1. Introduction
Aging is a pivotal factor for numerous diseases, including neurodegeneration,
obesity, diabetes, cardiovascular diseases, and metabolic disorders [1, 2].
Growth factors, nutrients, and energy metabolism are pivotal factors for cell
growth, development, and proliferation. Activation of the mechanistic target of
rapamycin (mTOR) promotes cell growth in response to favorable environmental cues
and is viewed as a master regulator of this response [3]. Many studies have shown
that mTOR signaling dysregulation is involved in age-related diseases, including
neurodegenerative diseases, diabetes, metabolic disorders, and cancer
[4]. mTOR signaling networks stimulate the
synthesis of nucleotides, proteins, and lipids and block autophagic catabolic
response at the post-translational and transcriptional levels [5]. The
PI3-K/Akt/mTOR signaling pathway is widely regarded as a central signaling axis
to regulate cell growth and proliferation, crucial metabolism processes,
apoptosis, and secretion [6]. Protein kinase B (PKB, also known as Akt) performs
its action as a central intersection between phosphoinositide 3-kinase (PI3-K)
and mTOR by phosphorylating various substrates. Considering its crucial role in
regulating vital cellular functions, dysregulation of PI3-K/Akt/mTOR is a
critical molecular event in mental illnesses [7]. Specifically, abnormalities
in PI3-K/Akt/mTOR signaling are involved in
Alzheimer’s disease (AD) [8]. Overactivation of PI3-K/Akt/mTOR signaling in the
brain is regarded as
an early
pathogenic event in AD and an essential candidate for pathophysiological
processes activated by -amyloid [8]. Evidence gathered also indicates that
insulin and IGF-1 can rescue and normalize the aberrant PI3-K/Akt/mTOR signaling
and protect against AD’s physiopathologic processes [9].
Recent studies focused on the regulators of longevity and health span showed
that strategies to delay aging are therapeutic
strategies for aging-related diseases such as AD [10, 11]. mTOR
inhibition [12] and autophagy enhancement [13] are regarded as crucial regulators
of longevity and health span, as well as the novel therapeutic strategies for
aging-associated diseases. mTOR functions as a nutrient sensor by regulating
“protective” autophagy programs [14]. Interestingly, activation of the mTOR
signaling pathway is related to AD [15]. The inhibition of mTOR is being
developed into a novel AD therapy [16].
Autophagy is a critical molecular mechanism in mediating the lifespan-extending
effects of dietary restriction and mTOR inhibition [17]. Autophagy is a normal
cellular process in which the lysosome degrades older cytosolic components due to
nutrient deprivation [18]. Many studies have shown that damage due to autophagy
occurs at the early stages of the AD process. Studies also showed that
autophagy performs a pivotal role in the production and metabolism of
A and AD progress [19]. As the self-degrading
process, autophagy is key in maintaining cellular homeostasis. Defects in
autophagy homeostasis are considered pivotal pathogenesis in shortening lifespan
and promoting multifarious aged-related diseases, including obesity, insulin
resistance, diabetes, dementia, atherosclerosis, and neoplasm. Preclinical
evidence supports autophagy modulators’ therapeutic promise to treat obesity and
metabolic diseases [20]. Recent work has shown that glucagon-like peptide-1
(GLP-1)-based therapeutic approaches may positively affect autophagy in
perivascular adipose tissue, thus improving obesity-related endothelial
dysfunction [21]. To explore the effects of GLP-1 in GLP-1/insulin/insulin-like
growth factor-1 (IGF-1) signaling pathway and the autophagic process, Candeias et
al. [22] evaluated the effect of GLP-1 GLP-1 mimetics, exendin-4 (Ex-4) on
insulin, and IGF-1, their downstream signaling and autophagic markers in brain of
the T2D rats [22]. The results showed that Ex-4 protects T2D rats against
hyperglycemia; insulin resistance enhances GLP-1 and IGF-1 levels in brain
cortical and subsequent signaling pathways. Ex-4 also regulated autophagy markers
(as mTOR, PI3K class III, LC3 II, Atg7, p62, LAMP-1, and Parkin).
Geniposide is a traditional Chinese medicine monomer isolated from the herb
Gardenia jasminoides. Its extensive pharmacological effects, including
anti-diabetes, anti-inflammation, antioxidation, neuroprotection, and
anti-asthma, have been noted [23]. The protective effects of geniposide in
neurodegenerative diseases have been of keen interest. A glucagon-like peptide-1
receptor (GLP-1R)-the dependent mechanism-protected geniposide [24, 25]. Further,
activation of PI3K/AKT signaling may also involve a geniposide-induced protective
effect [26]. Li et al. [27] showed that although geniposide was a useful
bioactive substance in treating AD, its toxicity was apparent at a dose higher
than 50 mg/kg/d. Dinda et al. [28] reviewed the therapeutic potential of
plant iridoids, including geniposide, in AD and Parkinson’s disease. Plant iridoids exhibit the
property of retarding the process of neurodegeneration in AD and Parkinson’s disease. Geniposide
performed its protective effects after passing the blood-brain barrier [29].
Plant iridoids, including geniposide, can ameliorate AD by increasing the
expression of PPAR-, and -secretase, insulin-degrading
enzyme, neprilysin, and decreasing the levels of A oligomers
(A) deposited in brain neurons. The molecular mechanism has been
extensively explored. It is suggested that plant iridoids, including geniposide, may: 1. Decrease
expression of GSK-3 and its receptor gene; 2. Improve the lysosomal
autophagy process by increasing the expression of LC3II, Beclin-1, and cathepsin
B genes for the clearance of A and neurofibrillary tangles (NFT); 3.
Enhanced expression of transporter proteins, such as P-glycoprotein and
low-density lipoprotein receptor-related protein-1, for the clearance of
A load from brain across the blood-brain barrier; 4. Enhanced expression
of PPAR- and ApoE proteins for clearance of A in ApoE mediated
pathway from the brain. Further, plant iridoids may decrease cognitive
impairment by enhancing the expression of synaptic proteins, such as SNAP-25,
BDNF, PSD-95, GAP-43 and SYP, to improve learning memory ability in AD. Some of
those plant iridoids, including geniposide, may improve the expression of
TH-positive neurons, GDNF, and Bcl-2 proteins by increasing the levels of
antioxidant enzymes, such as GSH-P and SOD, and down-regulate insulin/IGF
signaling by activating MEK. Furthermore, geniposide may enhance the expression
of autophagy-related LAMP-2A-protein for clearance of LB from dopaminergic
neurons in the PD brain via improving the lysosomal autophagy process.
Song et al. [30] pretreated differentiated SH-SY5Y cells or
primary hippocampal neurons with Schizandrol A and subsequently subjected the
cells to -amyloid peptides of 1-42 amino acids (A) and estimated the effect of Schizandrol A by testing
its effects on cell viability, apoptosis, oxidative stress, and autophagy.
Further, these investigators explored the molecular mechanism underlying this
effect by treating cells with an mTOR inhibitor (rapamycin) and a PI3K inhibitor
(LY294002) to analyze the role of the PI3K/AKT/mTOR pathway. Their results showed
that Schizandrol A effectively inhibited A-triggered increases
in apoptotic cell number and pro-apoptotic protein expression, reduction of
viable cells, as well as alterations in markers of oxidative stress. Also,
Schizandrol A enhanced LC3-II/LC3-I and Beclin-1 and reduced the expression of
p62. At the molecular level, they showed Schizandrol A rescued the
PI3K/AKT/mTOR-autophagy pathway dysregulation resulting from
A exposure.
Based on the overlapping functions between GLP-1 and mTOR inhibition, including
energy balance, AD protection and diabetes treatment, we hypothesized in an earlier study that mTOR
inhibition may mediate the protective effect of GLP-1 in AD [31]. Similarly,
Jiang et al. [32] explored molecular mechanisms underlying the effect of
GLP-1 to improve insulin signaling in ER-stressed adipocytes. These investigators
showed GLP-1 directly modulated ER stress response, in part, by inhibiting the
mTOR signaling pathway. Further, a study from our group showed that the
downregulation of mTOR signaling and enhancement of autophagy in APP/PS1 mice
mediated the effect of geniposide to protect against amyloid deposition and
behavioral impairment [33].
In this paper, we test the hypothesis that mTOR inhibition and autophagic
activity are key molecular events that control the protective effects of
geniposide against A in vitro.
2. Materials and methods
2.1 Chemicals and reagents
The SH-SY5Y cell line’s human neuroblastoma was obtained from
the Stem Cell Bank, Chinese Academy of Sciences.
Geniposide (purity 98%) was purchased from Aladdin Bio-Chem Technology
Company, LTD, Shanghai, PR China. A (CAT: 1932-2-15, Peptide
Sequence:
Asp-Ala-Glu-Phe-Arg-His-Asp-Ser-Gly-Tyr-Glu-Val-His-His-Gln-Lys-Leu-Val-Phe-Phe-Ala-Glu-Asp-Val-Gly-Ser-Asn-Lys-Gly-Ala-Ile-Ile-Gly-Leu-Met-Val-Gly-Gly-Val-Val-Ile-Ala)
was purchased from Qiangyao Biotechnology Company. Anti-LC3II antibody (CAT:
L8918) was purchased from Sigma, USA. Anti-mTOR antibody (CAT: ab134903),
anti-p-mTOR (Ser2448) antibody (CAT: ab109268), anti-Akt (Ser473) antibody
(CAT: ab81283), anti-Akt antibody (CAT: ab238477), anti-Atg7
antibody (CAT: ab133528), anti-Beclin1 antibody (CAT: ab210498), and anti-P62
antibody (CAT: ab210498) were purchased from Abcam, UK.
Anti-Bcl2 antibody (CAT: BS70205), anti-Bax
antibody (CAT: BS6420), -action antibody, and HRP-labeled Goat
anti-Rabbit IgG were purchased from
Bioworld Technology Company, Shanghai,
PR China. Fetal bovine serum was purchased from Cellmax technology Company. Beijing,
PR China.
2.2 Cell culture
SH-SY5Y cells (ATCC CRL-2266, Shanghai,
PR China) were cultured in DMEM/F-12 medium containing streptomycin (100
g/mL), penicillin (100 U/mL), and 10% heat-inactivated fetal
bovine serum at 37 C in a humidified incubator based on 5% CO and
95% air.
A
was dissolved in 100% 1, 1, 1, 3, 3, 3-hexafluoro-2-propanol (HFIP) to a
concentration of 1 mg/mL. This solution was incubated at room temperature (RT)
for 1 h and, after that, sonicated for 10 min. The HFIP/A solution was
subsequently dried down in a gentle stream of nitrogen, and the dried
A was resuspended in 1 mM DMSO. The preparation was incubated
for 12 min at RT and then pipetted and stored at -80 C. Before use, the
preparation was rapidly thawed, utilizing 0.1 M PBS, and a final
A concentration of 20 M was prepared. Neurons were
grouped into control; A treatment, the only treatment of
geniposide, and
A + geniposide treatment.
2.3 Cell viability (MTT) assay
The viability of SH-SY5Y cells was measured utilizing a 3, (4,
5-dimethylthiazol-2-yl) 2, 5-diphenyltetrazolium bromide (MTT) assay. Before
analysis, SH-SY5Y cells were seeded into 96-well density, and the cell density
was adjusted to 5,000 cells/well and incubated for 24 h before treatment. For
selecting an appropriate concentration of A, the cells were
treated with different concentrations of
A (0, 5, 10, 20, 40
M). Apparentcytotoxicity was seen in cells treated by 20,
40 M A and the concentration of 20 M
A was selected to conduct our study. Where indicated, cells
treated with 20 M A were also treated with
different concentrations of geniposide (0, 5, 10, 20, 40 M).
SH-SY5Y cells in various treatment groups (A only, geniposide
only, and A and geniposide) were treated 24 h. After this, MTT
was added to the culture media (0.5 mg/mL
final concentration) and incubated for 4 h at 37 C in a CO incubator. The culture medium was mixed with extraction buffer, and then
absorbance was measured at 490 nm after an overnight incubation utilizing a
microplate absorbance reader (Bio-Rad Instruments). Untreated cells were used as
controls, and cell viability was calculated using the formula:
Cell viability = A of a sample (treated by A, sole
geniposide and A + geniposide separately) / A of the
control sample
where A = absorbance.
2.4 Western blot
SH-SY5Y cells were lysed with RIPA protein lysis buffer containing 1 mM PMSF
(Beyotime Biotechnology, Shanghai, PR China) for 30 minutes after washing with cold
PBS. Total proteins in the supernatant were quantified using a BCA protein assay
(Beyotime Biotechnology, Shanghai, PR China) after centrifugation of the cell lysate
at 12000 r/min for 20 min at 4 C. Proteins were subsequently resolved in 10%
sodium dodecyl sulfate-polyacrylamide (SDS-PAGE) gels (Beyotime Biotechnology,
Shanghai, PR China) and transferred to polyvinylidene difluoride membranes (Beyotime
Biotechnology, Shanghai, PR China). Membranes were incubated in 5% BSA (TBST) for 2
h at room temperature and after that were incubated with primary antibodies
against Anti-LC3II, mTOR, p-mTOR, Akt, p-Akt, Atg7, Beclin1, and P62 overnight at
4 C. Membranes were subsequently washed and treated with horseradish
peroxidase-conjugated secondary antibody (1 : 5000) for 2 h at room temperature.
Proteins were visualized utilizing an enhanced chemiluminescence method, and
-actin was used as a loading control.
The protein bands were visualized using the Chemi-Doc XRS + imaging system
(Bio-Rad). The Western blots were subjected to quantification of the protein band
density using the Image Pro.
2.5 Statistical analyses
The results were expressed as mean SD. A one-way ANOVA analysis was used
to determine statistical significance. The contrast between multiple groups was
performed by one-way ANOVA based on SPSS 19.0 software, and the differences
observed were further analyzed by the least significant difference (LSD)-t-test.
A P-value of less than 0.05 was considered statistically significant.
3. Results
3.1 Geniposide reverses loss of cell viability induced by
A in SH-SY5Y cells
To investigate the effect of A on SH-SY5Y
cells, an MTT assay was conducted to quantify cell viability. Results indicated a
concentration-dependent effect of A on cell viability
(Fig. 1A). Lower doses of A (5 and 10 M) did not
affect cell viability, whereas higher concentrations of A
(20 and 40 M) had measurable effects on cell viability.
A (20 M) treatment significantly decreased the cell
viability to 61.8 4.1% versus control (100%). Based on these findings,
20 M concentrations of A were selected for further
study. Treatment of SH-SY5Y cells with various concentrations of geniposide did
not affect the cells’ viability versus untreated controls (Fig. 1B). However, a
concentration-dependent relationship of geniposide to protect against lost cell
viability following A exposure was observed (Fig. 1C).
Specifically, cell viability was restored from 61.8 4.1% in cells
treated with 20 M A to 64.1 3.2%, 70.0
3.2%, 74.8 4.3%, and 69.0 3.517% after different
concentrations (5, 10, 20, and 40 M, respectively) of geniposide.
20 M geniposide was selected for further studies based on the maximum
effect to improve viability induced by 20 M A
treatment.
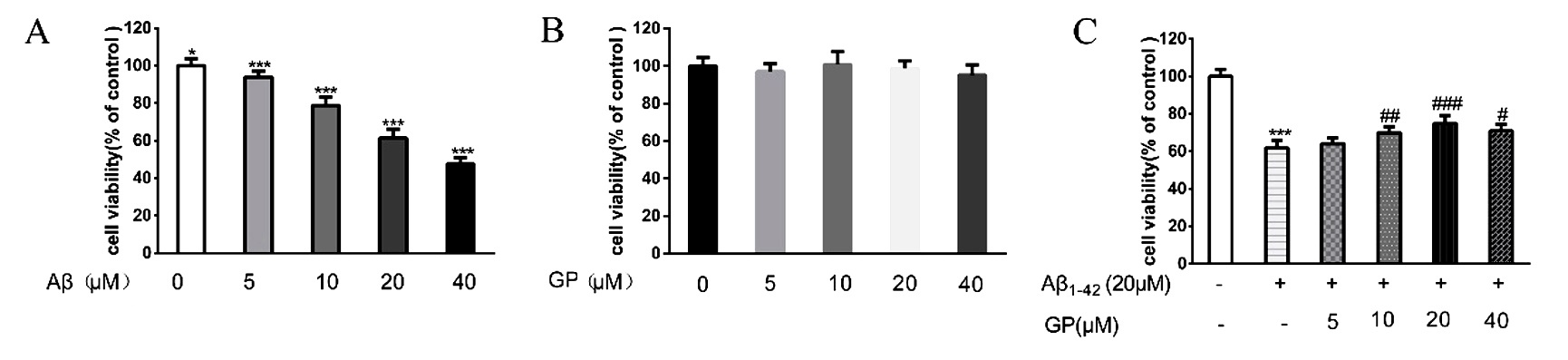 Fig. 1.
Fig. 1.
Geniposide reverses cellular toxicity induced by A in
SH-SY5Y cells. There was no significant difference in cell viability between the
cells treated by different concentrations of geniposide and control. Cells
treated with 20 M A for 24 hours. Cell viability was
measured utilizing an MTT assay. Values were denoted as mean SD.
***P 0.001, *P 0.05 vs. control. P 0.05, P 0.01,
P 0.001 vs. A treatment.
3.2 Geniposide protects against A by
downregulating mTOR signaling
mTOR signaling was upregulated in
the SH-SY5Y cells treated by
A phosho-AKT
(Ser473)/AKT ratio increased from
0.370 0.087 in control to 0.748 0.131 in SH-SY5Y cells treated by
A (Fig. 2A), and the phospho-mTOR (Ser2448)/mTOR ratio
increased from 0.476 0.076 in control to 0.907 0.160 in SH-SY5Y cells
treated with A (Fig. 2B).
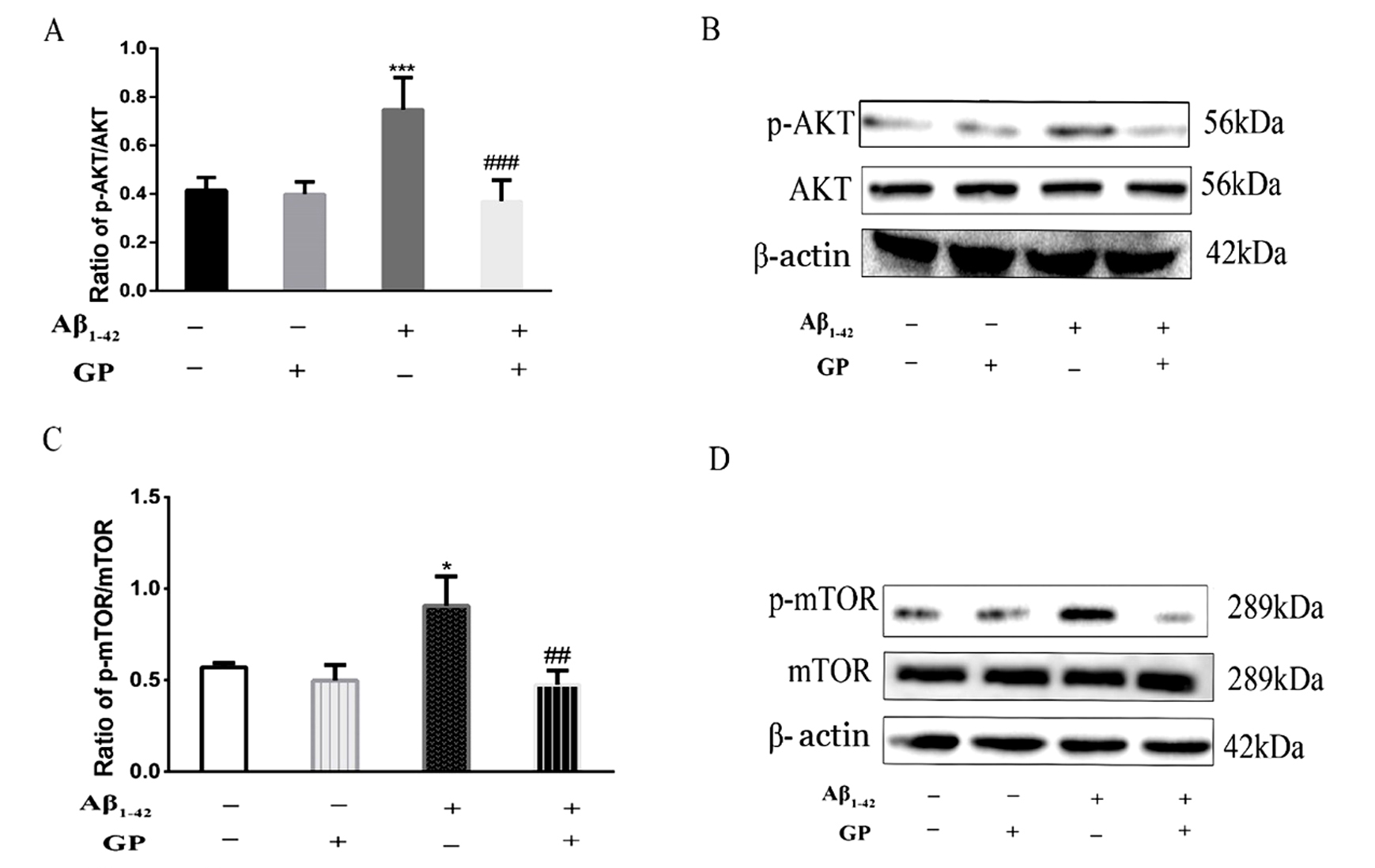 Fig. 2.
Fig. 2.
Changes in mTOR signaling in SH-SY5Y cells treated with
A and geniposide. Western blot analysis was conducted to
measure the phospho-AKT (Ser473)/AKT ratio, phospho-mTOR (Ser2448)/mTOR ratios in
treated SH-SY5Y cells. Geniposide inhibited increases in phospho-AKT (Ser473)/AKT
ratio and phospho-mTOR (Ser2448)/mTOR ratios induced by A.
-actin was used as an internal control. All results are presented as the
mean SD (n = 6). *P 0.05, **P 0.001 vs. control.
P 0.01, P 0.001 vs. A treatment.
Treatment of SH-SY5Y cells with geniposide only did not influence mTOR signaling
as the phospho-AKT (Ser473)/AKT ratio (0.400 0.050) as the phospho-mTOR
(Ser2448)/mTOR ratio (0.498 0.085) in the SH-SY5Y cells treated with
geniposide only were not statistically different from control cells. Geniposide
reversed mTOR signaling upregulation induced by A as the
phospho-AKT (Ser473)/AKT, and phospho-mTOR (Ser2448)/mTOR ratios were upregulated
in the SH-SY5Y cells treated by A. Specifically, we measured a
0.415 0.052 in phospho-AKT (Ser473)/AKT ratio (Fig. 2A) and a 0.570
0.0239 in phospho-mTOR (Ser2448)/mTOR ratio (Fig. 2B) after geniposide
treatment.
3.3 Geniposide protects against A toxicity by
enhancing autophagy
Autophagy was inhibited in SH-SY5Y cells treated by A (Fig. 3). Specifically, the LC3-II/LC3-I ratio decreased from
0.330 0.080 in control to 0.204 0.034 in SH-SY5Y cells treated with
A (Fig. 3A). Beclin1 decreased from 0.358 0.102 in
control to 0.131 0.044 in SH-SY5Y cells treated with A
(Fig. 3B), and Atg7 decreased from 0.806 0.241 in control to
0.317 0.142 in SH-SY5Y cells treated with A (Fig. 3C).
Finally, expression of p62 increased from 0.306 0.137 to
0.728 0.170 in SH-SY5Y cells treated with A (Fig. 3D).
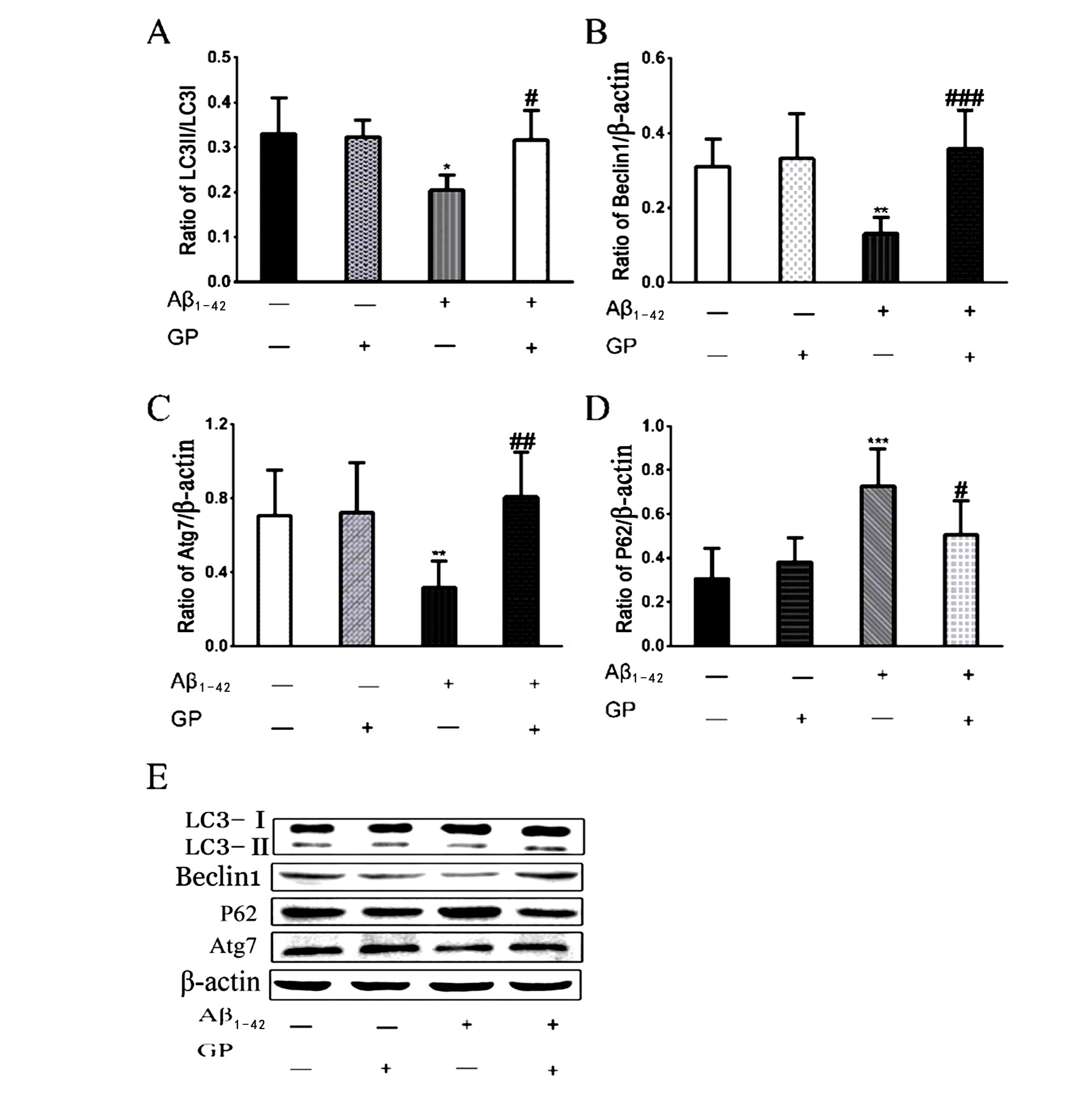 Fig. 3.
Fig. 3.
Changes in autophagy-related proteins in SH-SY5Y cells.
Western blot analysis of the LC3-II/LC3-I ratio, Beclin1, Atg7, and p62
expression was quantified by western blot. Geniposide increased the LC3-II/LC3-I
ratio and Beclin1 and Atg7 expression and decreased the expression of p62 induced
by A. -actin was used as an internal control. All
results are presented as the mean SD (n = 6). * P 0.05, ** P
0.01, ***P 0.001 vs. control. P 0.05, P 0.01, P 0.001 vs. A treatment.
The LC3-II/LC3-I ratio (0.323 0.038), and expression of Beclin1 (0.332
0.119), Atg7 (0.723 0.270), and p62 (0.383 0.108) in the
SH-SY5Y cells treated by only treatment of geniposide were not statistically
different from those measured in control cells, indicating that treatment of
SH-SY5Y cells with geniposide only did not influence autophagy-related signaling.
Geniposide did reverse the inhibition of autophagy induced by A. Specifically, geniposide treatment increased the level of LC3-II/LC3-I ratio to
0.317 0.066 (Fig. 3A), Beclin1 expression to 0.310 0.075 (Fig. 3B), and Atg7 to 0.705 0.247 (Fig. 3D). Similarly, geniposide treatment decreased the expression of p62 to 0.506
0.155 (Fig. 3C).
3.4 Geniposide protects against A by inhibiting
Apoptosis
Apoptosis was activated in the SH-SY5Y cells following treatment with
A. The Bax/Bcl-2 ratio was increased after a 24 hours treatment
with A (1.864 0.333) versus control (0.391
0.194) (Fig. 4). However, geniposide alone did not influence the Bax/Bcl-2 ratio
in SH-SY5Y (0.421 0 .140) cells treated with only geniposide. In contrast,
geniposide blunted apoptosis activation induced by A as the
Bax/Bcl-2 ratio fell dramatically to 0.499 0.185 in SH-SY5Y cells treated
with geniposide and A (Fig. 4).
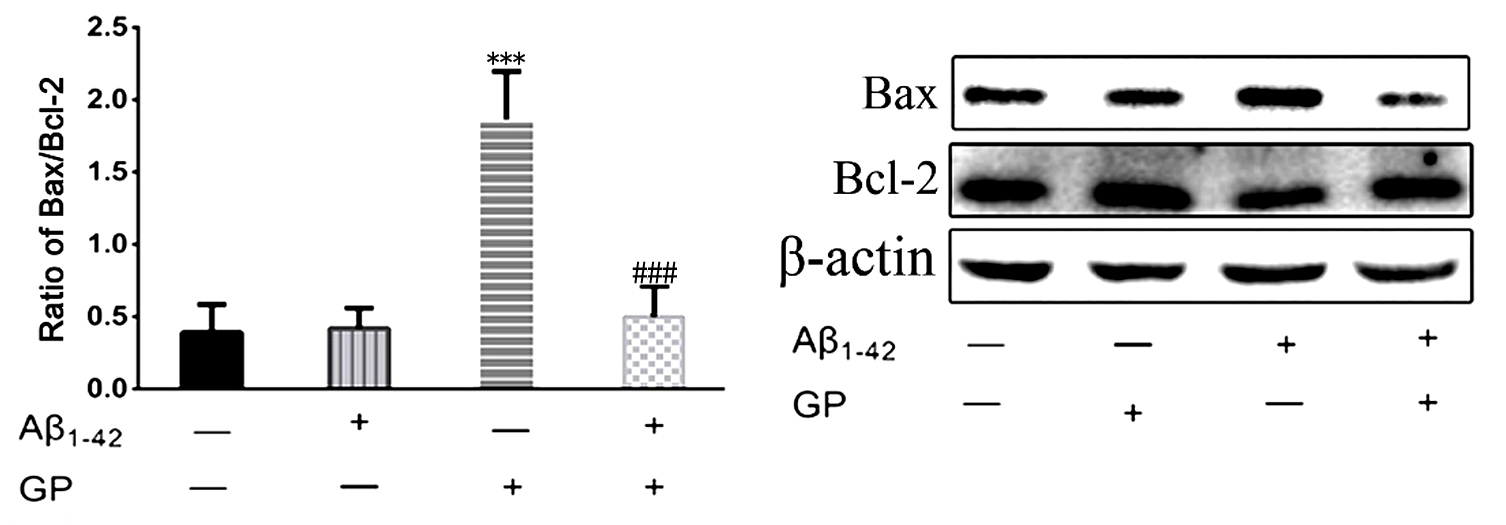 Fig. 4.
Fig. 4.
Changes in apoptosis-associated proteins in SH-SY5Y cells.
Quantitative western blot analyses of Bax and Bcl-2 expression were conducted.
-actin was used as an internal control. All results are presented as the
mean SD (n = 6). ***P 0.001 vs. control. P 0.001 vs
A treatment.
In sum, data gathered during this study provides evidence that geniposide can
protect against the toxic effects of A by inhibiting mTOR (Fig. 5).
Evidence supporting this conclusion comes from the observations that
phospho-AKT (Ser473)/AKT and phospho-mTOR (Ser 2248)/mTOR ratios were restored to
near control levels with geniposide, and geniposide enhanced autophagy by
increasing the LC3-II/LC3-I ratio, increasing expression of Beclin 1, Atg7, and
inhibiting expression of p62. Finally, we observed that geniposide blunted the
apoptotic response to A, as evidenced by measuring the
Bax/Bcl-2 ratio.
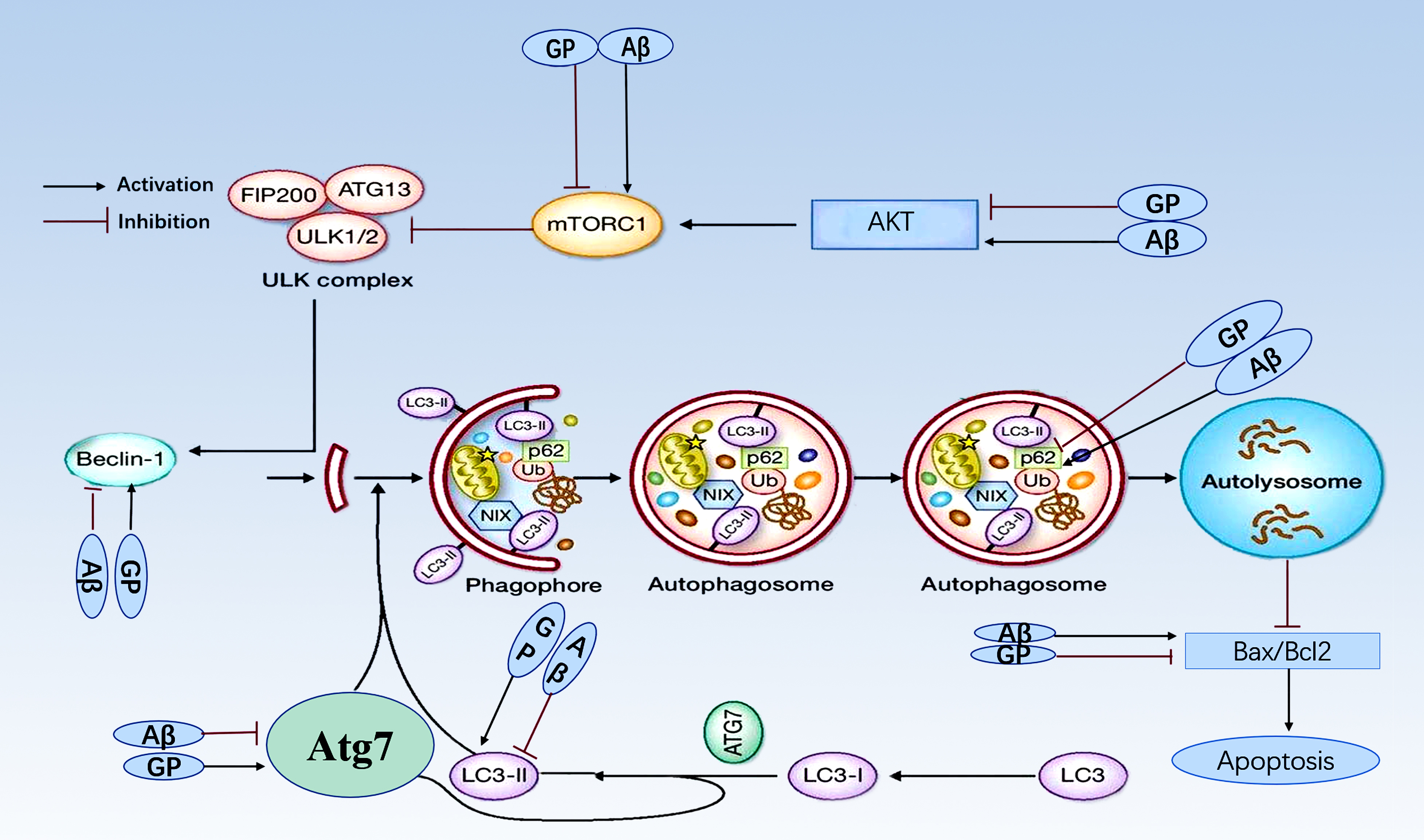 Fig. 5.
Fig. 5.
Molecular mechanism for geniposide protection. Geniposide
performs its protection against A toxicity by inhibiting mTOR
and enhancing autophagy. Geniposide reverses increase of AKT and mTOR induced by
to A. Geniposide reverses a decrease in the LC3-II/LC3-I ratio,
decreases expression of Beclin 1 and Atg, and increases expression of p62 induced
by A. Geniposide also blunts the A-induced
apoptotic response by reducing the Bax/Bcl-2 ratio.
4. Discussion
A prior study showed that geniposide-mediated protection against pathological
hallmarks of AD and behavioral impairment correlates with downregulation of
mTOR signaling and enhanced autophagy in APP/PS1 double transgenic mice [33, 34]. In the present study, we sought to determine the mechanism by
which geniposide prevents
A-associated
toxicity. Considering that geniposide can activate the glucagon-like-1 receptor
(GLP-1R) and adenylyl cyclase (AC)/cAMP signaling pathways and promotes
insulin secretion and inhibition of protein kinase A (PKA) [35], we hypothesized
that geniposide prevents A toxicity by inhibiting
the PI3-K/Akt/mTOR signaling pathway, and
enhances the autography as an agonist of the GLP-1 receptor. Our results showed
that geniposide protected SH-SY5Y cells against lost cell viability induced by
A. Further, we showed mTOR signaling was upregulated in the
SH-SY5Y cells treated by A. phosho-AKT (Ser473)/AKT ratio and
the phospho-mTOR (Ser2448)/mTOR ratio increased in SH-SY5Y cells treated with
A. Geniposide reversed mTOR signaling upregulation induced by
A. The phospho-AKT (Ser473)/AKT and phospho-mTOR (Ser2448)/mTOR
ratios were reversed after geniposide treatment. This finding implies that
inhibition of the PI3-K/Akt/mTOR pathway may be a pivotal molecular event
controlling geniposide’s ability to prevent the toxic effects of A.
Autophagy is a primary physiologic function for clearing abnormal
proteins within mammalian cells and contributes to protein homeostasis and
neuronal health. An autophagy deficit is found in early AD pathogenesis, and
autophagy plays a critical role in the formation and metabolism of
A [31]. In the present study, we
assessed autophagy by measuring the LC3-II/LC3-1 ratio, as well as Atg7, p62, and
Beclin1 expression utilizing western blotting in SH-SY5Y cell lines treated with
A. Our results showed that geniposide protected
against the cellular damage induced by A in
SH-SY5Y cells. Further, we showed that
geniposide reversed the LC3-II/LC3-I ratio
and repression of Atg7 and Beclin1 induced by A and
reversed the expression of p62 enhanced by A in SH-SY5Y
cells. The cytosolic form of LC3-I is converted to the
phosphatidylethanolamine-conjugated form (LC3-II) and binds to autophagosomes’
membranes [36]. Thus, the LC3-II/LC3-I ratio is an often-used marker for
autophagy in various tissues, including the brain [37]. We observed a decrease of
the LC3-II/LC3-I ratio after treatment of
SH-SY5Y cells with A, which suggests that A
damages the brain by, in part, inhibiting autophagy. The ratio was reversed after
the treatment by geniposide, indicating that geniposide protects against AD by
enhancing autophagy. Atg7 is an E1-like activating
enzyme that is down-regulated during aging [38] and is needed for the autophagic
conjugation system and formation of autophagosomes [39]. Similarly, the
expression of Beclin-1, an autophagy-associated gene, is also widely used to
reliably quantify autophagosome formation. There is a close relationship between
AD and Beclin1, as Pickford et al. [40] showed a decrease of
Becline 1 in the brain of patients with AD. Our results indicate that the
enhancement of autophagy-related proteins, including the LC3-II/LC3-I ratio,
Atg7, and Beclin-1, maybe a critical molecular event in the protective effects of
geniposide during the toxic response to A.
LC3B-II is a trustworthy indicator
for the formation of autophagic vacuoles, just as the lipidized form LC3B-I and
p62 are markers for autophagic flux as an
adapter for selective autophagy [41].
The degradation of p62 is widely utilized as a marker to monitor the autophagic
activity because p62 can directly bind to LC3 and is selectively degraded during
autophagy [42]. To estimate the effect of IL-4 on the formation of autophagic
vacuoles and promote autophagic flux in microglia, Tang et al. [43] measured LC3 B-II and p62 in microglia and
found an enhancement of LC3 B-II and an attenuation of p62
in microglia treated with IL-4. Song
et al. [44] showed that the treatment of selenium-enriched yeast
(Se-yeast) also significantly attenuated the levels of p62 accompanying an
increase of turnover of A and APP in AD mice.
Similarly, by these studies, we showed that geniposide lowered the expression of
p62, which was increased in SH-SY5Y cells treated by A.
In summary, we speculate that mTOR inhibition and enhancement of autophagy
induced by mTOR inhibition may be a critical molecular event in geniposide
mitigating A-induced toxicity.
Abbreviations
A, -amyloid;
AO, A oligomers;
AC, adenylyl cyclase;
AD, Alzheimer’s disease;
Atg7, autophagy-related gene 7;
Ex-4, exendin-4;
GLP-1, glucagon-like peptide-1; GLP-1R, glucogen-like peptide-1 receptor;
HFIP, 1, 1, 1, 3, 3, 3‐hexafluoro‐2‐propanol;
IGF-1, insulin-like growth factor-1;
mTOR, mechanistic target of rapamycin;
NFT, neurofibrillary tangles;
PI3K, phosphoinositide 3-kinase; PKA, protein kinase A; PKB (also known as Akt), Protein kinase B;
RT, room temperature.
Author contributions
Dong-Xing Liu, Yan-fang Chang were involved in the design and execution of the experimental job and the statistical analysis of the data. Di Zhang and Wei-min Hu contributed to the statistical analysis of the data and manuscript writing. Xiao-hui Wang and Lin Li were involved in the design and execution of the study. All authors contributed to the development of the manuscript and reviewed and approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Acknowledgment
We thank three anonymous reviewers for excellent criticism of the article.
Funding
Research project was supported by Shanxi Scholarship Council of China (2017- important 4), and by the Fund for Shanxi “1331 Project” Key Subjects Construction.
Conflict of interest
The authors declare no conflict of interest.
Availability of data and materials
The datasets analyzed during the current study are available from the
corresponding author on reasonable request.






