






 , Musarrat Husain Warsi 3, Nilesh Chandra 4, Reshma Parekar 5, Maneesha Khalse 5, Kamlesh Patel 5
, Musarrat Husain Warsi 3, Nilesh Chandra 4, Reshma Parekar 5, Maneesha Khalse 5, Kamlesh Patel 51 Department of Pharmaceutics, Delhi Pharmaceutical Science and Research University, 110017 New Delhi, India
2 Center for Advanced Formulation Technology, Delhi Pharmaceutical Sciences and Research University, 110017 New Delhi, India
3 Department of Pharmaceutics and Industrial Pharmacy, College of Pharmacy, Taif University, 21944 Taif, Saudi Arabia
4 Division of Discovery Research, Indian Council of Medical Research, 110029 New Delhi, India
5 Lupin Limited, 400055 Mumbai, India
Abstract
Generic dydrogesterone products are widely indicated in various conditions such as infertility, menstrual disorders, and the prevention of miscarriage. Moreover, the therapeutic equivalence of generic dydrogesterone products has never been explored, despite reports of interindividual variability in the bioavailability of dydrogesterone. Therefore, this study aimed to compare two generic formulations of dydrogesterone (A and B) by evaluating the respective quality, pharmacokinetics, and endometrial tissue concentrations of these formulations in rats.
Differential scanning calorimetry (DSC), X-ray diffraction (XRD), and the drug content were compared in the quality analysis. Meanwhile, dissolution and rat plasma kinetic profiles were compared to determine the in vitro and in vivo equivalence. Endometrial drug levels were assessed by liquid chromatography–tandem mass spectrometry (LC-MS/MS) and in vivo imaging.
The DSC and XRD results revealed that a higher percentage of the dydrogesterone in formulation A was amorphous, compared to formulation B, where the dydrogesterone was more crystalline. The difference in drug content between the formulations was not significant (p > 0.05), and the dissolution profile and plasma bioavailability of formulations A and B were similar (p > 0.05). LC-MS/MS analysis showed that dydrogesterone levels in the endometrial tissue of rats treated with formulation A were markedly higher compared to those treated with formulation B. The in vivo imaging results corroborated with the LC-MS/MS analysis, with nearly 43.0% higher dydrogesterone accumulation observed in the uterus of animals treated with formulation A compared to formulation B.
These data exhibited that therapeutic inequivalence may exist between the generic formulations of dydrogesterone; hence, the generic formulation should be carefully selected.
Keywords
- biological availability
- chromatography
- crystallinity
- dydrogesterone
- generic drug
- target tissue concentration
Dydrogesterone is a retro-progesterone, with improved physico-chemical
properties and oral bioavailability (~28%) compared to
progesterone [1, 2]. It is used for a variety of indications, including female
infertility [3], prevention of miscarriage [4], dysmenorrhea [5], endometriosis
[6], irregular menstrual cycles [7], and as a component of menopausal hormone
therapy [8]. Oral dydrogesterone is extensively used as an alternative to
micronized oral and vaginal progesterone with proven efficacy and safety and high
patient compliance [3, 9]. Until 2021, only the innovator product Duphaston
(Abbott Healthcare, Netherlands) was available in the Indian market, with annual
sales of US
Reports establishing bioequivalence between oral dydrogesterone and oral/vaginal progesterone are available [2, 3]. However, comparison of therapeutic equivalence between generics and evaluating the impact of quality of dydrogesterone on therapeutic equivalence has not been reported previously. It is well reported that dydrogesterone exerts its action through selective binding to progesterone receptors in the endometrium [1]; thus, we hypothesized that the determination of dydrogesterone concentration in endometrial tissue would be more relevant for establishing therapeutic equivalence of generic products. Thus, the present study evaluates the quality, pharmacokinetics, and endometrial tissue concentration of dydrogesterone following oral administration of two generic formulations of dydrogesterone to rats.
Generic tablet formulation of dydrogesterone, namely formulation A (Dydrogesterone Tablet 10 mg, Lupihope, Lupin Ltd., Mumbai, Maharashtra, India) and formulation B (Dydrogesterone Tablet 10 mg, DPSR University, Pushp Vihar, New Delhi, India). Pure dydrogesterone was provided as a gift sample by Lupin Ltd. (Mumbai, Maharashtra, India). Indocyanine green (ICG) was purchased from Tokyo Chemical Industry (Chennai, Tamil Nadu, India). All other reagents and solvents used were of analytical grade and were procured from Merck (Mumbai, Maharashtra, India).
Thermal properties of dydrogesterone formulations were investigated using a differential scanning calorimeter with TAC-7 thermal analysis controller with an intracooler-2 cooling system (DSC-7, Perkin Elmer, Waltham, MA, USA). About 5 mg of the samples were placed in perforated aluminum-sealed pans (50-L). The heat run was set from 40 to 200 °C at a rate of 5 °C/min, under a nitrogen environment.
XRD diffractograms of dydrogesterone formulations were recorded using a Panalytical Xpert Pro Diffractometer (PANalytical, Almelo, Netherlands) with a copper line as the source of radiation. The process parameters include 40-kV voltage, a 40-mA current, and a scanning rate of 0.02°/min over a 2° range of 3–40°.
To determine dydrogesterone content in tablet formulations, the tablets were weighed, crushed, and powder equivalent to 0.01 g of dydrogesterone, based on label claim, was weighed accurately and dissolved in 100 mL of methanol. The solution was filtered, and 5 mL of this solution was diluted to 100 mL with methanol. The absorbance of this solution was determined at 286 nm using a UV-spectrophotometer (UV-1800, Shimadzu, Kyoto, Japan).
Cumulative drug release as a function of time was evaluated using a USP type II
apparatus (Distek Inc., North Brunswick, NJ, USA). The rotation speed was 50 rpm,
and 900 mL of water maintained at 37
The study was conducted using healthy Sprague-Dawley female rats, 6–8
weeks old, weighing 250
For kinetics and endometrial tissue concentrations, ten rats were randomly and equally divided into two groups. Group A (n = 5) rats were treated with formulation A, and group B (n = 5) rats were treated with formulation B at a dose equivalent to 1.2 mg/kg of dydrogesterone. Briefly, tablets were weighed, powdered, and an amount equivalent to 1.2 mg/kg of dydrogesterone was suspended in purified water. The suspension was administered to the rats using an oral feeding sonde. The treatment was given 3 hours after the standard pellet diet (8 g), and no food was provided thereafter. Heparinized vacutainers were used to collect blood before treatment and at 0 h, 1 h, 2h, 4 h, and 8 h time points post-treatment from the jugular vein catheter. The samples were then centrifuged, plasma collected, and stored at –70 °C until analyzed by liquid chromatography mass spectrometry (LC-MS/MS). The plasma-concentration time profiles were plotted using Microsoft Excel, and the pharmacokinetic analysis was performed by non-compartmental analysis using the Excel add-in PK-Solver® (Redmond, WA, USA). The researcher who handled the animals was aware of the treatment, but the analyst who analyzed the samples was not aware of the same.
For endometrium tissue concentration, the animals were sacrificed at 8 h
post-dosing by intraperitoneal injection of 6.5% sodium barbital at a dose of 250
mg/kg. The abdominal wall was opened longitudinally along the midline, the uterus
was excised, and the entire endometrium was peeled off from the uterus [16]. The
extracted endometrium was cut, weighed, and homogenized in acetone. The solution
was collected after filtration from a 0.25 µm filter and analyzed for
dydrogesterone endometrium concentration (ng/mg) using a liquid extraction-based
liquid chromatography coupled with mass spectrometry (LC-MS/MS, TQ 8045 RX
Shimadzu, Kyoto, Japan). The method was developed for the quantification of
dydrogesterone in human plasma as reported previously [17]. The plasma samples
(0.1 mL) were extracted with methanol (0.4 mL) to precipitate proteins.
Thereafter, the plasma was filtered, evaporated to dryness under nitrogen, and
volume was made up using the mobile phase. The dydrogesterone was eluted in 1.5
minutes on a reversed-phase column (Kinetex, C18, 1.7 µm 100
For in vivo imaging, a total of nine rats were used. Rats were randomly and equally divided into three groups. Group A (n = 3) and group B (n = 3) rats were treated once orally with ICG-labeled formulations A and B, respectively. Briefly, tablets were weighed, powdered, and an amount equivalent to 1.2 mg/kg of dydrogesterone was suspended in 0.5 mL purified water to which 10 µg of ICG was added. The mixture was stirred and allowed to equilibrate for about 1 h. The mixture was filtered, and the filtrate was washed twice with 1.0 mL of water to remove free ICG. The filtrate was resuspended in 0.5 mL of purified water and administered to the rats using an oral feeding sonde. The control group (n = 3) received 1.0 mL of ICG aqueous solution. Rats were shaved and underwent a scan at baseline and post-8 h of dosing under isoflurane (1.0%, v/v) anesthesia (see supplementary material) by preclinical imaging system IVIS Lumina Series III (Perkin Elmer, Waltham, MA, USA) at excitation/emission wavelength 745/840 nm [18]. For analysis of the in vivo ICG localization, regions of interest (ROIs) were drawn over the uterus using Living Image 4.3.1 (Perkin Elmer, Waltham, MA, USA). The emission intensity of each sample corresponds to the drug concentration was expressed in radiant efficiency units.
Data were statistically described in terms of mean
Crystallinity of dydrogesterone in formulations was determined using DSC and XRD. The DSC thermogram showed a glass transition peak (59 °C–62 °C), owing to crystalline dydrogesterone in both the formulations. The enthalpy change for the glass transition peak was higher for formulation A compared to formulation B (Fig. 1).
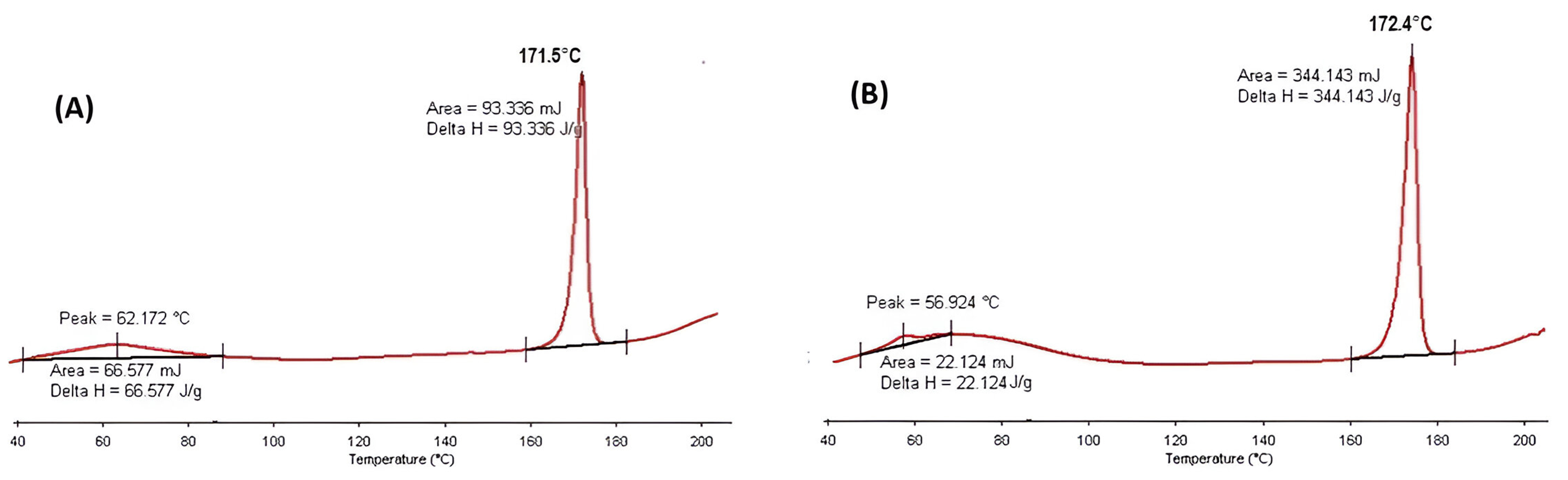 Fig. 1.
Fig. 1.
DSC thermogram of (A) formulation A and (B) formulation B of dydrogesterone.
A prominent endothermic peak at ~172 °C, possibly owing to the melting of dydrogesterone, was visible in the DSC thermogram of both the formulations (Fig. 1). The melting enthalpy of dydrogesterone was lower for formulation A compared to formulation B. The higher glass transition enthalpy and lower melting enthalpy suggest a higher percentage of amorphous dydrogeaterone in formulation A [19]. XRD was performed to confirm the difference in dydrogesterone crystallinity in the formulations (Fig. 2). In agreement with DSC, the XRD results revealed reduced 2-theta peaks for formulation A compared to formulation B. The results confirmed a higher percentage of amorphous dydrogesterone in formulation A. The difference in dydrogesterone crystallinity might be due to differences in API source, variation in API manufacturing, use of different excipients, or variation in tablet processing parameters. DSC and XRD studies revealed that dydrogesterone in formulations A and B differs in quality.
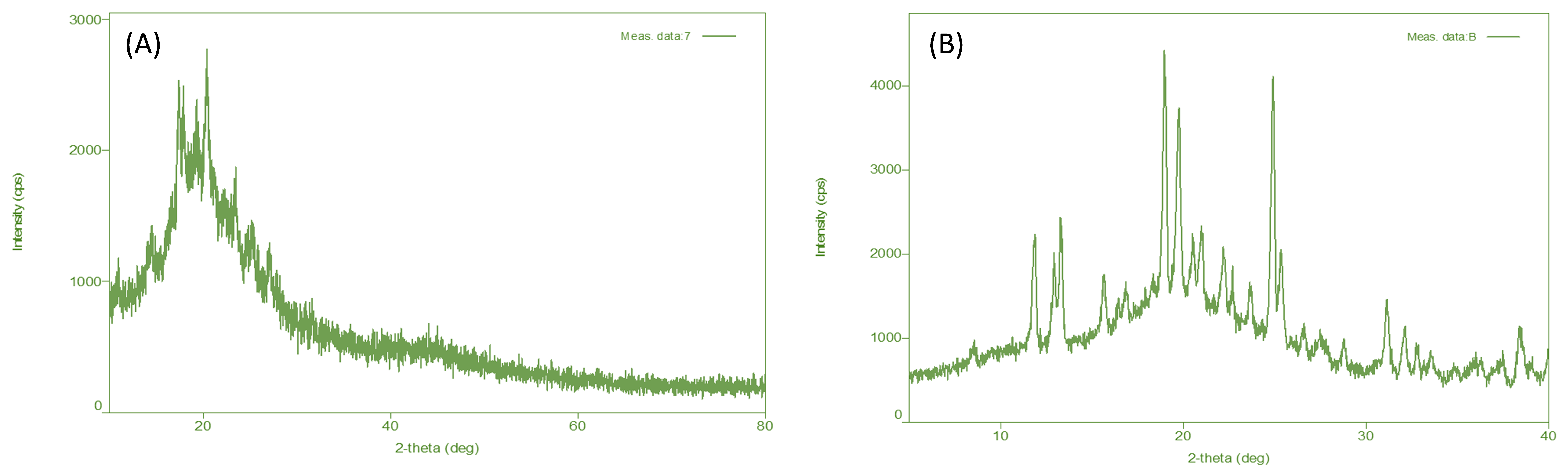 Fig. 2.
Fig. 2.
XRD spectra of (A) formulation A and (B) formulation B of dydrogesterone. XRD, X-ray diffraction.
The dydrogesterone content in formulation A was found to be 99.8
The dissolution profile of formulations A and B is shown in Fig. 3.
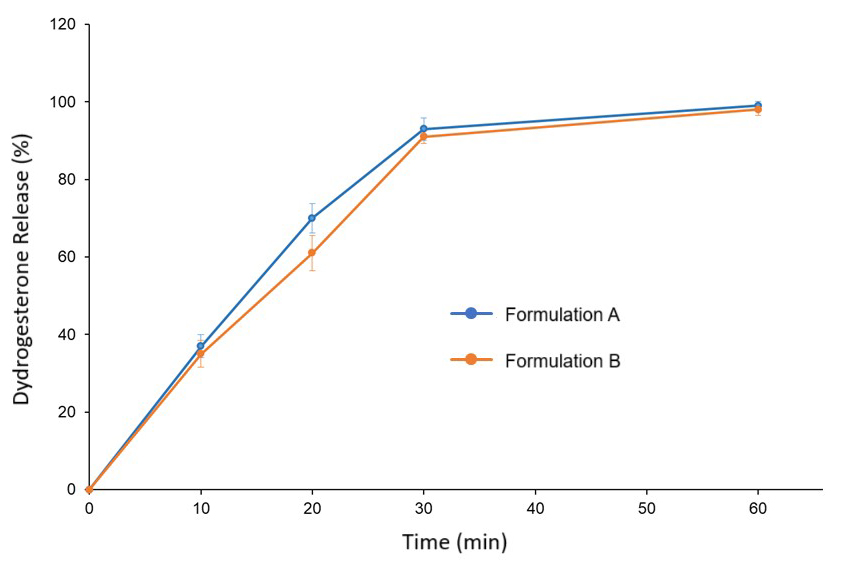 Fig. 3.
Fig. 3.
Dissolution profile of dydrogesterone formulation A (blue line) and formulation B (orange line). Error bars indicate standard deviation (n = 6).
The data demonstrated that the dissolution rate of formulation A was slightly
higher than formulation B; however, the profiles were statistically similar (f2
Similar observations were observed in vivo following oral
administration of formulations A and B to rats. The plot of dydrogesterone plasma
concentration versus time exhibited nearly comparable profiles for formulations A
and B (Fig. 4). The observed pharmacokinetic parameters are listed in Table 1.
The rats treated with formulation A demonstrated slightly higher (31.5
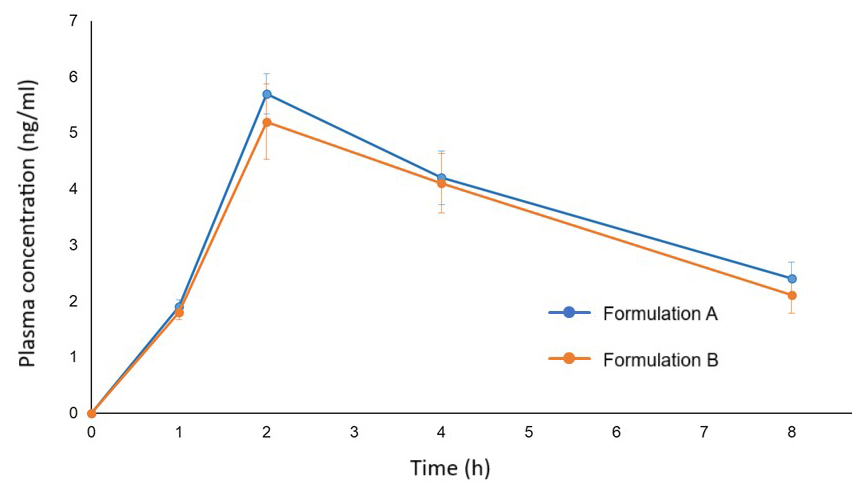 Fig. 4.
Fig. 4.
Plasma dydrogesterone concentration vs time plot following oral administration of dydrogesterone formulation A (blue line) and formulation B (orange line) to rats (n = 5). Error bars indicate standard deviation.
| Parameter | Formulation A | Formulation B |
| Cmax (ng/mL) | 5.7 |
5.2 |
| Tmax (h) | 2 h | 2 h |
| AUC (ng/mL/h) | 31.5 |
28.2 |
The data are presented as mean
The mean endometrial concentration of dydrogesterone determined by LC-MS/MS at 8
h post-dosing of formulations A and B was 39.5
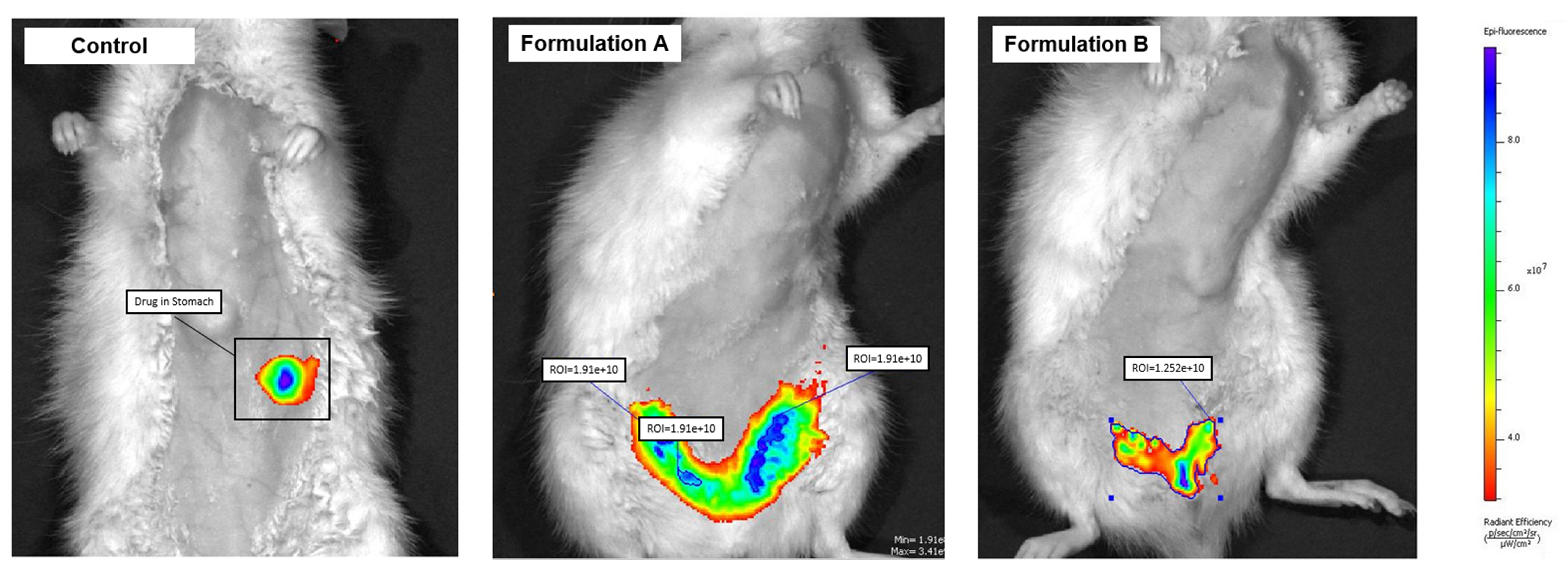 Fig. 5.
Fig. 5.
In vivo images of rats following oral administration of ICG-labeled dydrogesterone and dydrogesterone formulations to rats. ICG, indocyanine green.
The results clearly demonstrate that the quality of the dydrogesterone is a strong predictor of therapeutic outcome. The assumption that pharmaceutical equivalence predicts therapeutic equivalence does not hold for BCS class IV dydrogesterone, and drug concentration at the target site is more relevant to predict therapeutic equivalence. For generics to be truly bioequivalent, they should contain the same strength of API in an identical state of purity and quality, all within the same excipient. Further, therapeutic equivalence requires the integration of pharmacokinetics and pharmacodynamics; otherwise, it is risky to consider generic drugs as alternatives to the innovator.
Drug crystallinity, being an important quality parameter, was determined by DSC and XRD. It is well known that the degree of crystallinity negatively affects the solubility [19]. In DSC, the enthalpy change of a transition corresponds to the degree of crystallinity, and a lower enthalpy indicates a sharper peak and higher crystallinity. DSC thermogram demonstrated that formulation A, with a higher enthalpy of transition, was less crystalline than sample B. In addition, the lower melting enthalpy observed for formulation A indicates the requirement of less energy and presence of dydrogesterone in the liquid state available for solubilization in external media [19]. Further, the XRD results were in corroboration with DSC results, and formulation A showed reduced 2-theta peaks indicative of a lower degree of crystallinity compared to formulation B. The difference in the degree of crystallinity of dydrogesterone in formulations A and B may be due to a difference in API source, variation in API manufacturing, or variation in tablet processing parameters. DSC and XRD data revealed that the quality of dydrogesterone in two generic formulations, A and B, differs.
On the contrary, assay for purity showed that the dydrogesterone content in
formulation A was not significantly different (p
To establish therapeutic equivalence, dydrogesterone concentration in endometrial tissue following oral administration of formulations A and B was compared using LC-MS/MS analysis and in vivo imaging of ICG-labeled formulations. The property of dydrogesterone to rapidly distribute to the endometrium and bind with progesterone receptors expressed at the endometrium allowed quantification in the endometrium [1]. LC-MS/MS was used because it is capable of quantifying low dydrogesterone amounts in endometrial tissues [17]. ICG was selected for labeling because of its ability to bind with lipophilic dydrogesterone (log P ~4.53) through hydrophobic interactions [18]. Interestingly, both LC-MS/MS and in vivo imaging detected markedly higher dydrogesterone levels in the endometrial tissue of rats treated with formulation A compared to those treated with formulation B. However, the deviation in the dydrogesterone amount determined by LC-MS/MS and in vivo imaging was observed. These differences might be due to several factors, such as (a) differences in tissue distribution pattern of dydrogesterone and ICG-labeled dydrogesterone, (b) estimation of radiant in the uterus and not in the endometrium alone, and (c) labeling of the whole formulation instead of API.
In contrast with in vivo pharmacokinetics, the findings of endometrial dydrogesterone concentration showed that formulations A and B are therapeutically inequivalent. Similar observations were reported previously where generic products failed in vivo despite being pharmaceutical equivalents of the innovator [20, 21, 22].
The present study emphasized that the difference in drug crystallinity could be one of the factors affecting therapeutic equivalence. However, the involvement of the particle size of dydrogesterone, formulation excipients, and manufacturing process, which were not assessed owing to the use of commercial formulations, may not be ruled out. Like crystallinity, particle size could influence drug absorption and tissue distribution [23]. Nevertheless, formulation excipients and manufacturing process could impact the drug absorption and distribution directly [24] or by changing the drug crystallinity [25]. Reports suggested that the biopharmaceutical properties of the drug, blend properties, and formulation parameters, are paramount for achieving therapeutic equivalence, particularly for BCS class IV drugs [14, 23, 24, 25]. Interestingly, we observed differences in tissue distribution however, the dissolution and pharmacokinetics of the formulations A and B remained the same. The objective of the study, focusing on the impact of the dydrogesterone quality on therapeutic equivalence, allowed administration of the formulation as a suspension and the rat as a suitable animal model. Further, directly using the rabbit model without first proving the concept in rats is discouraged. Considering a pilot, proof-of-concept study, a small number of rats per group, and a simple methodology were adopted. Notably, the same dosing time, similar fed status, and endometrial quantification of the drug by LC-MS/MS and noninvasive imaging reduce the variance. Bioavailability was determined based on blood samples collected over a period of 8 hours, followed by euthanization of the animal and determination of the drug in endometrial tissue. The low standard deviations observed for Cmax and AUC, support the usage of fewer blood sampling time points. Further, due to the short half-life of dydrogesterone (~5 h), the drug concentration in the endometrial tissue was measured at 8 hours post-dosing [1]. Despite a similar outcome, the dydrogesterone concentration as measured by LC-MS/MS and imaging differed. This could be due to potential biases introduced by ICG labeling. Notwithstanding several challenges, non-invasive imaging is unquestionably useful for determining tissue distribution and medication deposition. The results highlighted the importance of assessment of drug concentration at the target site and explained the therapeutic failure of bioequivalent generic products.
In conclusion, therapeutic inequivalence may exist between generic dydrogesterone formulations; therefore, the formulation should be carefully selected. The experimental approach presented in the present study might be useful for obtaining insightful data on the benefits and potential problems surrounding the use of low-quality dydrogesterone. However, extensive pre-clinical studies are required to establish the exact reason for these observations.
All data points generated or analyzed during this study are included in this article, and there are no further underlying data necessary to reproduce the results.
GKJ, MHW, NC and RP conceptualized, designed, and planned the research study. GKJ conducted the experiments. MK and KP analyzed and interpreted the results and provided resources for the study’s conduct. MHW wrote the manuscript with support from GKJ, NC, and RP. MHW made figures, tables, and searched references. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript and have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The study obtained approval from the Institutional Animal Ethics Committee of DPSRU, New Delhi, under approval number IAEC/2024/19. All the animal procedures were carried out in accordance with the guidelines of the Committee for Control and Supervision of Experiments on Animals (CCSEA).
The authors extend their appreciation to Taif University, Saudi Arabia, for supporting this work through project number (TU-DSPP-2024-136). The authors are thankful to DSIR-DPSRU-CRTDH Centre for Advanced Formulation Technology for providing research facilities and Lupin Limited (India) for providing Lupihope formulation (formulation A) and technical support for the conduct of the study.
This research work was funded by Taif University, Saudi Arabia, Project No. (TU-DSPP-2024-136).
The co-authors, Reshma Parekar, Maneesha Khalse, and Kamlesh Patel, are employees of Lupin Limited, Mumbai, India. The judgments in data interpretation and writing were not influenced by this relationship. All other authors have no competing interests.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/IJP44211.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.