






1 Department of Neurology, The First Affiliated Hospital of Jinzhou Medical University, 121000 Jinzhou, Liaoning, China
2 Department of Neurology, Anshan Changda Hospital, 114001 Anshan, Liaoning, China
Abstract
The microglia-mediated neuroinflammatory response significantly contributes to neuronal damage following ischemic stroke by activating the signal transducer and activator of transcription 3 (STAT3) signal pathway. Rutin has been shown to exhibit STAT3-inhibitory properties, yet the effects of rutin on microglial polarization under ischemic context remain insufficiently understood. Therefore, this study aimed to investigate the effects of rutin on microglial activation and neuroprotection under oxygen-glucose deprivation (OGD) conditions and to elucidate the underlying mechanism.
Neuro-2a cells were subjected to OGD, and the subsequent conditioned medium (CM) was collected, and used to stimulate BV2 cells in vitro. The impact of rutin on microglial activation was assessed by analyzing cytokine profiles and microglial polarization. STAT3 phosphorylation was detected by Western immunoblot analysis, and the potential binding sites of rutin and STAT3 were determined through in silico molecular docking analysis. Neuron-microglia co-culture systems were employed to evaluate neuroprotective effects through viability and apoptosis assays.
Microglia cultured with CM exhibited increased pro-inflammatory cytokines and M1 polarization, which were reversed by rutin treatment. Additionally, rutin significantly promoted anti-inflammatory microglial polarization in a hypoxic condition through a decrease in phosphorylated STAT3 levels. These effects were demonstrated in a dose-dependent manner. Notably, OGD-challenged neurons exhibited enhanced survival and reduced apoptosis when co-cultured with rutin and CM-treated microglia.
Rutin offers neuroprotection by modulating microglial activation towards an anti-inflammatory phenotype, which is associated with the STAT3 pathway, underscoring the potential of rutin as a therapeutic agent for ischemic stroke and related cerebral injury.
Keywords
- rutin
- ischemic stroke
- microglia
- polarization
- STAT3
- neuroprotection
- oxygen-glucose deprivation
Ischemic stroke is the predominant form of stroke and accounts for approximately 85% of all stroke cases [1]. It occurs when cerebral blood flow is abruptly interrupted or severely reduced due to factors such as cerebral thrombosis, embolism or systemic hypotension. This disruption leads to oxygen and nutrient deprivation in brain tissue, resulting in neuronal injury or death [2]. Notably, in the acute phase of a stroke, the potential for extensive neuronal loss escalates with each passing minute. Therefore, immediate action to protect neurons is crucial in minimizing permanent damage and enhancing the prognosis for stroke patients [3].
Neurons are the fundamental functional units of the central and peripheral
nervous systems, exhibit complex mechanisms of injury under ischemic conditions.
A precipitous decline in blood flow can disrupt energy metabolism, leading to
secondary imbalances in sodium and potassium ion gradients and the activation of
various enzymes that cause intracellular structural damage [4]. Moreover,
ischemia and hypoxia induce the substantial production of free radicals and
trigger the immune system to release inflammatory mediators, potentially
culminating in the permanent death of neurons [5]. Following immune system
activation, microglia can rapidly respond to damage of neural tissue as the
primary immune cells of the central nervous system. These cells proliferate and
migrate to the affected area in the aftermath of injury, participating in damage
control and the repair process [6]. In the context of ischemic stroke, microglia
may exhibit either a pro-inflammatory M1 phenotype or an anti-inflammatory M2
phenotype [7]. Under stress conditions, the pro-inflammatory phenotype releases
inflammatory cytokines, such as tumor necrosis factor alpha (TNF-
Significant advancements have been made in research on modulating the
inflammatory phenotypes of microglia as potential targets for inhibiting
neuroinflammation and promoting neuronal protection and there are several key
molecules and signaling pathways observed to influence this phenotypic
transformation, including Toll-like receptors, Nuclear Factor Kappa B
(NF-
Rutin is a naturally occurring flavonoid compound ubiquitously found in a variety of plants such as buckwheat, citrus fruits, apples, grapes and tea. It is noted for its low toxicity and high biocompatibility, which make it a candidate for therapeutic applications [15]. As a potent antioxidant, rutin has demonstrated neuroprotective properties primarily through the inhibition of neuroinflammation [16]. Importantly, rutin was reported to be able to inhibit the expression of STAT3 in glioma cells and microglia, highlighting its potential in modulating inflammatory pathways within the central nervous system [17]. Furthermore, rutin has been shown to promote the polarization of microglia towards an anti-inflammatory M2 phenotype under conditions of retinal ischemia-reperfusion injury by inhibiting the JAK/STAT3 signaling pathway [18]. Despite these promising findings, the regulatory effects of rutin on microglial activity in the context of cerebral ischemia, as well as its subsequent impact on neuronal cells, remain poorly understood.
Consequently, this study employed an oxygen-glucose deprivation (OGD) model via in vitro experiments using neurons and microglia to investigate the role of rutin on microglia and neurons under OGD conditions. By elucidating the specific neuroprotective mechanisms of rutin, our study contributes to the development of therapeutic approaches targeting microglial phenotype regulation and offers novel insights into the management of ischemic brain injury.
This study was performed at The First Affiliated Hospital of Jinzhou Medical University from February, 2023 to January, 2024.
The Neuro-2a (N2A) mouse neuroblastoma cells (iCell-m040) and mouse microglia BV2 cells (iCell-m011) were procured from iCell Bioscience Inc. (San Francisco, CA, USA) and cultured according to standard protocols. All cell lines were validated by STR profiling and tested negative for mycoplasma. The cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin and 100 µg/mL streptomycin (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) in a humidified incubator at 37 °C with 5% CO2.
The N2A cells were subjected to OGD for the conditioned medium (CM) collection [19]. Briefly, N2A cells were removed to glucose-free Earle’s Balanced Salt Solution (Gibco) and placed in an anaerobic chamber (Coy Laboratory Products, Grass Lake, MI, USA) with 1% O2, 5% CO2 and 94% N2 at 37 °C for 6 hrs before the supernatant was collected as CM. For cell treatments, BV2 cells were exposed to the CM for 6, 12, and 24 hrs to study the effects of the neuronal damage on microglial activation and response. To investigate the potential effects of rutin on BV2 cells, a series of rutin (PHL89270, Sigma-Aldrich, St. Louis, MO, USA) concentrations (0, 0.5, 1, 2.5, 5, 10, and 20 µM) were used to treat BV2 cells for 24 hrs. Based on the cell viability post-treatment, safe concentrations of rutin were selected for further experiments. At rutin concentrations of 0.5, 1, and 2.5 µM, BV2 cells were treated with CM or 0.05% dimethyl sulfoxide as a vehicle control for 24 hrs. Cells without additional treatment were served as the control.
Microglial BV2 cells were grown in CM, with or without the addition of rutin for a duration of 24 hrs. Following treatment, the medium was removed and replaced with normal culture medium. The N2A neurons were exposed to OGD conditions for 3 hrs prior to replenished with normal culture medium and positioned beneath the microglial cell inserts (Dow Corning, Corning, NY, USA). After maintaining the co-culture for 12 hrs, the microglial cell inserts were removed to proceed with the subsequent evaluations of neuronal survival and apoptosis.
Cell viability was quantified using the CCK-8 assay (Dojindo Molecular Technologies, Inc., Kumamoto, Japan) according to the standard protocol provided by manufacturer. Cells were seeded in 96-well plates and allowed to adhere overnight. Following treatment, 10 µL of CCK-8 solution was added to each well containing 100 µL of culture medium. The plates were then incubated at 37 °C in a 5% CO2 atmosphere for 2 hrs. The absorbance of the formazan product was measured at 450 nm using a microplate reader (BioTek Instruments, Inc., Winooski, VT, USA).
Molecular docking of rutin to the STAT3 protein was performed to elucidate the potential binding interactions and conformation. The three-dimensional structure of STAT3 was retrieved from the Protein Data Bank [20] and the chemical structure of rutin was obtained from the PubChem database [21]. Rutin was energetically minimized using the MM2 force field in Chem3D (CambridgeSoft, Cambridge, MA, USA) to ensure the lowest energy conformation. The molecular docking simulation was conducted using AutoDock Vina (The Scripps Research Institute, La Jolla, CA, USA). The docking parameters were set to search a grid box defined around the catalytic chamber of STAT3, accommodating the ligand’s dimensions. A Lamarckian genetic algorithm was employed to explore possible binding poses and the pose with the highest affinity was selected for visualization, achieved using PyMOL (Schröinger, LLC, New York, NY, USA).
Western blot analysis was performed to detect the relative expression of polarization and signaling pathway-related biomarkers in BV2 cells under diverse conditions [22]. Protein lysates were prepared using RIPA buffer (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with protease and phosphatase inhibitor cocktails (Sigma-Aldrich, St. Louis, MO, USA). Protein concentration was determined by the BCA Protein Assay Kit (Pierce, Thermo Fisher Scientific, Waltham, MA, USA), following the manufacturer’s instructions. Equal amounts of protein were separated on SDS-PAGE gels and transferred onto PVDF membranes (MilliporeSigma, Burlington, MO, USA). The membranes were blocked in 5% non-fat dry milk in TBST for 1 hr at room temperature and incubated overnight at 4 °C with primary antibodies against CD86, CD206, p-JAK2, JAK2, p-STAT3, STAT3 and GAPDH, of which detailed information was listed in Supplementary Table 1. Subsequently, the membranes were washed and incubated with HRP-conjugated secondary antibodies (Cell Signaling Technology, Danvers, MO, USA) for 1 hr at room temperature. Protein bands were visualized using enhanced chemiluminescence detection reagents (GE Healthcare, Chicago, IL, USA) and imaged with a ChemiDoc Touch Imaging System (Bio-Rad Laboratories, Hercules, CA, USA). Band intensities were quantified using Image Lab software (Bio-Rad Laboratories, Hercules, CA, USA) and the data were normalized to the expression of GAPDH.
The RT-PCR was employed to quantify the expression levels of
inflammation-related cytokines. Total RNA was isolated from cell samples using
the TRIzol reagent (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA)
following the manufacturer’s protocol. The concentration and purity of RNA were
determined spectrophotometrically using a NanoDrop ND-1000 Spectrophotometer
(Thermo Fisher Scientific, Waltham, MA, USA). Reverse transcription was performed
to synthesize cDNA from the total RNA using the High Capacity cDNA Reverse
Transcription Kit (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA,
USA) according to the manufacturer’s instructions. Quantitative PCR
amplifications were then conducted using the PowerUp SYBR Green Master Mix
(Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) on a QuantStudio 5 Real-Time PCR System (Applied
Biosystems, Thermo Fisher Scientific, Waltham, MA, USA). The cycling conditions were set as follows: Initial uracil-DNA
glycosylase activation at 50 °C for 2 min, followed by dual-lock DNA
polymerase activation at 95 °C for 10 min and then 40 cycles of
denaturation at 95 °C for 15 sec, with annealing and extension at 60
°C for 1 min. Primer sequences specific for mouse
IL-10, IL-6 and TNF-
Apoptosis was assessed by Annexin V/Propidium Iodide (PI) staining using the
Annexin V-FITC Apoptosis Detection Kit (BD Biosciences, San Jose, CA,
USA) according to the manufacturer’s instructions [23]. Cells were harvested
following treatment and washed twice with PBS. Subsequently, the cell pellet was
resuspended in 1
The graphical representations and statistical computations were conducted by
GraphPad Prism (Version 8.0; GraphPad Software, San Diego, CA, USA). The
distribution of data was initially appraised for normality utilizing the
Shapiro-Wilk test, depending on which One-way Analysis of Variance (ANOVA) with
subsequent post hoc analysis via Tukey’s test or the Kruskal-Wallis
test, followed by Dunn’s Multiple Comparisons test were used. Quantitative
variables were reported as Mean
Following exposure to CM derived from N2A neurons subjected to OGD, BV2
microglia revealed significant upregulation of the pro-inflammatory cytokines
IL-6 (Fig. 1B) and TNF-
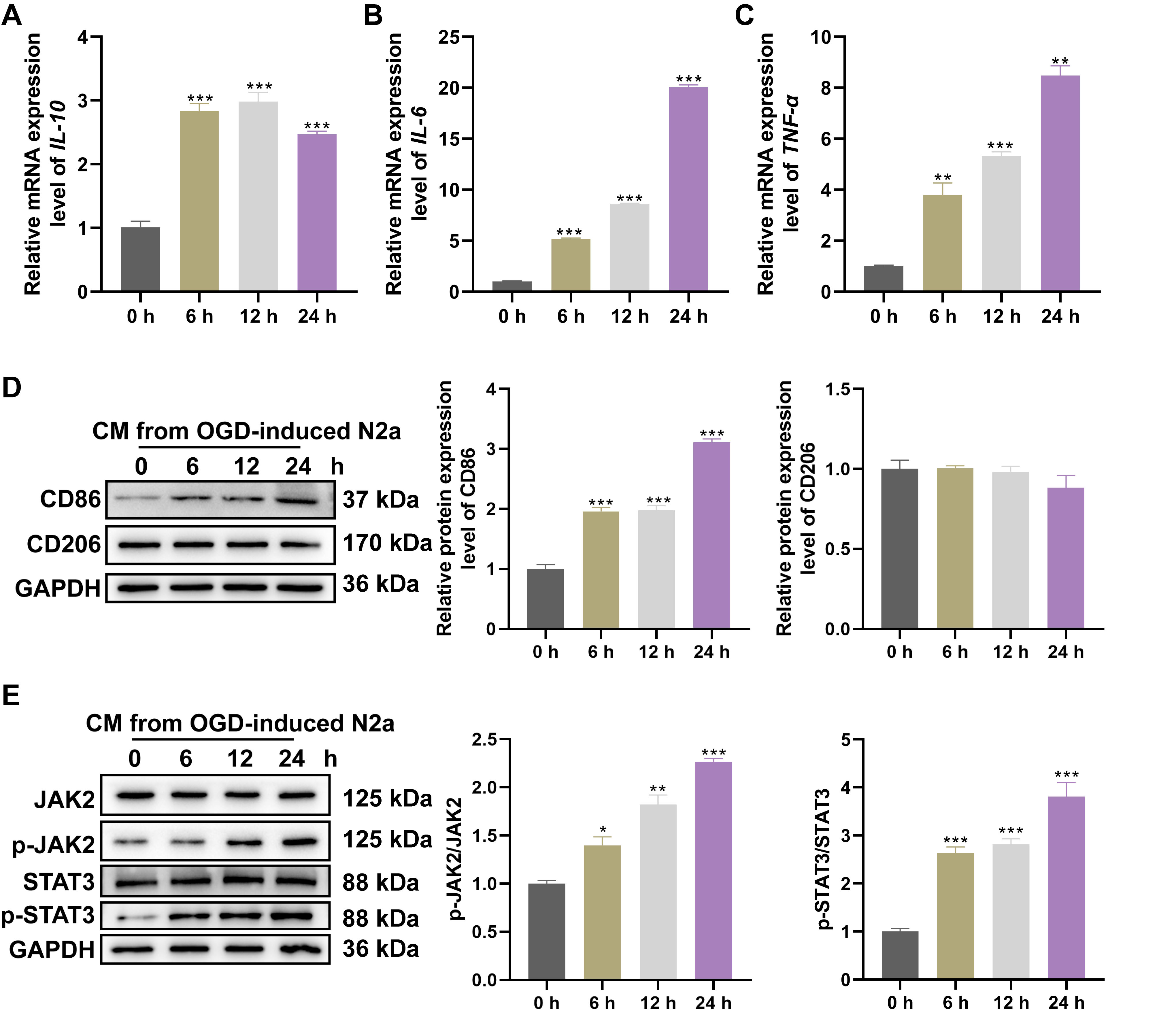 Fig. 1.
Fig. 1.
CM collected from OGD neuron induced microglia polarization and
inflammatory responses via JAK2/STAT3 activation. After stimulating BV2 cells
with CM collected from N2A cells exposed to OGD for 0, 6, 12 and 24 hrs, relative
levels of (A) IL-10, (B) IL-6, (C) TNF-
Rutin is a glycosylated bioflavonoid, whose molecular skeleton consists of multiple phenolic hydroxyl groups, an ether linkage and a disaccharide moiety. The molecular formula of rutin is expressed as C27H30O16, with a molecular weight of 610.5 g/mol (Fig. 2a). In the molecular docking, rutin is shown to occupy the catalytic chamber of the STAT3 protein, surrounded by key amino acid residues including Lys631, Gln633, Pro715 and Thr721 (Fig. 2b). The compound’s positioning suggests potential inhibitory effects on the protein’s activity by the direct interaction with these residues.
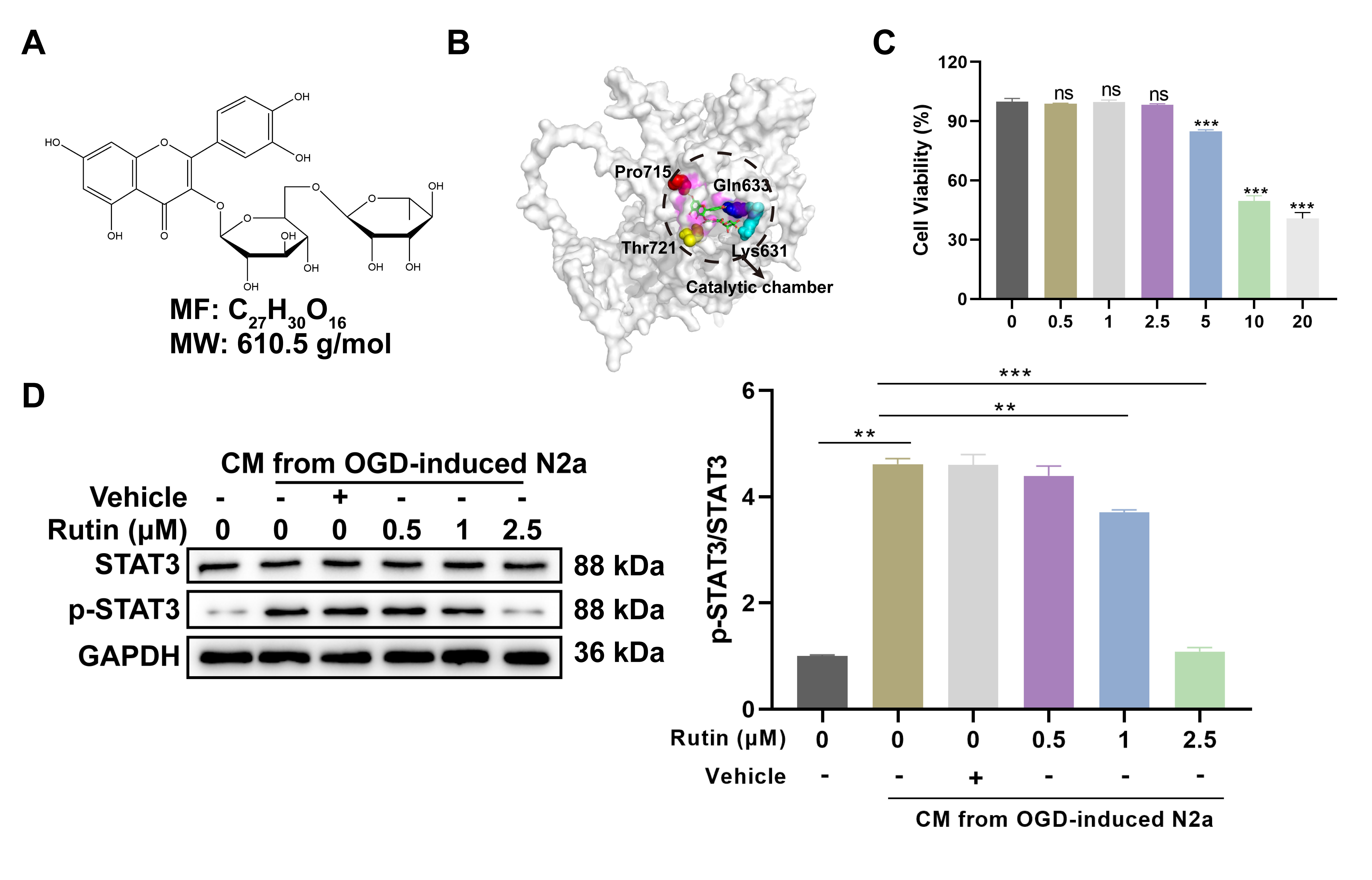 Fig. 2.
Fig. 2.
Rutin inhibits STAT3 activation-induced by CM from OGD-treated
neurons in microglia. (A) Chemical structure of rutin (C72H30O16)
with a molecular weight of 610.5 g/mol. (B) Molecular docking of rutin and STAT3
protein. (C) After 24 hrs incubation with various concentrations of rutin (0,
0.5, 1, 2.5, 5, 10 and 20 µM), cell viability assay of BV2 microglial cells
was evaluated and (D) Western blot analysis was conducted to determine the STAT3
phosphorylation in BV2 cells treated with non-toxic concentrations of rutin (0,
0.5, 1, 2.5 µM) for 24 hrs with CM stimulation. Data are presented as Mean
To evaluate the cellular toxicity of rutin, BV2 microglial cells were treated with a gradient of rutin concentrations for 24 hrs prior to the CCK-8 assay. The concentrations of rutin above 2.5 µM resulted in notable decrease in cell viability (Fig. 2c), thus guiding the selection of non-toxic concentrations (0, 0.5, 1 and 2.5 µM) for subsequent assays. Subsequently, these dosages of rutin were used to treat BV2 cells with CM from OGD-treated N2A neurons and western blot analysis revealed a dose-dependent inhibition of STAT3 phosphorylation by rutin. The highest inhibitory effect was observed at 2.5 µM rutin, which significantly abrogated the CM-induced increase in STAT3 activation (Fig. 2d). These results suggest that rutin can effectively counter the activation of the STAT3 pathway at non-toxic concentrations, probably by directly interacting with crucial residues and disrupting protein function.
Treatment with 2.5 µM rutin in BV2 microglial cells exposed to CM from
OGD-treated N2A neurons was associated with significant alterations in
inflammatory cytokine profiles and microglial activation markers. Real-time PCR
analysis demonstrated a marked increase in the expression of the
anti-inflammatory cytokine IL-10 following rutin treatment compared to
control and vehicle-treated groups (Fig. 3A). Additionally, the addition of rutin
reversed the upregulation of pro-inflammatory cytokines IL-6 (Fig. 3B)
and TNF-
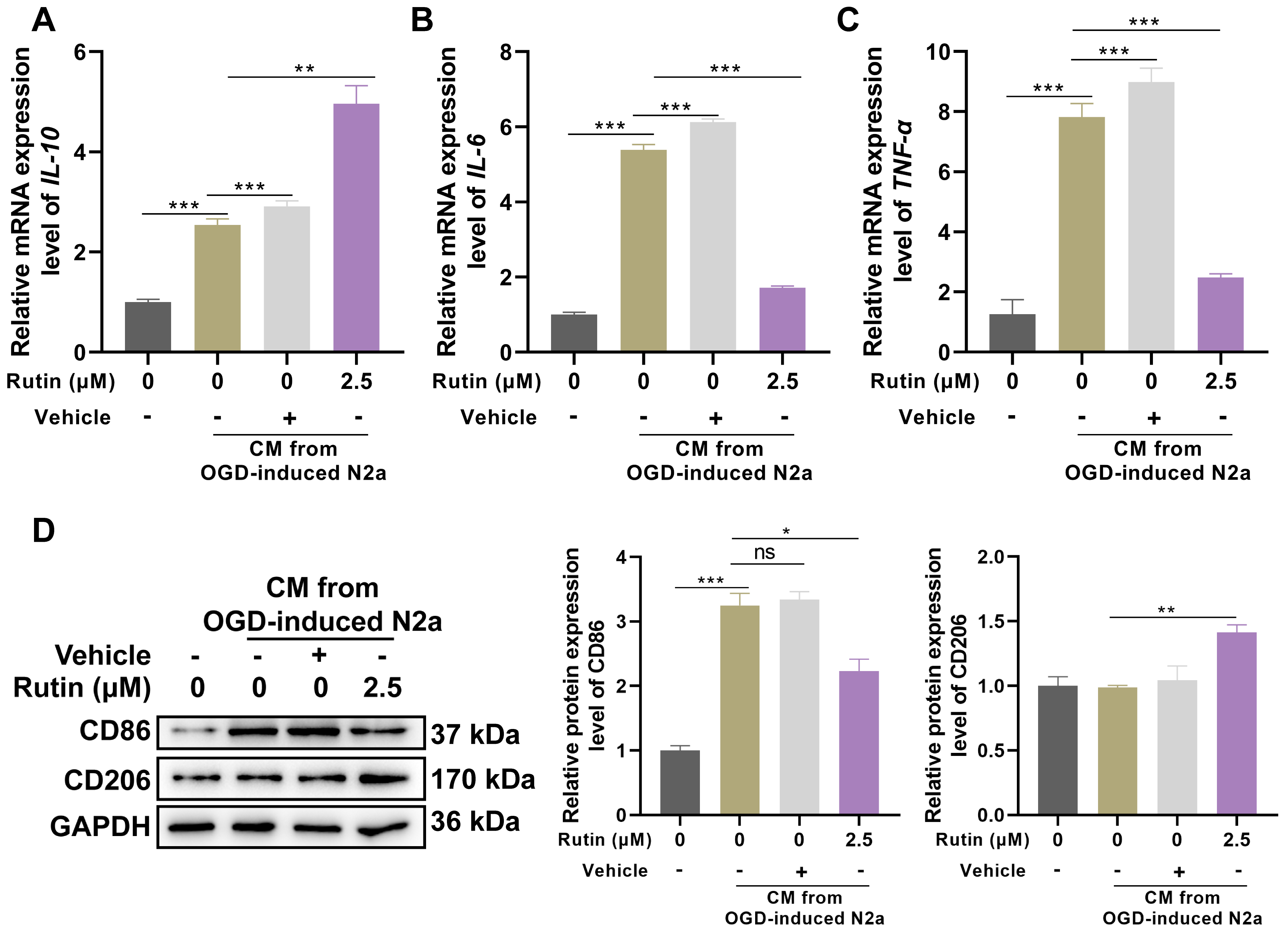 Fig. 3.
Fig. 3.
Rutin suppresses the inflammation and polarization of microglia
that respond to CM from OGD-treated neurons. BV2 cells were exposed to CM in the
presence or absence of 2.5 µM rutin for 24 hrs, (A–C) Real-time PCR
analysis was used to detect the expression of IL-10, IL-6 and
TNF-
The microglia were treated with CM alone, CM supplemented with vehicle or rutin for 24 hrs, followed by coculture with OGD-challenged neurons. After a 12 hrs period of co-incubation, the viability and apoptosis of neurons were determined (Fig. 4A). Cell viability was quantified using CCK-8 (Fig. 4B), which indicated a significant reduction in neuronal survival following exposure to OGD, with further decrement when neurons were cocultured with CM-exposed microglia. However, the detrimental effects on neuronal viability induced by CM-treated microglia were notably mitigated by the addition of rutin, as evidenced by a higher cell survival rate in the CM+rutin group compared to the CM+vehicle group. Moreover, apoptosis assays using Annexin V/PI staining (Fig. 4C,D) corroborated these findings. Neurons cocultured with CM and vehicle-treated microglia (CM+vehicle) demonstrated elevated levels of apoptosis compared to those observed in neurons maintained with normal microglia. Remarkably, the CM+rutin treatment significantly reduced the apoptotic cell count compared to the CM+vehicle treatment. These results demonstrated that rutin effectively mitigates the neurotoxic effects of pro-inflammatory microglia on neurons subjected to OGD.
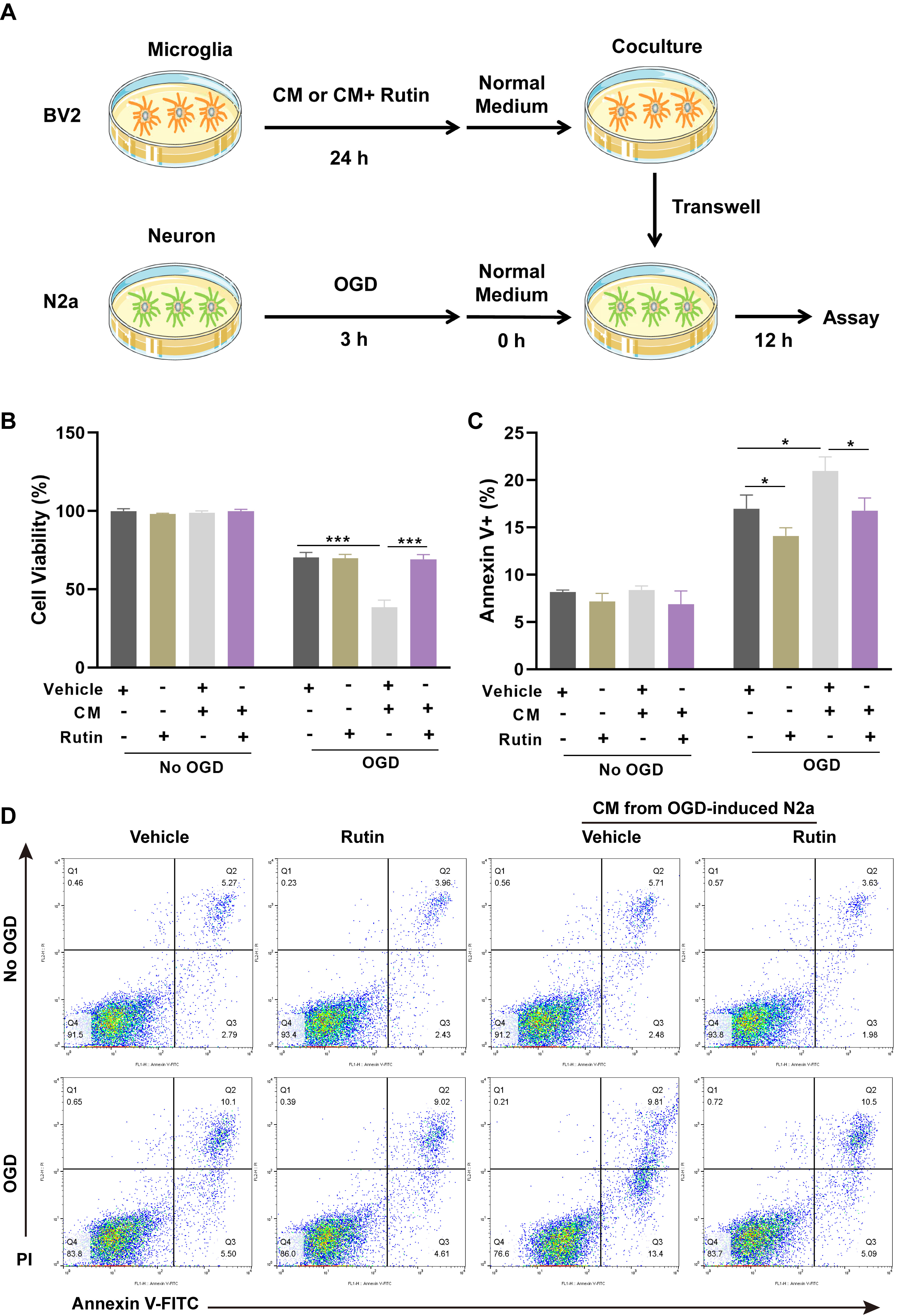 Fig. 4.
Fig. 4.
Rutin abolishes the neurotoxic effect of pro-inflammatory
microglia in cocultured neurons. (A) Schematic representation of the
experimental schedule for co-culturing BV2 microglial cells and OGD-challenged
N2A neurons. Microglia were treated with CM alone or supplemented with vehicle or
rutin for 24 hrs, followed by a 12 hrs co-incubation with OGD-treated neurons.
(B) After co-incubation, the cell viability of neurons was detected by CCK-8
assay. (C) Apoptosis in neurons was demonstrated by Annexin V/PI staining and (D)
Annexin V-FITC apoptosis assay. Data are presented as Mean
Ischemic stroke constitutes a significant health and socioeconomic burden, wherein the mitigation of neuronal cell death due to cerebral ischemia and hypoxia is crucial for alleviating the impact of the disease and enhancing patient prognosis [24]. This study delineated in vitro the pivotal role of rutin in modulating the STAT3 signaling pathway, thereby reducing neurotoxic effects under OGD conditions through the promotion of an anti-inflammatory phenotype in microglia. The insights into the mechanisms of rutin’s neuroprotective effects underscore its potential as a therapeutic agent for ischemic brain injuries and other neuroinflammatory disorders. These findings contributed to the understanding of cellular mechanisms that could be targeted to develop effective treatments for ischemic stroke and related conditions.
The OGD condition is extensively employed in in vitro studies of
ischemic stroke models to simulate the hypoxic and energy-deficient environment
encountered by brain tissue during ischemic events [25, 26]. By depriving cultured
neuronal cells of oxygen and glucose, CM effectively replicates the critical
conditions of ischemic stroke, allowing for the examination of how microglia
respond to signals released from neurons under ischemic-like conditions. In this
study, the exposure of microglia to CM resulted in a time-dependent activation of
inflammatory responses and a tendency toward a pro-inflammatory phenotype, which
was evidenced by the significant upregulation of pro-inflammatory cytokines such
as IL-6 and TNF-
The STAT3 is activated in response to extracellular signals and subsequently translocate to the nucleus to directly regulate the transcription of specific genes such as Bcl-2 and Bcl-xL to modulate cell death and MCP-1 involved in immune surveillance [27, 28]. Given the understanding of STAT3’s role in modulating the activity of microglia and its implications in neurological diseases, the development of therapeutic strategies targeting the STAT3 pathway has emerged as a focal point of research. Small molecule inhibitors of STAT3, such as WP1066 and S3I-201, have been tested in various studies to evaluate their neuroprotective effects in conditions such as cerebral ischemia, encephalitis, and neurodegenerative diseases [29, 30, 31]. The inhibition of STAT3 activation reduced the production of pro-inflammatory cytokines and mitigated neuroinflammation. The regulatory effects of certain natural compounds on STAT3 have also been highly esteemed due to their low toxicity and high biocompatibility compared to purely synthetic drugs. For instance, curcumin can alleviate spinal cord dysfunction brought by hypoxia and ischemia by inhibiting the JAK2/STAT3 signaling pathway [32]. Moreover, resveratrol also exerts a protective effect on neurodegenerative diseases by inhibiting the activation of microglia via upregulates the phosphorylated STAT3 [33]. Consistently, a reduction in the levels of phosphorylated STAT3 was also observed following treatment with rutin in our study, further substantiating its role in reducing STAT3 activation. Molecular structure analysis and docking suggest that the significant molecular size and complex structure of rutin allow it to occupy the catalytic chamber of the STAT3 protein, potentially altering the chemical properties or spatial configuration of key amino acid residues such as Lys631, Gln633, Pro715 and Thr721 and then inhibiting STAT3 activity and blocking its ability to activate pro-inflammatory genes. Furthermore, the multiple phenolic hydroxyl groups and other functional groups within the rutin molecule may form hydrogen bonds, hydrophobic interactions or van der Waals forces, enhancing the stabilization of its binding with the STAT3 protein. The strong binding of rutin to STAT3 at the molecular level effectively blocks the pro-inflammatory phenotypic transformation of microglia and facilitates their polarization towards an anti-inflammatory phenotype. This was evidenced in this study by a notable decrease in the expression of CD86, alongside a significant increase in the M2 phenotype marker CD206 [22]. Notably, a concentration of 2.5 µM rutin achieved a marked inhibition of the inflammatory response in microglia, providing a dose reference for related in vitro experiments.
Furthermore, when microglia treated with rutin were co-cultured with neurons under ischemic conditions, there was a significant enhancement in neuronal survival and a reduction in apoptosis levels, indicating rutin’s potential to mitigate microglia-mediated neurotoxicity. Therefore, the inhibition of STAT3 by rutin not only reduces the production of pro-inflammatory factors but also indirectly protects neurons from damage induced by inflammatory mediators. These findings were consistent with the neuroprotective effects of rutin observed both in nerve cell lines and in animal models [34, 35, 36]. Current results further confirm the pivotal role of the STAT3 pathway in microglia in this process.
The present study also has certain limitations. Although, the protective effects of rutin on ischemic neuronal injury was demonstrated in vitro, the efficacy and mechanisms of rutin in vivo remain to be validated through further animal experimentation. Specifically, comprehensive investigations into the bioavailability, dose-response relationships and long-term safety of rutin are imperative for a more complete understanding of its therapeutic potential and implications for clinical use.
This study illustrated that rutin effectively attenuates the activation of the JAK2/STAT3 signaling pathway in microglia exposed to CM from OGD-treated neurons, thereby inhibiting the pro-inflammatory phenotype and promoting a shift towards an anti-inflammatory state, which is conducive to neuronal survival and reducing apoptosis. These findings suggested that rutin holds substantial promise for enhancing the overall resilience of neurons to ischemic conditions via mitigating microglia-mediated neurotoxicity, offering a viable strategy for the treatment and management of ischemic stoke and diseases characterized by cerebral ischemia.
This study elucidates the neuroprotective role of rutin in mitigating microglia-mediated neuroinflammatory responses under ischemic conditions, specifically through its inhibitory effects on the STAT3 signaling pathway. Our findings enhance understanding of how rutin modulates microglial activation, shifting it from a pro-inflammatory to an anti-inflammatory state under conditions of oxygen-glucose deprivation, which subsequently offers potential benefits for attenuating the neurotoxic effect on neurons in an ischemic context. These provide a basis for using rutin as a non-toxic, anti-inflammatory and neuronal protective intervention aimed at improving treatment outcomes and the quality of life of patients with ischemic stroke and related cerebral injuries.
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
CL: Conceptualization, Investigation, Data Curation, Formal analysis, Validation, Writing - Original Draft; SHL: Conceptualization, Investigation, Data Curation, Formal analysis, Validation; YJJ: Conceptualization, Investigation, Data Curation, Formal analysis, Validation, Writing - Review & Editing. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by Liaoning Medical University Principal Fund (No. XZJJ20130218).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/IJP44190.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.