

1 Kunming Medical University & The First Affiliated Hospital, Kunming, 650500, P. R. China
2 Kunming Medical University, Kunming, 650500, P. R. China
# These authors contributed equally.
Abstract
Astrocytes, one of the most abundant and heterogeneous types of glial cell in the brain and spinal cord, are responsible for various essential functions in the healthy central nervous system, including maintaining the blood brain barrier integrity, regulating neuron differentiation and supporting, nourishing, protecting, insulating and repairing neurons. They also fulfill a range of other homeostatic maintenance functions. Astrocytes are activated after traumatic brain injury. They then exhibit heterogeneous gene expression and changes in morphology, proliferative capacity and various functions in response either acute or chronic brain injury and associated secondary brain injury. Some biomarkers and imaging tools have been used to monitor astrogliosis after traumatic brain injury. Initially, morphological characteristics and the physiology of astrocytes are reviewed. Subsequently, alterations of astrocytes are described, which includes both the complex mechanisms and roles of reactive astrocytes. The roles of biomarkers and signaling pathways following traumatic brain injury have been summarized as well as the morphological and functional changes in astrocytes. In the latter case, by considering astrocytes as therapeutic targets of traumatic brain injury, the mechanisms of the latest drug treatments are explained. This review highlights the beneficial effects of astrogliosis according to some recent findings, which provides new insights for the treatment of traumatic brain injury.
Keywords
- astrogliosis
- traumatic brain injury
- reactive astrocytes
- biomarker
- functional considerations
Traumatic brain injury (TBI), is a devastating cerebral injury and is a leading cause of death and disability in developed countries (Park et al., 2008). It occurs when an external force injures the brain and is commonly classified based on severity. In the US, the fatality rate is about 21%, usually occurring within 30 days after TBI while more severe TBI carries a mortality rate of 30- 50% (Brown et al., 2008). People worldwide seek treatment for TBI as a result of falls, traffic accidents, sports and weapon-related activities,among others, but clinical treatment options are limited (Pekny and Nilsson, 2005). TBI can also be categorized into primary and secondary injury. The initial injury often gives rise to the development of secondary injuries, such as inflammation, neurological dysfunction, cellular death, oedema, ischaemia, oxidative stress and excitotoxicity, all of which worsen the primary lesion and lead to the activation of biochemical, metabolic and functional pathways including astrocyte activation (Li et al., 2017).
Astrocytes are believed to be activated by cellular fragments released from dying or damaged cells within a lesion and surrounding central nervous system (CNS) areas (Karve et al., 2016). Astrocyte activation due to trauma, other CNS injuries, or diseases are known as astrogliosis, and are comprised of an abnormal increase in astrocyte number, gene expression, morphology and scar formation (Barres, 2008). Ultimately, astrogliosis leads to positive (e.g., synapse formation, neural repair) and negative (e.g., inhibition of axon regeneration) effects.
The mechanism following TBI remains unclear. Also, there are relatively few studies about how to avoid deleterious effects while treating TBI by targeting astrocytes. As TBI cannot be entirely prevented, it is necessary to investigate biological, physiological and pathological development of astrocytes for clinical treatment.
Astrocytes are a group of cells with heterogeneity, which have not been classified according to their anatomical location, molecular composition, type of neurotransmitter used or their response to diverse brain traumas (Chaboub and Deneen, 2012). Astrocytes occur more than five times more frequently than neurons. Many types of astrocytes exist in the CNS, including fibrous and protoplasmic glial cells, and numerous anatomical differences have been found between them. Fibrous astrocytes (located in the brain's white matter), exhibit strong interfascicular processes, whereas, protoplasmic glial cells (found in grey matter) are more predominant, contain more organelles and exhibit highly branched tertiary processes. Both of them are morphologically complex with star-shaped appearances and radially project a multitude of repeatedly branching variable diameter cellular processes that exhibit terminal footplate protuberances. These end-feet support and maintain the blood-brain barrier (BBB) which itself is constituted by the endothelial cells of brain microvessels. Additionally, adjacent to the pia mater, these two types of astrocytes extrude processes that run perpendicular to the pia to form the Pia-glial membrane.
Glial fibrillary acidic protein (GFAP) is the major intermediate filament (IF) protein of astrocytes. It participates in the formation of the cytoskeleton and maintainenance of its tensile strength. The morphological heterogeneity of astrocytes can be observed by three-dimensional reconstruction with antibodies against GFAP where the branched, tubular processes are the main manifestation of this heterogeneity. Astrocytes are a general term for a group of cells whose morphology differs depending on their subtypes, molecular diversity, and anatomical location (Hu et al., 2016). For example, glial processes appear in the cortex less regularly than in the hippocampus. The protein connexin-43 (Cx43) is not expressed by neurons and is strictly excluded from oligodendroglia, microglia and NG2 cells (Theofilas et al., 2017). For this reason, a connexin-43-GFP reporter mouse has recently been adopted to highlight the heterogeneity of astroglial cell morphology. Research into mechanisms of astrocyte morphology has shown that the dynamic recombination of actin cytoskeleton is controlled by the small GTPase of the Rho family and this is now generally believed to be the cause of cell morphology change. Although, some cytokine and cell adhesion factors are known to participate in morphological changes of astrocytes as well. CD44 is an integral membrane glycoprote which functions as an adhesion molecule for epithelial cells (Kondo et al., 1998). The overexpression of CD44 results in a larger cell size and astrocytes appear to exhibit a flat shape, while heparin-binding epidermal growth factor (HBEGF) alters the shape of astrocytes to radial glial-like phenotypes and induces expression of IF Further, RT-qPCR analysis has indicated that HB-EGF affects the expression of the Notch signaling pathway gene to play a role in the morphological heterogeneity of astrocytes (Puschmann et al., 2014).
Astrocytes in healthy CNS are considered to be the main homeostatic cells. Studies have reported that these cells are responsible for the maintenance of internal homeostasis, including ion, neurotransmitter, metabolic, synaptic, and organ homeostasis. They also regulate blood flow and microcirculation. The cell membrane of brain endothelial cells (BECs) and perivascular end-feet of astrocytes ensure the integrity of the BBB (Yamamizu et al.,2017). Astrocytes play an important role in the homeostatic regulation of the cerebral microenvironment due to their secretion of chemical mediators capable of rapid regulation of endothelial cell permeability (Abbott, 2002; Verkhratsky et al., 2017).
Astrocytes fulfill various homeostatic functions involving potassium (K+), chloride (Cl-), hydron (H+), sodium (Na+), and calcium (Ca2+). K+ efflux is associated with excessive neuronal activity, and K+ in extracellular space is consequently moved into astrocytes by active transport that mediates local uptake to buffer extracellular K+. This process is enhanced by the Kir4.1 channel in a VGLUT1-dependent manner (Cai et al., 2011), the Na+-K+-Cl--cotransporter-1 (NKCC1) (Amadeo et al., 2018), the Na+-K+-ATPase pump (Du et al., 2016) and Cx43 (Lykke et al., 2017). The molecular mechanism of maintaining Cl- ion homeostasis by astrocytes includes GABA receptor anion channels (Fraser et al., 1995) and ClC-2. Ca2+ ion homoeostasis relies on a Na+/Ca2+ exchanger 1-3 (NCX1-3) (Verkhratsky et al., 2012) and plasmalemmal Ca2+ pump (PMC) and is controlled by the coordination of IP3R and store-operated Ca2+ channels (Blaustein et al., 2002; Liu et al., 2017; Reyes et al., 2012; Sakuragi et al., 2017). Neurotransmitter homeostasis refers to glutamate, GABA, glycine, and adenosine.
Astrocytes are activated by the cellular debris and inflammatory mediators that follow injury of and disease within nerves. Following TBI, mechanical stress acts on astrocytes to activate mechanically sensitive caution channels, through which extracellular Ca2+ influxes and initiates the release of astrocyte adenosine triphosphate (ATP) by either autocrine or paracrine mechanisms. Eventually, ATP promotes activation of multiple signaling pathways in and between cells and induces the release of endothelin-1 (ET-1), matrix metalloprotein-9 (MMP-9) and glutamate (Ahmed et al., 2000). The mechanism for activation of astrocytes is driven by complex signalling interactions, including (amongst others): ion channel and gap junctions, purinergic receptor, excitotoxic neurotransmitters and perturbations in calcium homeostasis. These activation processes are also related to other cells such as microglia via IL-1, Tumor necrosis factor α (TNF-α) and the complement component 1q (Zhang and Xu, 2018).
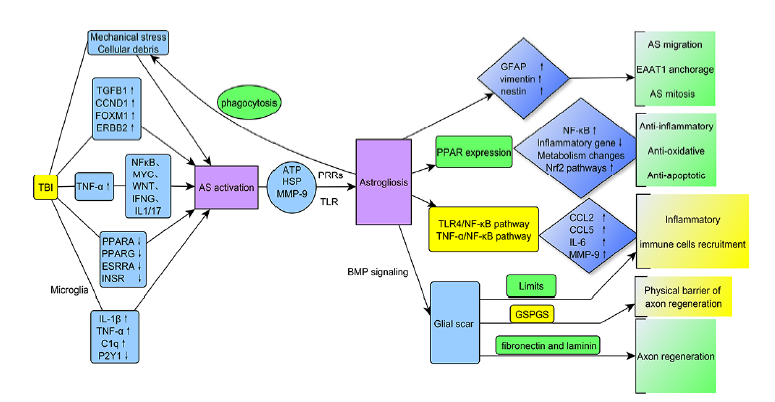
Astrocytes are activated and promote development of astrogliosis , which results in morphological and functional changes by regulation of multifunctional receptors, forming glial scar and making impact on multiple signaling pathways. Series of processes conclusively lead to the different effects on CNS following TBI (Fig. 1).
 Figure 1.
Figure 1.TBI induces astrocyte activity and astrogliosis, which includes regulation of multifunctional receptors, formation of glial scar tissue and impacts multiple signaling pathways.
Astrocytes respond to various forms of CNS injury and disease by a process referred to as reactive astrogliosis, a pathological hallmark of CNS structural lesions. Based on numerous studies, reactive astrogliosis has recently been defined by four characteristics: (1) Increased responsiveness of astrocytes to various injuries, diseases and subtle disturbances in CNS including molecular, cellular and functional changes. (2) Changes experienced by reactive astrocytes vary with the form and severity of the lesion. After TBI, mild to moderately reactive astrogliosis changes the expression of molecules thus changing their function. This is accompanied by varying degrees of cellular hypertrophy. In addition to the morphological and functional changes already mentioned, severely diffuse newly proliferated astrocytes appear. Along with tissue damage and inflammation forming dense areas of glial scar, serious reactions include newly proliferating astrocytes and other cell types, such as fibromeningeal cells and other glial cells, as well as deposition of collagen in the extracellular matrix. Astrocytes around a lesion produce many inflammatory mediators (cytokines and chemokines) that are damaging to neurons, which themselves can also recruit peripheral cells and activate microglia. Ultimately, these changes affect the local microenvironment to determine the extent of tissue damage and subsequent repairs. (3) Changes in reactive astrogliosis are regulated by complex intercellular and intracellular signaling molecules. (4) Changes experienced during astrocyte proliferation are not entirely hardwired but rather depend on surrounding neural and non-neuronal cells (Sofroniew and Vinters, 2010).
Particular serum biomarkers (See Table 1) and imaging tools can be employed to identify reactive astrocytes, assess and diagnose TBI (Halford et al., 2017).
 |
Yellow: Common physiological and reactive astrocyte biomarkers.
The developmental progression of astrocytes can be identified and visualized by their morphological criteria and the expression of specific proteins as characterized markers such as GFAP, vimentin, protein S100, glutamate transporter GLT1, glutamine synthetase (GS), Kir4.1 channel, water channel aquaporin 4 (AQP4) and Cx43 (Morel et al., 2014). Astrocyte precursors express intermediate proteins of vimentin, nestin, and synemin, while mature and differentiated astrocytes express only GFAP and vimentin.
Antibodies against GFAP, the most widely used immunostain, and the expression of GFAP significantly increased at three, six and twentyfour hours post-TBI (Huang et al., 2015). Compared with GFAP, the immune response cells of N-myc downstream regulated gene2 (NDRG2) are more abundant and more evenly distributed in the brain. For example, there are cells strongly positive for NDRG2 in the caudate nucleus, where GFAP rarely occurs. Vimentin is one of the proteins in IF that provides elasticity and is responsible for maintaining cytoskeletal integrity.
S100B has long been recognized as an important marker of TBI and is produced by activated astrocytes, which can further promote the activation of microglia, increase the production of inflammatory factors such as TNF-α, IL-1β, and Il-6 and cause long-term neurological dysfunction. Animal models have shown a significant increase in S100B levels in the brain lasting up to five days after TBI (Kabadi et al., 2015). Interestingly, proliferating cell nuclear antigen was detected as S100A6+ in astrocytes indicating that S100A6 (calcyclin) is a novel marker for astrocyte precursors (Yamada and Jinno, 2014). The differentiation of astrocytes can be visualized by glycogen metabolism and glycogen is a hallmark of mature astrocytes (Brunet et al., 2010). The marker that identifies the different subpopulations of astrocytes is also useful because the mechanisms that underly astrocyte diversity remain unclear and elucidating their functional heterogeneity is a difficult task. Fortuitously, a recent study has reported that a CS56-specific antibody could be used as a novel marker to detect astrocyte subpopulations (Okuda, 2018). Additionally, GS, a 38 kD breakdown product of aldolase C (ALDOC), ubiquitin carboxyterminal hydrolase L1 (UCH-L1), brain lipid binding protein (BLBP) and astrocytic phosphoprotein (PEA15) have been tested as candidates for TBI evaluation (Guida et al., 2017).
However, GS was was found to be an unpromising marker because of its variability in many neurological diseases, such as cerebral hypoxia, epilepsy and chronic neurodegenerative processes. Additionally, Studues have shown that there is no significant correlation between the expression of GS and GFAP (Bartlett and Li, 1990). UCH-L1 level wase elevated in cerebrospinal fluid (CSF) 24 hours post-TBI while blood levels were unchanged. During the first week after TBI, ALDOC levels were significantly raised, but stable. Levels of BLBP and PEA15 can be associated with injury progression. ALDOC, BLBP and PEA15 show a sharp increase in both severe and mild TBI blood and CSF tests; 25 kDGFAP-BDP appeared overnight, but was rarely present in mild TBI (Agoston and Shutes-David, 2017). Moreover, astrocytic Ngb expression has been observed in sub-acute and chronic TBI (Chen et al., 2015).
In addition to biomarkers, a variety of advanced imaging tools are also used to study astrogliosis. Manganese-enhanced magnetic resonance imaging (mMRI) can be employed during the subacute phase of TBI (7--14 days) when astrogliosis is characterized by crescent-shaped T1-weighted signal enhancements surrounding the impact core. Experiments have confirmed that the hyperintensive area is glial scar tissue, while the void is an impacted necrotic area (Talley Watts et al., 2015). MMRI employs manganese is an analog of calcium, dysfunction of which is associated with secondary insult in TBI. It was also discovered that diffusion tensor imaging (DTI), an advanced MRI technology, was more sensitive to microstructural abnormalities of the CNS than standard MRI. GFAP expression in the cortical gray matter of a TBI model was significantly and positively correlated with fractional anisotropy (FA) in the acute (1--7 days) and subacute (7--14 days) periods after TBI due to the increased content and directional cohesiveness of astrocytes.
In white matter, decreased FA indicates neuronal demyelination and axonal degeneration, while increased FA is associated with glial scar formation but not axonal regeneration. In the subacute phase, the mean kurtosis (MK) of the ipsi- and contralateral damaged regions was significantly increased, and MK improvement was inversely proportional to distance from affected areas. Immunohistochemical analysis showed that MK increase was related to the proliferation of reactive astrocytes (Zhuo et al., 2012). Thus, FA and MK in DTI can be used to study and monitor reactive astrogliosis.
Regrettably, no causal relationship has been found between diffusion magnetic resonance imaging (dMRI) at all posttraumatic time points and reactive astrocytes, so dMRI is less valuable for the study of astrogliosis (Zhuo et al., 2012). In addition to various MRI imaging modalities, the major imaging innovations for investigation of astrogliosis are two-photon imaging in vivo, positron emission tomography (PET) and live tissue imaging. Intracellular Ca2+ acts as a second messenger and its transient changes can be used as an indicator of the physiological activity of astrocytes at different times after TBI. On this basis, astrocytes can be imaged in vivo with a two-photon microscope (Shandra and Robel, 2019). Although SPECT has a higher diagnostic value in mild TBI than CT and MRI (Raji et al., 2014), it is a poor discriminator of astrogliosis. Translocator protein 18 kDa (TSPO) PET imaging has demonstrated that in addition to reactive astrocytes, microglia cells can also overexpress selectively binding TSPO radioligands after TBI (Lavisse et al., 2012), thus with SPECT it is difficult to elucidate astrocyte changes. Confocal microscope live tissue imaging can be used to explore interactions between reactive astrocytes and immune cells during the period of inflammatory response that follows TBI (Escartin and Murai, 2014). Overall, advanced imaging techniques, particularly different types of MRI, have led to new insights into the function of astrocytes.
The morphological changes of astrocytes following TBI include cell hypertrophy, synaptic elongation and increase in number (See Table 2). Palisading processes grow about one week after injury and glial scars are formed in severe cases (Bitner et al., 1987). Additionally, these changes are accompanied by both proliferation and migration of reactive astrocytes and elevation of GFAP that are related to the severity of the injury (Fix et al., 1996). Acute and diffuse lesions without tissue damage cause slight and reversible hypertrophy of astrocytes and in this case, reactive astrogliosis is limited to local areas without proliferation. More persistent and more severe lesions result in more pronounced cell hypertrophy, increased growth and neurite extension toward the injured site, along with astrocyte proliferation and migration. In highly reactive astrogliosis, glial scars are predominant. Astrocytes appear to begin hypertrophy 2--3 days after injury and, accompanied by glial scar formation, remain hypertrophic up to seven days after injury. Reactive astrogliosis can be maintained for up to 60 days. The multiple processes of hypertrophic astrocytes project to an injured area, but as the distance from the injury site increases hypertrophy decreases. Finally, brain tissue without substantial damage up-regulates GFAP but shows no signs of hypertrophy. One factor that causes astrocyte hypertrophy is the endothelin B receptor (ETBR) (Rogers et al., 2003), while another major driver of hypertrophy and growth in the formation of scar-producing astrocytes is the cytoskeleton (microtubules, IF and actin filaments in microfilament).
 |
ETBR is a G-protein-coupled receptor that activates the phosphatidylinositol pathway. Its ligand ET boosts mitosis of cells, induces glutamate efflux and promotes dynamic change of actin filaments. ETBR is the major ET receptor type expressed on astrocytes in in vitro (Rogers et al., 1997). Experiments in vitro have shown that treatment by ET1 induces the hypertrophy and proliferation of normal astrocytes and the infusion of bosentan (a mixed ETAR and ETBR antagonist) causes a significant decrease of astrocyte hypertrophy in damaged nerve tissue. The up-regulation of ETBR is a potential cause of astrocyte hypertrophy after TBI, but its specific signaling pathway is still unclear (Rogers et al., 2003). In addition to inducing astrocyte hypertrophy, ETBR is also involved in astrocyte proliferation, and loss- and gain-of-function studies and promoter activity assays (Li et al., 2010) have shown that Jagged1/Notch1 signaling enhances phosphorylation signaling and transcription. The level of signal transducers and activators of transcription 3 (STAT3) indirectly increases ETBR expression and its ligand (ET1) promotes mitosis of astrocytes.
It has further been confirmed that the Notch1-STAT3-ETBR axis is one of the pathways that promotes reactive astrogliosis proliferation following brain injury (LeComte et al., 2015) and astrocyte proliferation instead of migration can be detected on day 5--7 after injury. GFAP-positive astrocytes were found to proliferate on day one, three and seven after injury, and the number of proliferating astrocytes reached their peak on the first day after injury. The DIX domain containing 1 (Dixdc1) and transforming control tumor protein (TCTP) have also been found to be involved in the regulation of astrocyte proliferation after TBI. Dixdc1 is a positive regulator of the Wnt signaling pathway and is upregulated in reactive astrogliosis (Lu et al., 2017). TCTP is a ubiquitous and highly conserved protein that plays a broad role in the regulation of cell proliferation, growth, apoptosis and cell cycles and regulates astrocyte proliferation as well as migration. Inhibition of TCTP also reduced migration of astrocytes, as the recombination of microtubules and F-actin is disrupted after siRNA transfection (Ren et al., 2015).
More severe TBI occurs in secondary cerebral edemas that account for most early deaths after TBI. Edema can be caused by the destruction of ionic homeostasis due to the incapacitation of astrocytes after TBI. Zhang et al. (2010) have shown that because extracellular glutamate levels increase after brain injury, microglia can release cytokines and directly contact astrocytes. Additionally, trauma significantly up-regulates the activity of neuronal NKCC and astrocyte AQP4 is significantly elevated after TBI, all of which can lead to cerebral edema. A study of the distribution of AQP4 in the CSF of patients who suffered TBI showed a significant increase of AQP4 in these patients (Lo Pizzo et al., 2013). So, some investigators (Kimelberg, 2005) believe that AQP4 is also the cause of astrocyte edema.
Tensile strength and cell polarity are important for morphological changes in astrocytes and a scratch damage model is commonly employed to trigger and estimate astrocyte polarization and migration towards the wound area (Magdalena et al., 2003b). A major tensile force originates from the dynamic reorganization of actin filaments and cell polarity is derived from microtubule-induced Golgi reorientation. Such directional extension towards the movement leading edge of cell movement allows astrocytes to migrate to a wound (Magdalena et al., 2003a). The key molecule for directed elongation of astrocytes is the small GTPase Cdc42, which regulates the arrangement of microtubule tissue centers (MTOCs) and Golgi through the mPar6-PKCzeta signaling pathway. Cdc42 polarizes transport of cytoskeletal regulators towards the leading edge to force microtubules to the tip of the protrusion; thereby, elongating the processes of reactive astrocytes (An et al., 2016; Robel et al., 2011). Significant up-regulation of IF in reactive astrogliosis performs the function of maintaining cell polarity and orientation of both motion and nuclear localization. IF plays a major role in the formation of reactive astrocytes and glial scars, so it is believed to be the most relevant cytoskeletal component. In short, the various components of the cytoskeleton are complexly involved in astrocyte alterations, an extension of processes and migration to the injured site (Schiweck et al., 2018).
Dead neurons release pro-inflammatory mediators such as IL6, chemokines CCL2, CCL5 and ATP. Also, dead cells directly contacting neurons can cause neuronal apoptosis. The basic process of limiting tissue damage is that of astrocyte collection and phagocytosis of dead cells after TBI (Castejon, 2015). Time-lapse experiments show that exposure to free-floating cell cadavers induce neuronal apoptosis, whereas, neurons that migrate through astrocyte-aggregated dead cell populations are unaffected. An in vitro model of brain injury showed that astrocytes actively collect and phagocytose whole dead cells, thereby protecting healthy neurons from bystander cell death (Loov et al., 2012).
The expression levels of EAAT1 and EAAT2 in one study of 36 TBI patients was found that the number of GFAP-positive astrocytes decreased within the first 24 hours of TBI (van Landeghem et al., 2006). But after that, in the white matter, EAAT1 positive cells increased by a factor of 39 within 24 hours (Beschorner et al., 2007; Dorsett et al., 2017). These transporters were widely distributed in the lesion area, injury-adjacent area and injury-remote areas of the brain (Beschorner et al., 2007; van Landeghem et al., 2006). Reactive astrocytes absorb glutamate from the synaptic cleft, not only reducing excitotoxicity but also providing cells with the substances required for neuronal metabolism. Following astrocyte injestion, some of the glutamate is converted to glutamine as a precursor for neurotransmitter fabrication while some is oxidized to produce ATP (Yan et al., 2017).
Mechanical injury can induce secondary damage following diffuse brain injury that includes destruction of axonal membrane, Ca2+ inflow and calpain-mediated hydrolysis of cytoskeletal protein-ii-spectrin (McGinn et al., 2009). Mitochondrial swelling and/or axoplasmic transport dysfunction may then occur and eventually lead to the disconnection of the swollen axon from the distal segment. This secondary traumatic axonal injury is associated with local cerebral blood flow and flow-metabolism decoupling (Harris et al., 2012). In fact, under an energy crisis, the brain can use alternative metabolic substrates, such as lactic acid and pyruvate, and reactive astrocytes can regulate the synthesis of lactic acid, ketone bodies and free fatty acids (FFA) by expressing PPAR (Carpenter et al., 2015; Iglesias et al., 2017). This process involves the upregulation of glycolytic enzymes (PKFFB3 and GAPDH) and lactate transporters (GLUT1 and McT1/4) and also the adjustment of target genes relevant to pyruvate and lactate (adipoq, LPL, acadl and angptl4) (Cai et al., 2018). Reactive astrocytes can also provide metabolic energy support for neurons by releasing glycogen into the ECM (Falkowska et al., 2015). As for the regulation of blood flow, neural signals and the supportive microenvironment, they are regulated by “neurovascular units’’ composed of microvessels, astrocytes and neurons (Abbott et al., 2006; Adriani et al., 2017). Additionally, PPAR inhibited NFκB and activated Nrf2 to reduce inflammation and produce antioxidant respectively (Iglesias et al., 2017). This rapid reaction is achieved through GT-Pase, which is a promoter and regulator of structural change of the Rho family. Ultimately, the changes to reactive astrocyte structure affect both the development, function and plasticity of synapses and the interactional ability between neurons and astrocytes (Krencik et al., 2017). Thus, reactive astrocytes can prevent secondary axonal injury following TBI by water regulation, ionically, neurotransmitters, antioxidants, cytokines and chemokines (Liu et al., 2017).
Increased superoxide anion, hydrogen peroxide, oxygen free radicals, and nitric oxide (NO) cause brain neuronal apoptosis and death after TBI, while reactive glial cells protect neurons from oxidative stress by releasing superoxide dismutase (SOD) and ascorbic acid. Iitsuka et al. (2012) found that when LPS stimulated astrocytes, SOD activity on the cell surface decreased to nearly half within 12--24 hours, indicating SOD was released from the astrocytic surface into the neural matrix. This is beneficial to the antioxidant activity of neurons. Pituitary adenylate cyclase activating peptide (PACAP) in reactive astrocytes increases catalase activity and prevents the accumulation of reactive oxygen species (ROS) by activating PAC1-R receptors and protein kinase A (PKA), protein kinase C (PKC) and MAP kinase signaling pathways (Douiri et al., 2016). Glutamate stimulates astrocytes to release ascorbic acid after TBI, which is a well-known antioxidant. Additionally, it also enters neurons and cells to inhibit glucose consumption and stimulate lactate transport (Castro et al., 2009). The oxidized form of ascorbic acid is dehydroascorbic acid (DHA), which has been found to prevent cell death and reverse mitochondrial dysfunction after exposure to H2O2 (Stanley et al., 2012). Some results have shown that DHA significantly increases the activity of glutathione peroxidase and glutathione reductase one hour after exposure to H2O2, which reverses the decrease of glutathione level and significantly reduces ROS production by the pentose-phosphate pathway (PPP) within 15 hours (Garcia-Krauss et al., 2016; Kim et al., 2005). These results suggest that astrocyte--neuron interactions serve as a basic mechanism for the recycling of ascorbic acid and participate in the antioxidant defense of brain tissue (Garcia-Krauss et al., 2016) (Table 3).
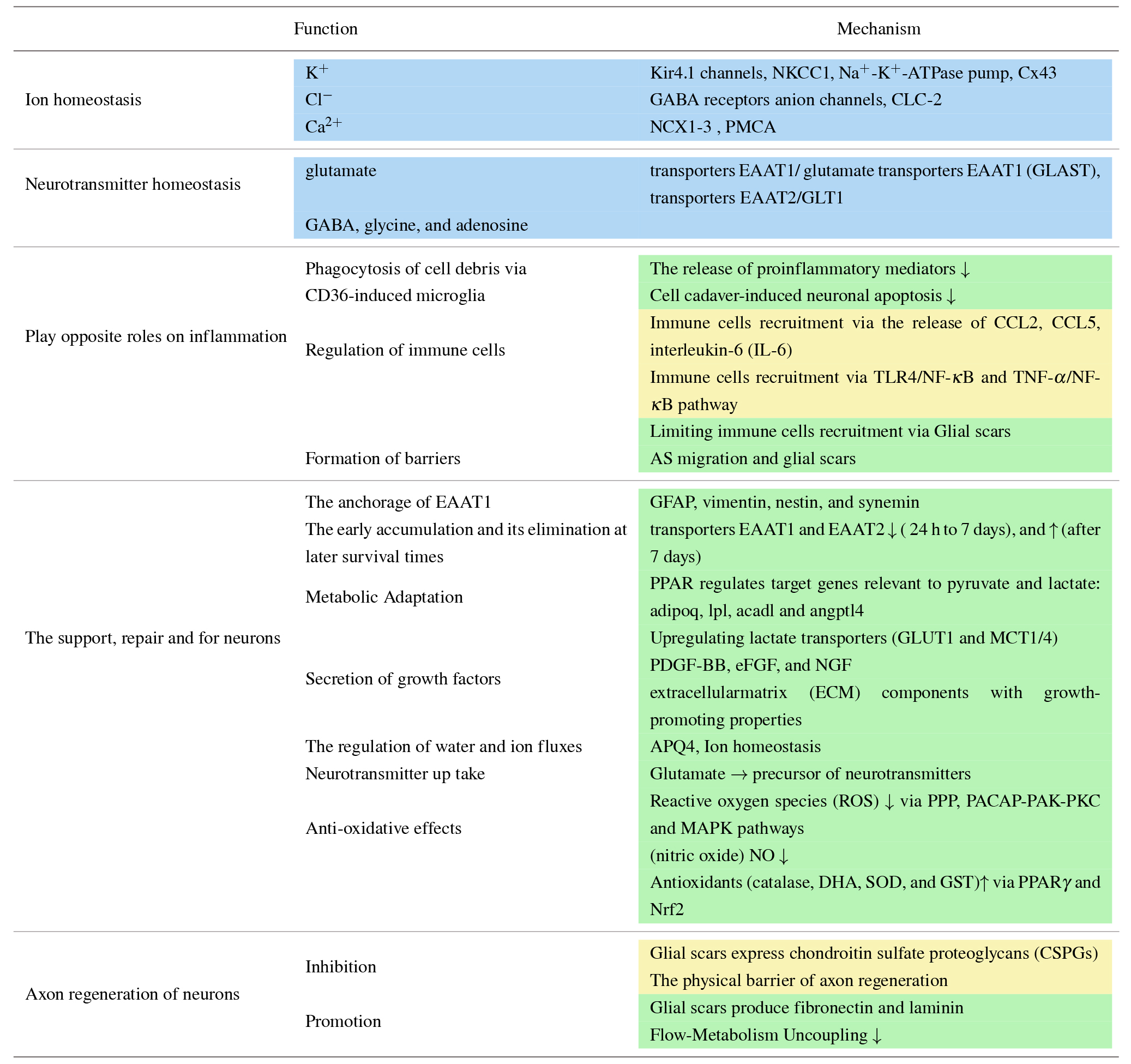 |
Blue: Common functions of physiological and reactive astrocytes.
Yellow: Beneficial change of reactive astrocytes after TBI.
Green: Harmful change of reactive astrocytes after TBI.
Damaged cells release, among other molecules, heat shock protein, MMP-9 and ATP to many pattern recognition receptors (PRRs) that are expressed on the cell surface or interior of astrocytes, such as Toll-like receptors (TLR), that identify various types of TBI damage-associated molecular patterns and then activate astrocytes at the peak level of their inflammatory response. ATP induces the activation of a p2x7-dependent inflammasome via the pannexin one channel (Corps et al., 2015; Russo and McGavern, 2016). The release of MMP-9 increases astrocyte exposure to pro-inflammatory cytokines such as IL-1β by activating the ERK pathway (Ralay Ranaivo et al., 2011). Reactive astrocytes then produce an immune response that releases cytokines and chemokines and activates the migration of both microglia and astrocytes to the injury site to produce an anti-inflammatory response to TBI. Meanwhile, neutrophils and monocytes enter the brain through the meningeal vessels and choroid plexus and initiate a wider range of inflammatory effects (Makinde et al., 2017). Neutrophils resist infection while releasing ROS, which causes tissue damage. Importantly, neurons also express TLRs, which allow them to respond to TLR-driven inflammatory responses and become damaged (Lozano et al., 2015). Also, inflammation damages the absorption of glutamate by astrocytes, resulting in excitotoxic harm, with further aggravation of nerve damage (Cekanaviciute and Buckwalter, 2016). Astrocytic aggravation of inflammation is also manifested in biochemical and metabolic signals. In vitro, the activity of NF-κB, the expression of pro-inflammatory cytokine and the release of MMP9 activity were up-regulated in reactive astrocytes 24 hours after TBI (Pan et al., 2012). The antioxidant response element (ARE) depletes Nrf2 during oxidative stress, activating the DNA binding activity of NF-κB in astrocytes and downstream pro-inflammatory cytokines. This causes increased the DNA binding activity of CD24 and NF-κB in astrocytes and downstream pro-inflammatory cytokines. All of these processes induce more serious inflammation (Li et al., 2014, 2015).
Alternatively, reactive astrocytes avoid excessive inflammation through two mechanisms. Either the glial scar limits the migration of inflammatory cells into the CNS or chemokines regulate the activation of white blood cells. Indeed, CCL2 not only recruits blood-derived macrophages that lead to neuroinflammation and denaturation after trauma but also inhibit the stimulation of astrocytes by IL-1β. It is by these means reactive astrocytes reduce their production of pro-inflammatory cytokines, thus to some extent protecting nerve cells.
Glial scar tissue is a physical boundary formed by the composition of ECM and active astrocytes after proliferation and migration. The glial scar encloses the injured area and forms a physical barrier against the expansion of inflammation and injury. It distributes along the interface of all blood vessels and becomes located both at the junction between the brain parenchymal cells and the BBB's endothelial cells and at peripheral cells. Since WBC must enter the CNS from the bloodstream through the endothelial cells, when present, glial scars restrict the entry of excessive white blood cell (WBC) into the CNS (Cekanaviciute and Buckwalter, 2016).
It is generally believed that the glial scar forms an inhibiting barrier for axonal regeneration. Additionally, the blood supply is affected by glial scar hyperplasia. Glial scars also release axonal growth inhibitors, such as proteoglycans and tenascins and ephrinb1 in astrocytes competes with ephrin-b1 in neurons, all of which inhibit new synapse formation. Neurons that grow under the atypical astrocytic cell pattern are malnourished, but their survival is not affected (Liu et al., 2015; Nikolakopoulou et al., 2016). However, there is an alternative view that in the early period of TBI glial scars repair and support axon regeneration with nerve growth factor (NGF), glial nerve growth factor (GDNF) and glial cell maturation factor. Here, through gene-targeted functional deletions in adult mice, it has been found that preventing or removing astrocyte scars does not lead to spontaneous regrowth of sensory or serotonin axons. In contrast, RNA sequencing showed that after lesion clearance, reactive astrocytes expressed and constantly delivered axon-specific growth factors through the formation of glial scars, stimulating regeneration of robust sensory axon (Li et al., 2018).
The treatment of various aspects of astrocyte-mediated responses is very promising (See Table 4), but the mechanism of astrocytic action is complex and mutually influential with targets. The latter means that one mediator can influence the different interactions and different mediators can influence the same interactions, the result is a variety of either beneficial or harmful effects. It is difficult to completely avoid side effects and achieve multitarget treatments. Blocking L- or N-type voltage-gated Ca2+ channels (VOCCs) prevents reactive astrocyte responses. After TBI there is a pathological increase in intracellular Ca2+, which is the mechanism that triggers cell dysfunction and death. It has been observed that Ca2+ channels are up-regulated after astrocytes are stimulated by LPS or mechanical trauma and Ca2+ enter the astrocytes through VOCC. Ca2+ increases the expression and phosphorylation of GFAP, a necessary step in binding Ca2+ to IFs and fulfilling the morphological transformation of active cells. Therefore, L-VOCC and N-VOCC inhibitors directly reduce the activation of reactive astrocytes by attenuating biochemical changes in resting astrocytes (Cheli et al., 2016; Shahlaie et al., 2013). Additionally, cell cycle inhibitors, such as the cyclin-dependent kinase inhibitor aflatoxin, reduces cyclin D1 during astrocyte proliferation and glial scar formation. Up-regulation reduces astrocyte scar formation and inhibits both neuronal death and cellular injury of reactive glia (Cernak et al., 2005). After TBI, the plateletderived growth factor receptorβ (PDGFR) inhibitor (AG1296) can be used to inhibit the activity of astrocytes and the formation of the glial scar (Pei et al., 2017). Some pathways are also involved in the protection of astrocytes. Resveratrol (RV) protects astrocytes from TBI by inhibiting apoptosis and autophagic cell death. TBI induces cell death via the ros/GSK-3β/mitochondria signaling pathway, while RV inhibits GSK-3β-mediated autophagy and apoptosis to increase cell survival. This suggests that RV may be a potential therapeutic agent for the treatment of TBI (Lin et al., 2014). A more aggressive approach to protect astrocytes from autophagy has been demonstrated where by pre-treatment with gastrodin (Wang et al., 2016), a significant decrease in the classical markers of autophagy, LC3-II, P62 and Beclin-1, was seen. This indicates that gastrodin protects astrocytes by inhibiting their autophagy (Wang et al., 2016).
 |
Urinary trypsin inhibitor (UTI) also protects astrocytes by maintaining their membrane integrity. When astrocytes are treated with UTI immediately or within 30 minutes of TBI, the percentage of LDH (which impairs mitochondrial membrane) released by astrocytes is significantly reduced (Lamprecht et al., 2017). More promising therapeutic targets include STAT3 and P2Y1 receptors, both of which are involved in the entire process of astrocyte response to TBI, which involves a large number of neural pathways. The expression of p-STAT 3 immediately increased after ischemia in TBI, peaked at three hours and then decreased. Immunohistochemical studies have shown activation of the JAK2/STAT3 pathway in neurons and astrocytes three hours after TBI. STAT3 inhibits small GTPase RhoA, thereby controlling the tension of actin filaments, the migration of active astrocytes and the accumulation of leukocytes. During glial scar formation, animal experiments have found that STAT3 is involved in secretion of the key glial scars protease MMP2 (Renault-Mihara and Mukaino, 2017).
The phenotype change of astrocytes, mediated by microglia, can be initiated by down-regulating the P2Y1 receptor; additionally, this receptor attenuates brain edema and reactive glial changes in a model of TBI by enhancing astrocyte mitochondrial metabolism (Talley Watts et al., 2013). In the TBI mouse, inhibition of the astrocyte-specific P2Y1 receptor promotes scar formation (Liao et al., 2012). Activation of P2X7 and P2Y1 in astrocytes separates damaged brain regions from surrounding healthy tissue and allows reactive astrocytes to synthesize neurotrophic factors and proteins involved in neuronal repair (Franke and Illes, 2014). The most attractive aspect of astrocytes’ potency is that astrocytes can directly generate human neural cells and provide a possibility for the regeneration of neuronal cells because of both their strongly proliferative nature and closely related lineage to neurons (Gao et al., 2017). Additionally, the following therapies are also in full development: RIP3 knock out (KO) (Liu et al., 2018), unc-51-like kinase 1 (Ulk1) KO (Wei et al., 2018), L-N-iminoethyl-lysine (Arnberg et al., 2012), angiotensin II: Sartans (Villapol et al., 2015), TAK-242 (Feng et al., 2017) and FOXO3a (Cui et al., 2011).
Dead neurons release excitotoxic glutamate, K+, ATP, and proinflammatory mediators and induce secondary axonal damage after TBI. Astrocytes have neurotrophic and supportive effects on nerve tissue under physiological conditions, but after TBI their functions also change. Typically, reactive astrocytes provide glutamate elimination, antioxidant protection, neuronal metabolic substrates, K+ buffering and growth factors to support and repair neurons. Reactive astrocytes also suppress inflammation by phagocytotic activity, regulating immune cells and forming physical barriers around damaged tissue. However, the recruitment of astrogliosis peripheral immune cells aggravates inflammatory damage in the CNS and to a certain extent glial scars restrict axonal regeneration (Gyoneva and Ransohoff, 2015; Ohtake and Li, 2015). So the reactive astrocyte can be referred to as a doubleedged sword in TBI. By taking advantage of protective reactions and simultaneously inhibiting harmful effects, it may be possible to control the deterioration induced by inflammation and protect dynamic synaptic growth in the brain. Morphological changes, proliferation and migration also occur with reactive astrocytes, which can be identified and monitored by biomarkers and imaging tools, thus providing a basis for a large number of therapeutic studies.
Glial cells were discovered more than 100 years ago, but their role in the treatment of TBI remains to be fully explored. The reason for this situation is in part the complex mechanism of reactive astrocytes after TBI. Further, the hetrogeneity of reactive astrocytes is not fully understood as it exists, not only within the same species but also between humans and rodents and even in the same individual. As various diseases occur in the CNS, glial cells undergo significant changes in morphology, quantity, and a series of neuro-molecular changes to perform various functions such as compensation, regeneration and repair. These processes are also accompanied by changes in methods of communication between central neurons. Nonsynaptic volume transmission is considered to be the most important information transmission mechanism for repair processes after TBI (Bach-y-Rita, 2001).
An awareness is required that reactive astrogliosis does not only appear in TBI and cannot be generalized to only characteristic changes such as cell hypertrophy, increased proliferation, scar formation and increased expression of GFAP and precursor cell markers, including vimentin and nestin. Astrocytes are originally morphologically and functionally heterogeneous cells that may exhibit an increased heterogenaity after being subjected to pathological activation by different types and degrees of damage and the different injury-protective mechanisms of the body. However, there is currently neither definition nor classification of the different types of reactive astrocytes, merely two descriptions as either A1 atypical or A2 typical astrocytes. A1 astrocytes activated by IL-1α and TNF C1q do not promote axon formation and exhibit strong neurotoxicity. Unlike A1 reactive astrocytes, A2 reactive astrocytes are activated by ischemia and strongly promote neuronal survival and tissue repair (Bi et al., 2013; Liddelow and Barres, 2017; Liddelow et al., 2017). Typical astrocytes support neuronal attachment and synaptic outgrowth, while atypical astrocytes inhibit neuronal regeneration. Further, atypical astrocytes have a special growth morphology, high cell density, a small number of oligodendrocytes in the superficial layer of the cortex and they occupy a smaller growth area than that of typical astrocytes (Liu et al., 2015; Wanner et al., 2008).
Something in common of these two classification methods is dividing reactive astrocytes into neuro-beneficial and neurotoxic types, which reinforces the so-called two-sidedness and complexity of reactive astrogliosis. However, the activation mode, biochemical characteristics, definition, and characterization of these cells still lacks comprehensive theoretical explanation. Using a single-cell gene expression profile is a more objective approach to defining reactive astrocytic subpopulations. Otherwise, it is not possible to explain why diseases completely unrelated to TBI can exhibit the same characteristics as reactive astrocytes, such as, for example, the fact that normal aging induces both A1-like astrocyte reactivity and Alexander disease (AxD), a disease of astrocytes. The latter is caused by heterozygous mutations in GFAP. Due to the accumulation of cytoplasmic protein content, such as Hsp and GFAP, it also exhibits astrocyte hypertrophy, a large increase of GFAP and is harmful to neighboring neurons (Clarke et al., 2018; Sosunov et al., 2013). In summary, investigation of the dual roles and heterogeneity of reactive astrocytes provides many important insights into the pathology and ideas for the treatment of TBI.
This work was supported by the National Natural Science Foundation of China [Grant numbers 81060101], Yunnan Provincial Science and Technology Department Funds [Grant No 2018FE001(-015)], National Innovation Funds for College Students [201710678006], Funds of Kunming Medical University [2017-JY-Y-014].
The authors declare no competing interests.