



 , Gang Li 2,*
, Gang Li 2,*
1 Department of Pharmacy, Inner Mongolia Medical University, 010110 Hohhot, Inner Mongolia Autonomous Region, China
2 Department of Pharmacology, Inner Mongolia Medical University, 010110 Hohhot, Inner Mongolia Autonomous Region, China
Abstract
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by β-amyloid (Aβ) plaque deposition, tau pathology, and mitochondrial dysfunction. Recent studies have found that the AMP-activated protein kinase (AMPK)/PTEN-induced kinase 1 (PINK1)/Parkin pathway plays a key role in the pathogenesis of AD by regulating energy metabolism, mitophagy, and neuroinflammation. As a core regulator of energy metabolism, AMPK enhances the efficiency of mitochondrial autophagy by phosphorylating PINK1, while inhibiting β-site amyloid precursor protein cleaving enzyme (BACE1) and reducing Aβ production. The PINK1/Parkin pathway selectively clears damaged mitochondria and alleviates oxidative stress and neuronal damage. This article systematically reviews the molecular mechanism of the AMPK/PINK1/Parkin pathway and its multi-target regulatory role in AD, and discusses the therapeutic strategies based on this pathway, to provide new directions for AD drug development.
Keywords
- Alzheimer’s disease
- AMPK
- PINK1
- Parkin
- mitophagy
- tau protein
- β-amyloid
Alzheimer’s disease (AD) is the most common type of dementia worldwide, with
clinical manifestations such as memory loss and cognitive dysfunction [1] and
pathological features such as deposition of
Adenosine monophosphate-activated protein kinase (AMPK) is a heterotrimeric
complex composed of three subunits:
PINK1, a mitochondria-localized serine/threonine protein kinase, also known as Parkinson disease 6 (PARK6), contains a mitochondrial targeting sequence at its N-terminus; the middle section has a kinase structure with kinase activity; the C-terminus contains a conserved region whose function is not yet fully understood. PINK1 is located on the outer mitochondrial membrane and plays a role in protecting cells from mitochondrial stress, acting as a probe for mitochondrial damage. Usually, PINK1 enters the mitochondria through the outer mitochondrial membrane and is subsequently cleaved by mitochondrial proteases and degraded in the mitochondrial matrix. However, when the mitochondrial membrane potential is disturbed (because of mitochondrial damage), the transport and cleavage of PINK1 is blocked, resulting in its accumulation and Ub molecules phosphorylation on the outer mitochondrial membrane [9]. The Ub molecule can interact with the Really Interesting New Gene 1 (RING1) structure in Parkin to activate Parkin, acting as a receptor molecule for Parkin recruitment. It is transferred from the cytosol to the mitochondria. This promotes the ubiquitination of mitochondrial outer membrane proteins and the activation of the ubiquitin proteasome system. In addition, studies have shown that PINK1 can also directly recruit autophagosomes and initiate weaker mitophagy [10].
The Parkin gene, also known as Parkinson Disease 2, Autosomal Recessive Juvenile (PARK2), is located at 6q26 on chromosome 6 [11, 12]. It comprises an N-terminal ubiquitin-like domain (UBL), a C-terminal RING1-IBR-RING2 domain, and a RING0 domain, and is part of the interring ring domain family of ubiquitin ligases [13]. The UBL domain can interact with other proteins to regulate the activity and localization of Parkin. The RING1-In Between Really Interesting New Gene fingers (IBR)-Really Interesting New Gene 2 (RING2) domain constitutes the E3 ubiquitin ligase activity center of Parkin and catalyzes the ubiquitination of substrate proteins. Following PINK1 activation, phosphorylated Parkin is selectively recruited to the surface of damaged mitochondria, where it attaches to substrate proteins in the outer mitochondrial membrane through its E3 ubiquitin ligase, forming ubiquitin chains that ubiquitinate outer mitochondrial membrane proteins [14]. These ubiquitinated substrate proteins recruit autophagy-related proteins (e.g., P26, Nuclear Dot Protein 52 kDa (NDP52)) to mediate mitochondrial coating by autophagosomes, which in turn initiates mitophagy and ultimately degrades damaged mitochondria.
Beyond its role in mitochondrial fission and ULK1 activation, AMPK critically intersects with the PINK1/Parkin pathway to orchestrate mitochondrial quality control and cell survival. AMPK directly phosphorylates PINK1 serine residues, stabilizing the kinase and enhancing its activity to initiate Parkin-mediated mitophagy [9]. This AMPK-PINK1 axis facilitates an “energy stress-induced mitochondrial repair” mechanism for targeted removal of damaged organelles. Under energy deprivation, AMPK and PINK1/Parkin function synergistically to eliminate dysfunctional mitochondria, thereby preserving cellular energy homeostasis.
As can be seen in Fig. 1, AMPK further amplifies PINK1/Parkin signaling indirectly through mammalian Target Of Rapamycin Complex 1 (mTORC1) inhibition [15]. By phosphorylating Tuberous Sclerosis Complex 2 (TSC2) and activating the Tuberous Sclerosis Complex 1 (TSC1)/TSC2 complex, AMPK suppresses mTORC1 activity. This relieves mTORC1-mediated repression of autophagy initiation and concurrently activates the transcription factor Transcription Factor EB (TFEB). Nuclear TFEB upregulates autophagy- and lysosome-related genes, enhancing cellular clearance capacity. Critically, targeted activation of the AMPK-PINK1 node addresses the dual AD pathologies of bioenergetic failure and impaired mitochondrial clearance, offering a potentially safer alternative to multi-target therapeutics.
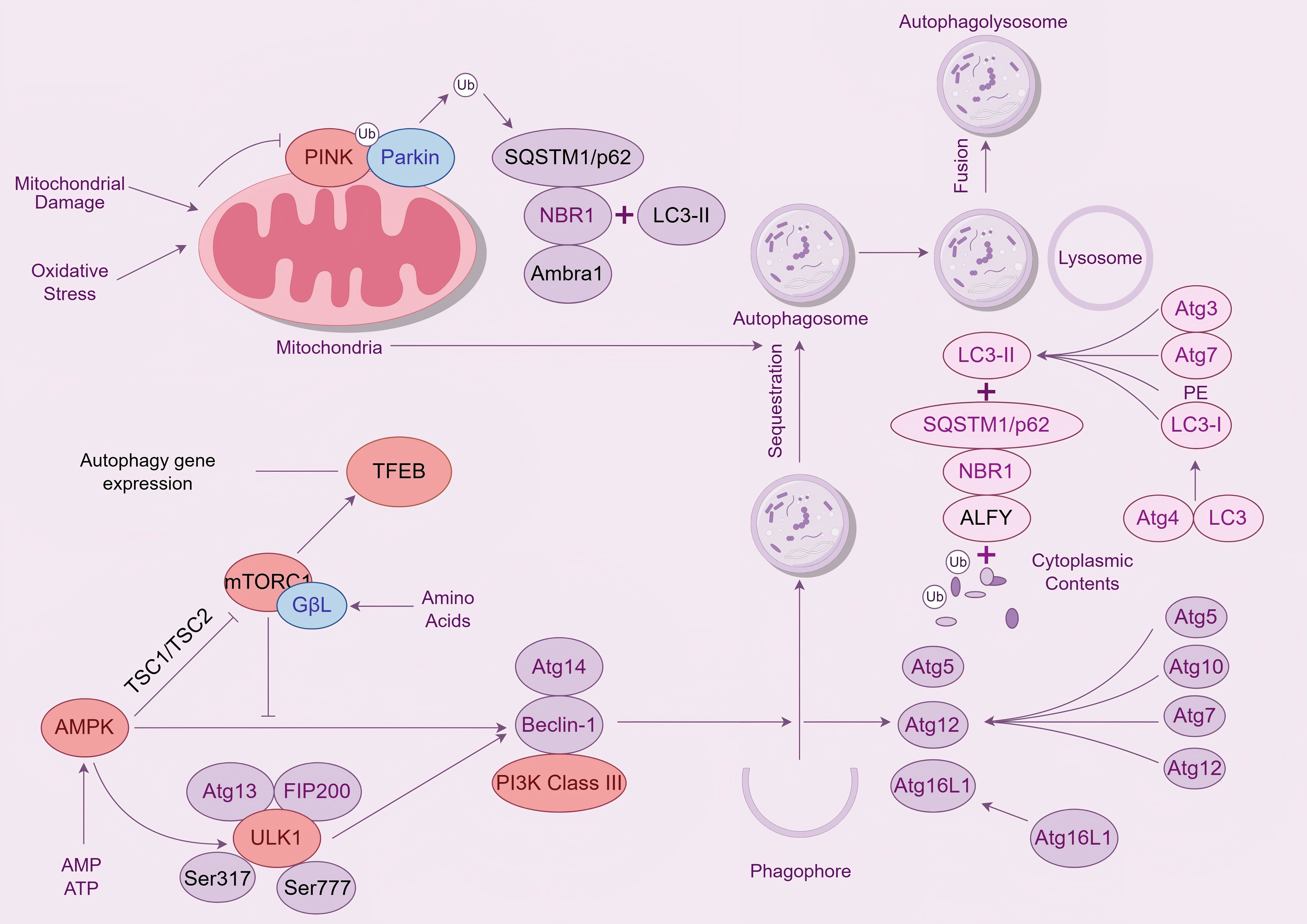 Fig. 1.
Fig. 1.
AMPK/PINK1/Parkin signaling pathway. Legend: The diagram shows the regulation of mitophagy. When mitochondria are damaged by oxidative stress, PINK1 and Parkin are activated, initiating the process of mitophagy. Simultaneously, the mTORC1 signaling pathway can sense nutrient signals such as amino acids and inhibit the expression of autophagic genes. The AMPK signaling pathway, on the other hand, is activated when energy is insufficient, promoting the formation of autophagy-related protein complexes. Autophagy-related proteins, such as members of the Atg family, play an important role in the formation of autophagosomes, and LC3 is processed to form LC3-II, which binds to proteins such as SQSTM1/p62 and participates in the encapsulation of mitochondria and other substances by autophagosomes. Autophagosomes fuse with lysosomes to form autophagolysosomes, which degrade mitochondria. AMPK, AMP-activated protein kinas; PINK1, PTEN-induced kinase 1; LC3, Microtubule - associated protein 1 light chain 3; SQSTM1, Sequestosome 1; TFEB, Transcription Factor EB; ALFY, Autophagy-linked FYVE protein; NBR1, NBR1 autophagy cargo receptor; TSC1, TSC complex subunit 1; ULK1, unc-51 like autophagy activating kinase 1.
In the brains of patients with AD, mitochondrial dysfunction leads to the accumulation of damaged mitochondria, which cannot produce energy normally and also release large amounts of ROS, further exacerbating oxidative stress and neuronal damage. Fig. 2 shows the relationship between AMPK/PINK1/Parkin pathway and AD pathology. Activation of the AMPK/PINK1/Parkin pathway can promote mitophagy, remove damaged mitochondria in time, reduce ROS production, and thus protect neurons from oxidative stress damage. In mouse models of AD, activation of the AMPK/PINK1/Parkin pathway could significantly increase the level of mitophagy, improve mitochondrial function, and alleviate cognitive impairment.
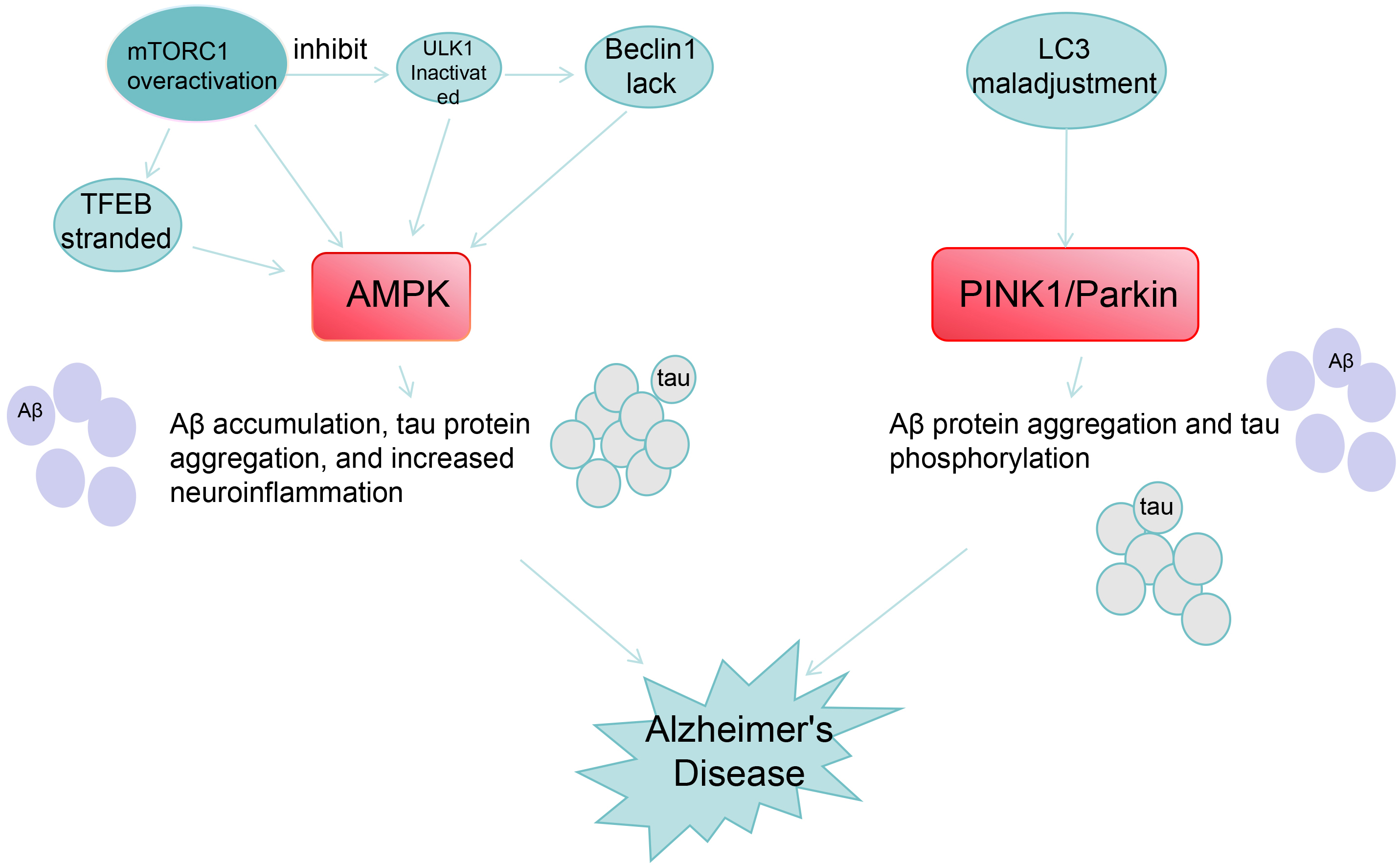 Fig. 2.
Fig. 2.
Relationship between AMPK/PINK1/Parkin and AD pathological
processes. Legend: This diagram shows the molecular mechanisms of two pathways
associated with the pathogenesis of AD: AMPK pathway: Overactivation of
mTORC1 inhibits ULK, leading to Beclin1 deficiency and TFEB retention, acting on
AMPK to triggers A
AMPK mainly regulates A
AMPK reduces tau hyperphosphorylation by inhibiting Glycogen Synthase
Kinase-3
AMPK inhibits the interference of the Nuclear Factor kappa-light-chain-enhancer
of Activated B cells (NF-
Since the occurrence of neurological diseases such as AD is closely related to mitophagy dysfunction, the regulation of mitophagy may have a beneficial effect on the treatment of such diseases. Fig. 3 below is a timeline of studies on the treatment of AD through the AMPK/PINK1/Parkin pathway, which can be traced back to as early as 2004.
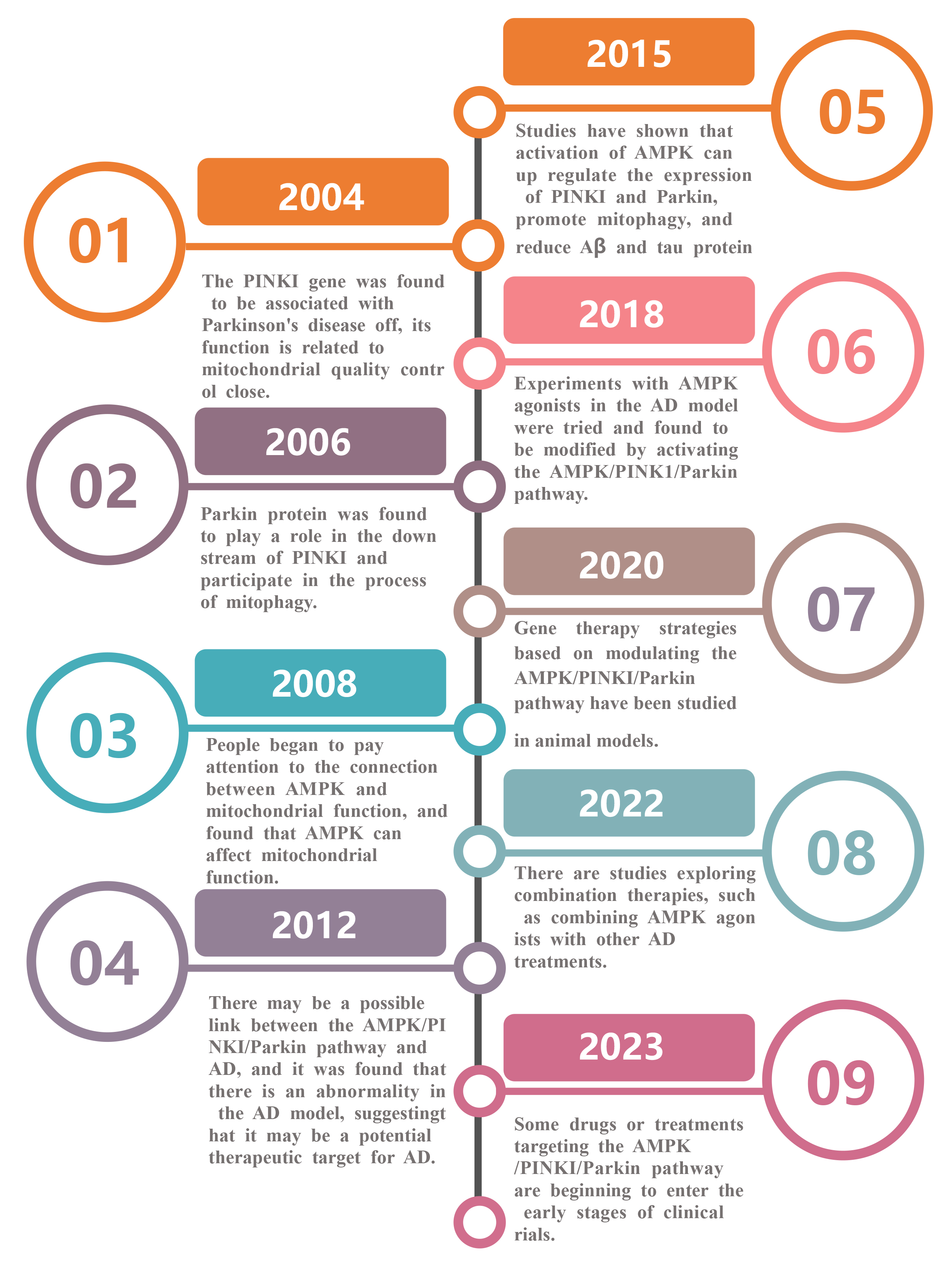 Fig. 3.
Fig. 3.
Timeline of studies on the role of AMPK/PINK1/Parkin pathway for the treatment of AD. Legend: This is a timeline of the progress of research on the AMPK/PINK1/Parkin pathway, from the discovery of the role of PINK1 gene in Parkinson’s disease (2004) to the elucidation of the role of various proteins in the pathway, exploring the connection with mitochondria and AD, conducting animal model studies, and attempting combination therapy, until the progress of drugs or therapies to the early stage of clinical trials can be achieved (2023).
AMPK can simultaneously regulate A
Cell experiments have shown that the AMPK agonist AICAR can increase the expression of pAMPK and pULK1 in APP transgenic cell line 20E2, regulate the expression of division- and fusion-related proteins, and improve mitophagy and mitochondrial dynamics abnormalities resulting from APP gene transfection [27]. Although specific AMPK agonists are yet to be developed, the use of AMPK activators to promote mitophagy and ameliorate AD may be a feasible treatment option in the future.
A
PINK1/Parkin enhancers mainly include natural compounds (e.g., urolithin A [29]) and small molecule compounds (e.g., trehalose), which can improve mitophagy and alleviate pathological features of neurodegenerative diseases by activating the PINK1/Parkin pathway. For example, MTK-458 can directly stimulate PINK1 and accelerate mitochondrial degradation and has entered phase I clinical trials. Kinetin (a molecule known to modulate splicing) [30] acts as a kinase modulator that activates Cdc2-like kinase 1 (CLK1). This activation phosphorylates arginine-serine-rich domains within splicing factors, leading to enhanced Parkin activity and subsequently enabling efficient recognition of damaged mitochondria. Some protein kinases such as casein kinase 2 (CK2), which can phosphorylate PINK1 and regulate its stability and activity, also regulate this pathway. The cytokinin kinetin N6-furfuryladenine, an adenosine-derived plant hormone, enhances PINK1 autoactivation, promotes Parkin recruitment to depolarized mitochondria, and blocks mitochondrial motility in axons [31]. Experimental studies have found that rapamycin, an mTOR inhibitor, can also activate mitophagy, enhance the downstream PINK1/Parkin pathway, reduce neuronal loss, and improve cognitive impairment [15].
Overexpression of the gene encoding
Through the comparison of the three drugs in Table 1 [18, 33, 34, 35], we can see that metformin, in AD-related trials, has mainly targeted patients with mild cognitive impairment (prodromal stage), and its phase II pilot study showed a positive effect on memory; the latest MAP trial is an adaptive design phase II/III study. AICAR is currently only an experimental AMPK activator, with no trials conducted so far on humans with AD; MTK-458, a PINK1 activator, is still in preclinical research for treatment of neurodegenerative diseases. The above content is mainly based on public clinical trial registrations and research reports, and no reports indicate that AICAR or MTK-458 have undergone human trials targeting AD.
| Drugs | Clinical research phase | Treatment window | Safety analysis | References |
| Metformin | Phase II/III | In the prenatal period or the stage of mild cognitive impairment (aMCI). | Common gastrointestinal reactions (diarrhea, nausea in 10% of patients). | Luchsinger et al. [34] |
| Very rare lactic acidosis. | Tahmi and Luchsinger [35] | |||
| Rare vitamin B12 deficiency. | ||||
| AICAR | Preclinical stage | No human data: early intervention may theoretically be effective. | No human safety data; Overactivation of AMPK can be detrimental (e.g., neuronal damage). | Hu et al. [18] |
| MTK-458 | Preclinical stage | No human data. | No human safety data; To be evaluated before a clinical trial. | Hertz et al. [33] |
AMPK, Adenosine monophosphate-activated protein kinase; AICAR, 5-Aminoimidazole-4-carboxamide ribonucleotide.
A combination of AMPK activators (e.g., AICAR) and PINK1/Parkin activators
(e.g., MTK-458 [33]) have been investigated in animal and cell model experiments.
AICAR can increase the intracellular AMP/ATP ratio, activate AMPK, and promote
energy metabolism and autophagy initiation. MTK-458 can directly stimulate PINK1
and accelerate mitochondrial degradation. The combination of the two can improve
the overall energy status of cells on the one hand, enhance mitophagy on the
other hand, and effectively remove the accumulation of misfolded proteins and
damaged mitochondria in the AD model, reduce neurotoxicity, and improve neuronal
function. Gene therapy can also be used in combination with drugs to overexpress
the PINK1 or Parkin genes and generate AMPK activators. The combination of the
two can reduce the formation of A
The AMPK/PINK1/Parkin pathway is intertwined with multiple other signaling
pathways to form a complex regulatory network. Therefore, strategies to avoid
affecting other normal physiological functions while leveraging the regulation of
this pathway in AD treatment are necessary. Overactivation of AMPK may interfere
with pathways such as mTOR and inhibit synaptic protein synthesis. Moreover, most
AMPK activators (e.g., AICAR) have poor blood-brain barrier penetration.
Excessive inhibition of BACE1 can affect physiological processes such as
myelination. The therapeutic effect in AD is phase-dependent, with early
targeting of the AMPK-PINK1 axis preventing mitochondrial injury. However,
mitochondrial damage may be irreversible at later stages and requires A
With the aging of the global population, maintaining g healthy life expectancy
has gradually become important, especially in older adults with degenerative
mental illnesses such as AD. The AMPK/PINK1/Parkin pathway plays an important
neuroprotective role in AD by regulating mitophagy, energy metabolism, and
neuroinflammation. Therefore, it has great potential for the treatment of
neurodegenerative diseases. AMPK enhances PINK1/Parkin-mediated mitophagy by
directly regulating PINK1 and activating ULK1, thereby alleviating A
YJ and GL are responsible for the conception and design of articles, collection and collation of research materials, and drafting the manuscript. YJ and GL are responsible for the revision of the manuscript, quality control and review of the manuscript, and overall supervision and management. Both authors contributed to editorial changes in the manuscript. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We would like to express our gratitude to all those who assisted with the writing of this manuscript, and to the peer reviewers for their opinions and suggestions.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.