induce ERK1/2 phosphorylation in astrocytes [51, 52], and E2 can reverse hyperoxia-induced phosphorylation of ERK1/2 in astrocytes [53].
Although it was shown that E2 activates ERK1/2 and Akt signaling to prevent cell death in oligodendrocyte precursor cells [8], we only found downregulation of GSK-3b under DHEA treatment after 6 h in OLN-93 cells. This might be due to differences in cell culture, experimental setting or even the used oligodendrocyte cell type.
Our findings show that the generated signaling of E2 + DHEA treatment in C6 cells has no synergistic effects. Thus, although the same signaling route is addressed, the combined effect on this particular pathway does not potentiate cell survival. On the other hand, we observed improved cell survival after E2 + DHEA treatment in OLN-93 cells. Here we found significant differences in the early generated signaling of E2 or DHEA treatment compared to E2 + DHEA cells but never both at the same time.
The observed changes in the receptor densities do not affect the occurrence of
synergism. Furthermore, two neuroprotective treatments that initiate the same
signaling pathway might not cause synergism since the full signaling capacity was
already reached by one of the stimulants alone. If two neuroprotective treatments
initiate different or differently timed signaling, synergism might occur more
quickly. Synergistic effects of DHEA and E2 as shown in OLN-93 cells are not
likely to occur in preterm infants due to the receptor affinities of human
receptors that will not lead to predominant AR activation by FZS (e.g., DHEA:
ER-
Auto- and cross-regulation are essential mechanisms for amplifying hormone signals, regulating hormone activities by negative feedback mechanisms, and coordinating hormone effects in a quick and tissue-specific manner [28]. In general, autorepression’s most crucial function is to restore homeostasis by downregulating the receptor as the hormones increase [28]. This effect could not be found in either of the cell types studied within the observation period. Future work would require investigating how the severe deficiency of estrogens and progesterone, together with the large number of adrenal androgens (fetal zone steroids), affects the receptor composition in the premature infant after birth and in the long term.
16OH-DHEA, 16
SH did the investigation, formal analysis and writing (original draft preparation). DS und CZ did investigation and writing (review & editing). JR did the investigation, formal analysis and writing (review & editing). BR did supervision and writing (review & editing). AV and MH did conceptualization, supervision and writing (review & editing).
Not applicable.
The authors thank Prof. Dr. Christiane Richter-Landsberg (University of Oldenburg, Germany) for the gift of cell line OLN-93 and Monika Hoyer for technical assistance.
The study was based on internal funds.
The authors declare that they have no competing interests. Given his role as the Review Board Member of JIN, Prof. Matthias Heckmann had no involvement in the peer-review of this article and has no access to information regarding its peer-review.
Not applicable.
Raw data is available from the authors upon reasonable request.