




 , Xue Bai 1,2,*
, Xue Bai 1,2,*1 Department of Radiation Oncology, Nanfang Hospital, Southern Medical University, 510515 Guangzhou, Guangdong, China
2 The First School of Clinical Medicine, Southern Medical University, 510515 Guangzhou, Guangdong, China
Abstract
Cancer-associated fibroblasts (CAFs) are key components of the tumor microenvironment that drive tumor growth, survival and therapeutic resistance. Although CAFs have long been viewed as a single stromal population, accumulating evidence indicates that they occupy diverse metabolic states shaped by local nutrients and tumor-derived signals. Recent studies across cancer types have described several recurring metabolic programs. In this review, we summarize five dominant CAF metabolic states as a functional, context-dependent framework: glycolytic, oxidative, fatty acid–oxidizing, lipid-rich, and amino acid–remodeling. These states reflect how CAFs adapt to local metabolic conditions. Collectively, CAF metabolic programs are linked to tumor support and immunosuppressive features, highlighting stromal metabolism as a potential therapeutic vulnerability.
Graphical Abstract

Keywords
- cancer-associated fibroblasts
- tumor microenvironment
- metabolic reprogramming
- glycolysis
- oxidative phosphorylation
- lipid metabolism
- amino acids
Tumors are complex ecosystems in which cancer cells, stromal cells and immune cells compete for nutrients while also engaging in metabolic cooperation [1, 2]. Metabolic reprogramming is a hallmark of cancer and supports the biosynthetic and energetic demands of rapid proliferation [3]. Nearly a century ago, Otto Warburg reported that many tumor cells preferentially rely on glycolysis even in the presence of oxygen, a phenomenon termed the “Warburg effect” [4]. This shift can supply metabolic intermediates required for tumor growth and division.
However, cancer metabolism is not confined to malignant cells. Stromal populations within the tumor microenvironment (TME) also undergo metabolic changes to support tumor progression [5]. Among them, cancer-associated fibroblasts (CAFs) are abundant and highly versatile. Rather than serving only structural roles, CAFs actively shape the metabolic landscape of tumors [6].
Recent studies show that CAFs occupy multiple metabolic states rather than a single uniform phenotype [7, 8]. In response to local nutrient availability and tumor-derived cues, CAFs can adopt glycolytic, oxidative, fatty acid–oxidizing, lipid-rich or amino acid–remodeling states. These specialized programs enable CAFs to supply metabolites, modulate immune activity and remodel the extracellular matrix (ECM). In this review, we synthesize recent work on CAF metabolic diversity and discuss its implications for tumor progression and immune evasion.
As a primary source of energy, glucose metabolism represents a central axis of CAF metabolic reprogramming [9]. In the following sections, we discuss how CAFs reprogram glycolysis and oxidative phosphorylation (OXPHOS) to support tumor–stroma metabolic coupling.
CAFs arise from several stromal origins, including resident normal fibroblasts (NFs), mesenchymal stem cells and pericytes, contributing to marked phenotypic and functional heterogeneity [10]. Metabolic reprogramming of CAFs is especially evident in glucose utilization, which is closely linked to tumor progression.
Under hypoxic conditions, perivascular CAFs adapt to metabolic stress by increasing glycolytic activity, which can help maintain redox balance and energy production [11, 12, 13]. This pattern reflects a Warburg-like phenotype and is commonly used to define the glycolytic CAF (glyCAF) state (Association/Inference). In pancreatic ductal adenocarcinoma (PDAC), single-cell RNA sequencing identified a CAF subset with high expression of glycolytic genes, whereas neighboring tumor cells show greater reliance on OXPHOS [14].
However, not all CAFs adopt a glycolytic phenotype. Metabolic heterogeneity exists across tumor types and within individual lesions. In contrast to glyCAFs, CAF subsets with an OXPHOS phenotype have been supported by functional assays across PDAC, prostate cancer, OSCC, and BCa (bladder cancer), showing high spare respiratory capacity and increased ATP production (Mechanistic) [15, 16, 17, 18].
Despite their oxidative metabolism, these CAFs still support tumor progression (Fig. 1a) (Mechanistic). In some contexts, upregulation of pyruvate carboxylase allows tumor-derived lactate to support nonessential amino-acid synthesis. These metabolites can then support CAF collagen production [17]. The accumulation of dense ECM may further exacerbates hypoxia and dampen immune activation [19]. In addition, oxidative CAFs release paracrine cytokines that influence both cancer and immune compartments. Among these factors, IL-8 is associated with increased PD-L1 expression in tumor cells and immune-evasive features [20]. IL-6 is linked to reduced T-cell and NK-cell cytotoxicity and dampened antitumor immunity [15]. Furthermore, hepatocyte growth factor (HGF) secreted by oxidative CAFs activates MET-dependent signaling in cancer cells, promoting tumor growth and therapeutic resistance [21].
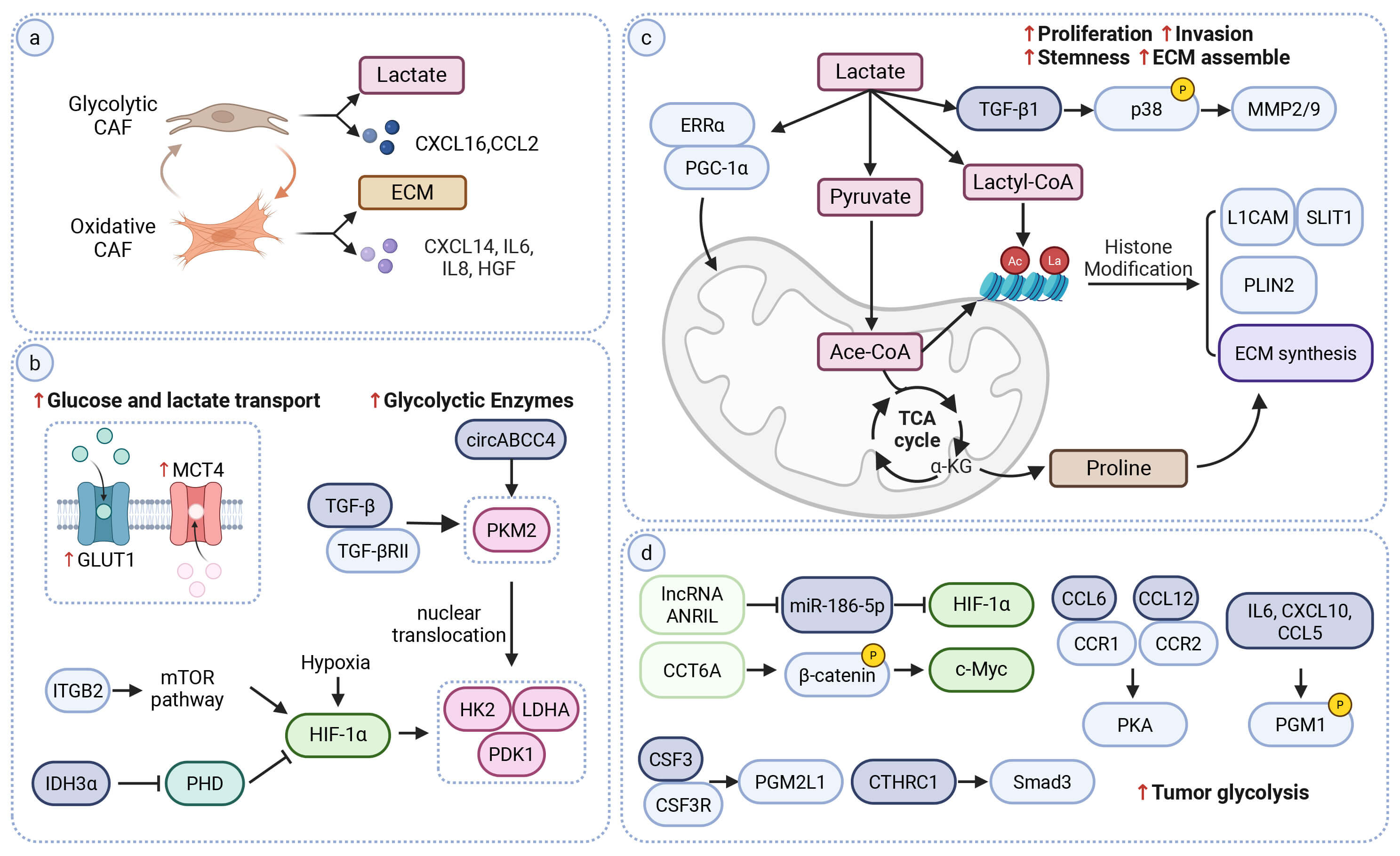
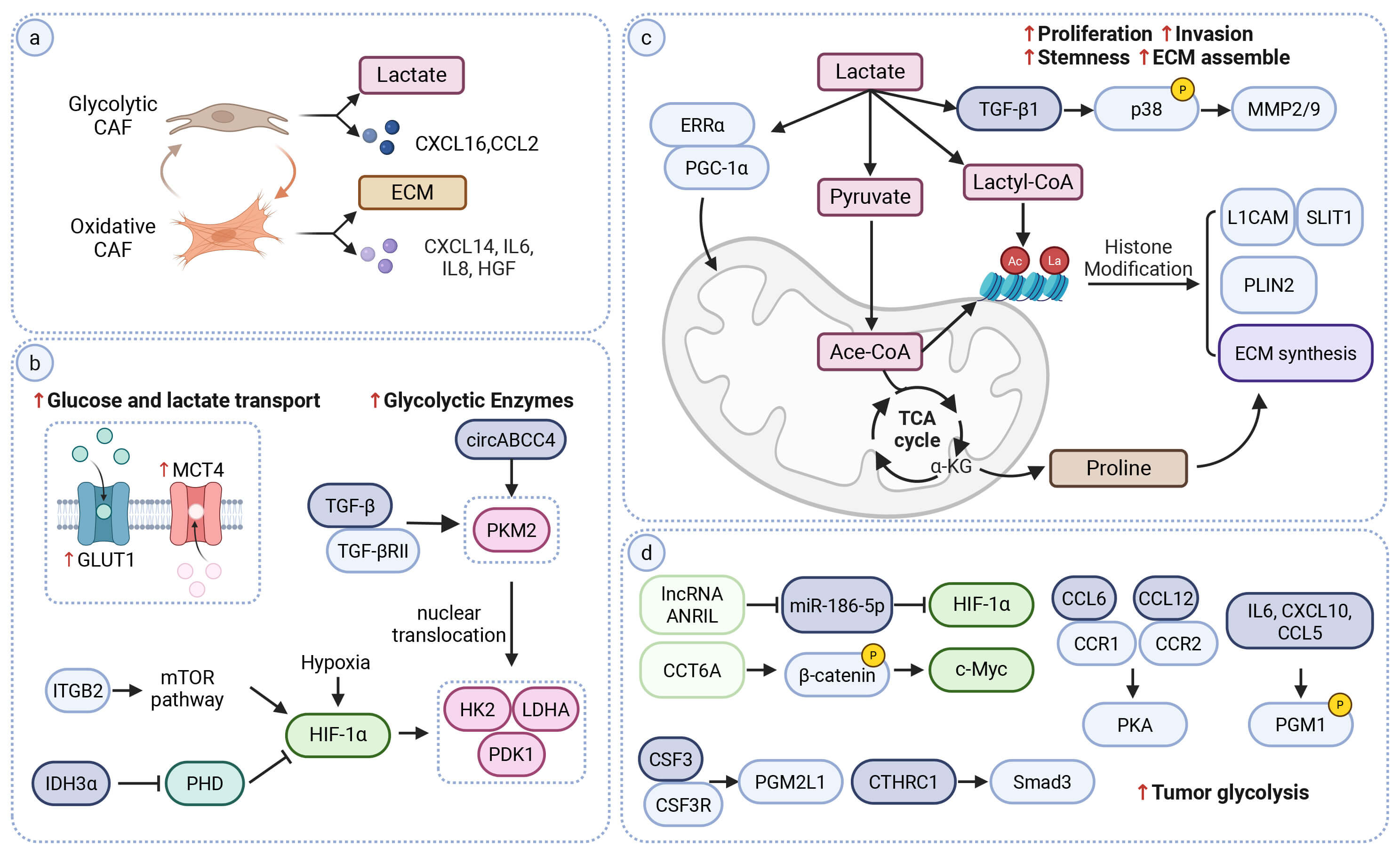 Fig. 1.
Fig. 1.
Metabolic and signaling mechanisms of CAF-associated glucose
reprogramming and tumor–stroma coupling. (a) Overview of CAF glucose metabolic
states. Glycolytic CAFs (glyCAFs) show increased glycolysis and lactate export,
and are associated with chemokines such as CXCL16 and CCL2. Oxidative CAFs show
high OXPHOS programs and are associated with factors including IL-6, IL-8, CXCL14
and HGF. These states correlate with tumor-supportive, immune-modulatory and
extracellular-matrix (ECM) remodeling features. (b) Regulatory nodes linked to
glycolytic activation in CAFs. Increased GLUT1 and MCT4 are associated with
enhanced glucose uptake and lactate export. Integrin-related pathways (e.g., ITGB2–mTOR) are
linked to HIF-1
Glycolytic and oxidative CAF states are metabolically coupled with tumor cells. At the same time, CAF metabolism remains highly flexible and adjusts to the local microenvironment [22]. These features suggest that CAF glucose metabolism may exist along a continuum from highly glycolytic to predominantly oxidative states. Microenvironmental cues often determine where CAFs fall along this axis. Hypoxia and nutrient deprivation tend to push CAFs toward a glycolytic state [23]. This metabolic plasticity highlights the need for context-specific analyses of CAF metabolism across different tumor settings (Table 1).
| States | Dominant pathway engagement | Key molecular markers | Environmental context | Primary functional output |
| Glycolytic | Glycolysis | GLUT1, MCT4, HIF-1 |
Hypoxia, high glucose demand | Aerobic glycolysis and lactate export |
| Oxidative | Oxidative phosphorylation | Elevated mitochondrial respiration, high spare respiratory capacity | Lactate-rich or oxygenated niches | Collagen synthesis, ECM remodeling |
| FAO | Fatty-acid oxidation | Fatty-acid uptake and mitochondrial |
Low-glucose conditions | Energy conservation, CAF activation |
| Lipid-rich | Lipogenesis | Lipogenesis enzymes (ACLY, FASN, SCD1), Lipid droplet accumulation | Oncogenic signaling or growth-factor stimulation | Lipid metabolite provisioning, angiogenesis |
| AA-remodeling | Amino-acid metabolism | Glutamine, arginine and tryptophan metabolic enzymes (GLS, GS, ASNS, PYCR1, NNMT, CD73, TDO2, ARG1/2) | Amino acid scarcity, mechanical stiffness | Nutrient shuttling, immune suppression |
ACLY, ATP citrate lyase; ACSL, acyl-CoA synthetase long-chain family; ARG1/2,
arginase 1/2; ASNS, asparagine synthetase; CPT1A/C, carnitine
palmitoyltransferase 1A/1C; FASN, fatty acid synthase; GLS, glutaminase; GS,
glutamine synthetase; HIF-1
Mechanistically, glyCAF-associated features are supported by multiple regulatory
pathways (Fig. 1b) (Mechanistic). CAFs upregulate GLUT1 to increase glucose
uptake and MCT4 to facilitate lactate efflux [24, 25]. A central node in this
reprogramming is HIF-1
This network also intersects with PKM2 signaling. TGF-
In parallel, TGF-
Glycolytic CAFs are a major source of extracellular lactate. Tumor cells can import lactate and use it to support mitochondrial OXPHOS, creating a directional metabolic flow from the stroma to the tumor [35]. 13C-glucose isotope tracing together with mechanistic experiments supports stromal-to-tumor lactate utilization, showing that CAF-derived lactate is taken up by tumor cells and fuels the TCA cycle (Mechanistic/Flux-supported) [36, 37].
Once considered a simple by-product of glycolysis, lactate is now recognized as
both a metabolic substrate and a paracrine signal (Fig. 1c). Tumor cells take up
lactate mainly through MCT1, which can reinforce stromal–tumor metabolic
coupling [38]. Across tumor types, CAF-derived lactate has been linked to
activation of multiple oncogenic pathways (Mechanistic). In ovarian cancer, it
promotes tumor-cell proliferation and migration through
TGF-
Lactate also contributes to epigenetic and metabolic remodeling in tumor cells. In PDAC, CAF-derived lactate induces histone lactylation. This modification increases the expression of neural invasion–related genes such as L1CAM and SLIT1 and promotes perineural invasion [41]. In prostate cancer, lactate uptake expands the acetyl-CoA pool and increases histone acetylation. This change upregulates lipid-metabolism genes such as PLIN2 and reshapes fatty-acid, which may enhance tumor invasiveness [42]. Histone acetylation also increases chromatin accessibility at ECM- related loci [43].
Recent pan-cancer spatial multi-omics studies indicate that CAF metabolic states
are spatially patterned. This niche-level organization is referred to as spatial
metabolic heterogeneity [44]. In oral squamous cell carcinoma (OSCC), fibroblasts
within glycolytic niches show a HIF-1
In hepatocellular carcinoma (HCC), spatial metabolomics further delineated a tumor–immune–stromal interface enriched in lactate-producing CAFs [46]. In this region, elevated lactate is associated with CCL2-dependent recruitment of tumor-associated macrophages (TAMs) and has been linked to M2-like polarization [46, 47]. CXCL16 has also been implicated in reduced CD8+ T-cell presence in the tumor core [48]. Collectively, these observations support that spatially confined glyCAF-like niches can coincide with stromal barrier features and immunosuppressive contexture (Fig. 1a) (Association/Inference).
Translational Implications: These findings suggest that lactate transport and glycolysis-linked signaling in CAF-rich niches may represent actionable vulnerabilities [35]. MCT1/MCT4 blockade (e.g., AZD3965) offers a strategy to disrupt stromal-tumor coupling. High stromal MCT4 expression may serve as a stratification marker to enrich for responders [25]. In tumors with lactate-rich niches accompanied by immunosuppressive features, these observations support evaluating metabolic inhibitors in combination with immunotherapy.
Beyond direct metabolite exchange, CAF-derived paracrine signals form networks that couple stromal and tumor metabolism. Through soluble factors and extracellular vesicles (EVs), CAFs engage multiple signaling pathways that influence tumor glycolysis and OXPHOS (Fig. 1d).
Among these signals, glycolysis-amplifying feedback loops are well
characterized. In bladder cancer, CAF-secreted CXCL14 binds CCR7 on tumor cells
and activates STAT3, leading to upregulation of HK2 and LDHA and increased
glycolysis. The resulting rise in lactate promotes NF-to-CAF conversion and
sustained CXCL14 production, which can strengthen chemoresistance [49]. In lung
cancer, CTHRC1+ CAFs stimulate TGF-
Additional CAF-derived mediators, including IL-6, CSF3, WNT5a, and CRMP2, act on tumor cells and activate regulators such as HIF-1
In addition to soluble cytokines, CAF-derived EVs deliver metabolites, enzymes
and noncoding RNAs that further shape tumor-cell metabolism. For example,
exosomal lncRNAs such as ANRIL, LINC01711, and NNT-AS1
have been reported to stabilize HIF-1
Lipid metabolism is another major axis of CAF metabolic plasticity that supports tumor growth and survival [59]. Evidence across tumor types shows that CAFs undergo lipid metabolic reprogramming. This reprogramming gives rise to two major lipid-related CAF phenotypes: fatty acid–oxidizing (FAO) CAFs, which rely on FAO to meet their energy demands, and lipid-rich CAFs, which accumulate and mobilize lipids to support tumor cells.
To limit competition with cancer cells for nutrients, some CAFs shift their
energy reliance from glycolysis to FAO, thereby maintaining metabolic balance and
supporting tumor progression (Fig. 2a) (Mechanistic). For example, in HCC, CAFs
express higher levels of the fatty-acid transporter CD36 than adjacent NFs.
Blocking CD36 reduces CAF proliferation, migration and activation markers such as
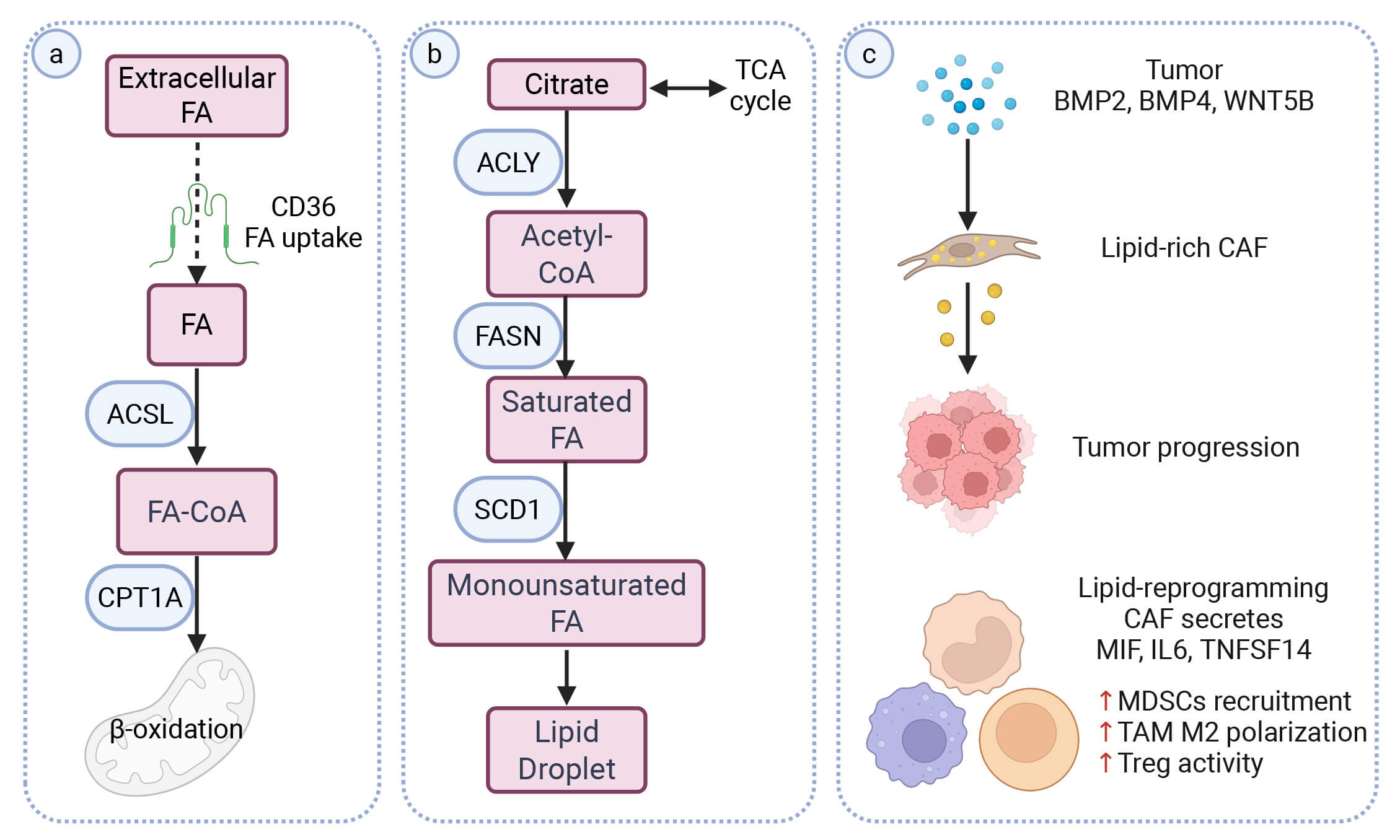 Fig. 2.
Fig. 2.
CAF-driven lipid metabolic programs and immune
contexture. (a) Fatty-acid uptake and
In contrast to FAO CAFs, lipid-rich CAFs accumulate lipid droplets (LDs) (Fig. 2b). These LDs function as an intracellular lipid reservoir. The stored lipids can be mobilized through lipolysis or lipophagy and exported from CAFs to tumor cells [7] (Mechanistic/Flux-supported). Lipid-rich CAFs arise through tumor-driven metabolic reprogramming and the accumulated mobilized lipids support the secretion of pro-tumorigenic factors (Fig. 2c) (Mechanistic). In KRAS-mutant CRC, tumor-derived BMP4 and WNT5B have been shown to drive the formation of lipid-rich CAFs that release VEGFA, HGF and other mediators to promote angiogenesis and tumor progression [62]. In SETD2-deficient PDAC, tumor-derived BMP2 induces lipid-laden CAFs with increased ABCA8a expression [63]. Lipids transferred from these CAFs increase acetyl-CoA in tumor cells and enhance H3K27 acetylation, which is associated with increased BMP2 production and a positive feedback loop [63].
A related circuit operates during pre-metastatic niche formation in CRC. Tumor-derived exosomal HSPC111 activates ACLY in hepatic fibroblasts, raising acetyl-CoA and H3K27 acetylation and inducing CXCL5 expression. CXCL5–CXCR2 signaling then promotes HSPC111 release from tumor cells, thereby reinforcing liver metastasis [64]. In OSCC, CAFs also upregulate ACLY to support de novo fatty-acid synthesis and ECM production [65]. In LUAD and PDAC, CAFs can activate lipid-storage programs through SCD1 and FASN, generating lipid-rich CAFs that support tumor metabolism and growth [7, 66].
Beyond LD-associated lipid storage, CAFs also secrete soluble lipid mediators that can directly support tumor survival. (Table 1) CAF-derived lysophosphatidylcholines (LPCs) are taken up by tumor cells to replenish membrane phosphatidylcholines and relieve ER stress or they are converted by autotaxin (ATX) into lysophosphatidic acid (LPA) to promote proliferation [67, 68].
Beyond fueling tumor growth and invasion, CAF lipid metabolism also shapes the immune landscape of the TME (Fig. 2c). A FAO CAF subset with high CPT1C expression is enriched in immunosuppressive tumors and promotes immune suppression through a FAO–IL-6–M2 macrophage axis [69]. Other lipid-metabolic CAF states, which are not defined by FAO or lipid accumulation, also modulate immune responses. In steatotic liver disease–related HCC, lipid metabolic reprogramming strengthens CAF–Treg interactions through the TNFSF14–TNFRSF14 axis [70]. This shift is associated with reduced CD8+ T-cell activity and increased Treg frequency. It also correlates with diminished responsiveness to immunotherapy [70]. In HCC, CAFs expressing multiple lipid-related genes secrete MIF to recruit MDSCs via CD74, consistent with an immunosuppressive niche [71]. Lipid metabolism in CAFs can also create conditions that favor antitumor immunity. In CRC, exogenous ketone supplementation suppresses KLF5 in CAFs and lowers their secretion of CXCL12. This reduction in CXCL12 limits the recruitment of immunosuppressive cells and improves responses to anti–PD-1 therapy [72]. Collectively, these studies illustrate the immunologic plasticity of CAF lipid metabolism and point to metabolic interventions as a potential strategy to improve antitumor immunity.
Translational Implications: Lipid-related CAF programs point to several potentially druggable nodes, including fatty-acid uptake (e.g., CD36) and lipogenesis pathways (e.g., ACLY/FASN) [60, 66]. Patients may be stratified by stromal lipid accumulation and CD36-high CAF signatures, ideally using spatial profiling to capture lipid-rich niches. These features provide a rationale to combine lipid-targeting strategies together with immunotherapy.
CAFs reprogram tumor-cell lipid metabolism through secreted factors and extracellular vesicles. In ALK-rearranged LUAD, CAF-derived HGF and neuregulin 1 (NRG1) activate AKT signaling in tumor cells, increasing de novo lipogenesis and reducing in ways that support survival and therapy resistance [73]. In aged melanoma, elevated IGFBP2 expression in CAFs similarly engages PI3K/AKT signaling, enhancing fatty-acid biosynthesis and driving a more invasive phenotype [74]. CAF-derived exosomes carrying miR-454-3p further protect tumor cells by suppressing ferroptosis [75]. In addition, CD10+ CAFs remodel lipid metabolism in cancer stem cells by degrading osteogenic growth peptide (OGP), an inhibitor of SCD1. Loss of OGP can increase SCD1-driven lipid desaturation, raising unsaturated lipid content, stemness and chemoresistance [76].
In summary, lipid metabolism forms a major axis of CAF function, integrating FAO, lipid-rich and secretory programs that shape tumor behavior and immune responses. Having discussed lipid-based interactions, we next summarize how CAFs remodel amino acid and nucleotide pathways to shape both nutrient flow and immunity.
Beyond glucose and lipid pathways, amino acid metabolism represents another major axis through which CAFs influence the tumor ecosystem [77]. Amino acid–remodeling (AA-remodeling) CAFs can preserve their own viability and help tumor cells adapt to amino-acid scarcity, while also shaping the immune landscape by modulating the availability of key amino acids required for effective antitumor immunity [78]. (Table 1)
This section focuses on metabolites that are nutrients and immune signals. To navigate these complex networks, we organize this chapter into three functional streams: (1) Nutrient Provision, focusing on how the glutamate–glutamine shuttle supports tumor mitochondrial metabolism (Section 4.1); (2) ECM Support, examining how aspartate and proline rewiring supports matrix synthesis and stiffening (Section 4.2); and (3) Immunometabolic Axes, highlighting how nucleotide (adenosine) and amino acid metabolites function as signaling molecules to shape the TME (Section 4.3).
A central feature of CAF metabolic reprogramming is their shift toward glutamine anabolism. Compared with NFs, CAFs upregulate glutamine synthetase (GS), allowing them to survive nutrient stress and secrete glutamine into the TME (Fig. 3a) [79] (Mechanistic/Flux-supported). The glutamine released by CAFs fuels tumor cells. In colorectal cancer ovarian metastases (oCRC), an RBP1+ myCAF subset expresses high levels of GS and converts glutamate into glutamine, which supports metastatic growth [80]. The NetG1–NGL1 axis reinforces physical contact between CAFs and tumor cells and may increase the efficiency of nutrient transfer [81]. NetG1+ CAFs release high levels of glutamine through GS and the vesicular transporter VGLUT1. Tumor-cell NGL1 helps maintain macropinocytosis, which supports uptake of these metabolites [81].
 Fig. 3.
Fig. 3.
Amino acid and nucleotide remodeling in CAFs and immune contexture. (a) Amino-acid remodeling centered on the glutamate–glutamine axis. CAFs can engage GS or GLS, depending on local nutrient conditions. Downstream, glutamate can be routed into several non-essential amino-acid pathways. These include aspartate/asparagine (AST/GOT, ASNS) and proline (PYCR1), which are linked to tumor support and ECM production. (b) Nicotinamide metabolism and stromal epigenetic coupling. CAF-associated NNMT converts NAM to MNAM while consuming SAM and generating SAH, consistent with reduced methyl-donor availability. In parallel, NAMPT-dependent conversion of NAM to NMN in tumor and myeloid compartments supports NAD+ regeneration. The relative balance of NMN and MNAM is associated with NAD+ availability, CD8+ T-cell cytotoxicity and CAF activation programs. (c) Immunometabolic axes linked to adenosine, kynurenine and arginine pathways. CAF-associated CD73 converts AMP to adenosine, which signals through A2A/A2B receptors and is linked to reduced CTL/NK activity. TDO2-mediated tryptophan catabolism generates kynurenine, which can activate AhR signaling and is associated with impaired DC function. ARG1/2-mediated arginine catabolism depletes arginine and generates ornithine-related metabolites, which are linked to TAM polarization and Treg expansion. Arrows indicate pathway direction; blunt-ended lines indicate inhibition. Red upward arrows denote increased expression/activity; blue downward arrows denote decreased expression/activity. Created in BioRender. Tu, W. (2026) https://BioRender.com/bvyjahf. AA, Amino acid; CTL, cytotoxic T lymphocyte; NK, natural killer cell; DC, dendritic cell.
Amino-acid transporters also shape this glutamate–glutamine shuttle. SLC1A1
brings extracellular glutamate into CAFs, where GS converts it to glutamine [82].
Glutamine is then exported through SLC7A5 and taken up by tumor cells, supporting
metabolic coupling [83]. Conversely, tumor-derived lactate and glutamate can
further stimulate glutamine synthesis in CAFs, forming a reciprocal
nutrient-exchange loop that sustains both stromal and tumor metabolism [79].
Tumor cytokines can further reinforce this coupling by inducing CAF expression of
LINC01614 [83]. This lncRNA can be packaged into exosomes and
transferred to tumor cells. In recipient tumor cells, LINC01614 binds
ANXA2 and p65 to activate NF-
Glutamate–glutamine metabolism also influences immune regulation within the TME. NetG1+ CAFs secrete IL-15, which suppresses NK-cell activation and cytotoxicity [81]. GS-high CAFs additionally release glutamine that is taken up by TAMs. This glutamine supports oxidative metabolism in TAMs and promotes their M2-like polarization, consistent with an immunosuppressive and tumor-promoting milieu [85].
Together, these findings highlight the glutamate–glutamine axis as an important metabolic and immunoregulatory hub that coordinates nutrient exchange, stromal activation and immune suppression within the TME.
CAFs also rewire the metabolism of other amino acids that support tumor anabolism and ECM production (Fig. 3a) (Mechanistic/Flux-supported). In BCa, a senescence-like TSPAN8+ myCAF subset upregulates glutaminase (GLS) to expand the intracellular glutamate pool and increases PYCR1 to drive proline synthesis. The enhanced glutamate supply also feeds aspartate-generating transamination pathways, supporting cancer stemness and therapy resistance [86]. Transporters involved in these pathways are also upregulated in CAFs. SLC1A3 facilitates aspartate transport, while SLC38A2 enhances asparagine transport, supporting amino-acid availability for stromal and tumor metabolic demands [87, 88, 89].
Stromal PYCR1 elevation promotes collagen production and ECM deposition, whereas its silencing reduces matrix formation and tumor growth by altering PDH-derived acetyl-CoA and glutamine-derived proline flux [90]. In prostate cancer, loss of p62 stabilizes activating transcription factor 4 (ATF4), which induces asparagine synthetase (ASNS) to increase asparagine production, buffer glutamine deprivation and support tumor proliferation [91]. CAF-specific ATF4 deficiency disrupts COL1A1 expression and glycine/proline enrichment, and impaired angiogenesis and collagen maturation [92].
Increased matrix stiffness activates mechanotransduction in CAFs and tumor cells. Under stiff conditions, CAFs show accelerated glutamine metabolism and increased aspartate export, as supported by isotope tracing (Mechanistic/Flux-supported) [78]. Tumor cells use stromal aspartate to support proliferation and release glutamate in return, which may alter CAF redox balance and further promotes ECM remodeling [78]. Together, these spatially coordinated amino-acid fluxes increased collagen production and tumor progression within the TME.
Crosstalk within the nicotinamide–NAD+ network shapes immune regulation (Fig. 3b) (Mechanistic). In CAFs, NNMT converts nicotinamide (NAM) into MNAM, consuming SAM and reducing NAD+ availability. By contrast, NAMPT in tumor cells and myeloid cells regenerates NAD+ through the salvage pathway [93]. This balance affects CD8+ T-cell activity: elevated NNMT/MNAM is linked to reduced cytotoxicity, whereas exosomal NAMPT can partially restore NAD+ levels and CD8+ function [94]. Beyond immunoregulation, high NNMT disrupts vitamin B₃ metabolism and alters histone methylation–acetylation balance, supporting CAF activation and fibrogenic programs [95].
Extracellular adenosine metabolism constitutes a key immunometabolic pathway in CAFs (Fig. 3c). As the dominant stromal source of CD73—an ecto-5′-nucleotidase that converts AMP to adenosine—CAFs drive local adenosine accumulation in the tumor microenvironment [96]. Elevated adenosine signals through A2A and A2B receptors on immune and stromal cells. This signaling is linked to reduced cytotoxic T- and NK-cell activity, increased Treg expansion, and impaired dendritic-cell activation, and it also correlates with pro-angiogenic and pro-fibrotic features [97, 98].
Tryptophan 2,3-dioxygenase (TDO2) converts tryptophan into kynurenine, generating a metabolically activated, pro-tumorigenic CAF state while promoting immune tolerance (Fig. 3c) [99]. Kynurenine can be exported from CAFs, potentially through transporters such as SLC7A5 and act on neighboring immune cells [100]. By activating the aryl hydrocarbon receptor (AhR), extracellular kynurenine has been reported to suppress dendritic-cell differentiation and function [99].
Arginine metabolism represents another immunoregulatory circuit that links stromal metabolism to T-cell suppression and matrix remodeling (Fig. 3c). Arginase-expressing CAFs (ARG1/2) convert L-arginine into ornithine and urea, depleting arginine in the TME and thereby suppressing effector T-cell activity [101]. Ornithine-derived metabolites, including proline and polyamines, support CAF proliferation, collagen biosynthesis and immune evasion [102, 103]. In ovarian and pancreatic cancers, ARG+ CAFs are associated with dense collagen deposition, immunosuppressive microenvironments and poor prognosis [104].
Translational Implications: Pharmacological inhibition of glutaminase (GLS) [86, 90], arginase (ARG1) [101, 104], and CD73 [96, 98] limit tumor anabolism and restores cytotoxic T-cell function. These pathways support evaluating metabolic interventions in combination with anti–PD-1/PD-L1 therapy [94, 97].
In addition to enzymatic amino-acid synthesis, CAFs obtain nutrients through macropinocytosis, an endocytic process that engulfs extracellular proteins and fluid [105]. This pathway is particularly relevant in nutrient-poor tumors such as PDAC. It can act as a survival strategy and support stromal–tumor metabolic cooperation. Under glutamine deprivation, elevated cytosolic Ca2+ activates CaMKK2–AMPK signaling in CAFs, which increases macropinocytic activity [106].
Macropinocytosis also contributes to CAF phenotypic stability. Under glutamine-limited conditions, active macropinocytosis prevents myofibroblastic CAFs (myCAFs) from converting into inflammatory CAFs (iCAFs), helping preserve the myCAF phenotype and constrain IL-6–driven inflammation [107]. When macropinocytosis is inhibited, myCAFs shift into iCAFs and this change is associated with a more inflammatory stroma and increased T-cell infiltration.
Collectively, these pathways reveal a multi-layered stromal amino-acid network that coordinates metabolic and immune remodeling. Through integrated control of adenosine, kynurenine, nicotinamide and arginine metabolism, CAFs establish an immunosuppressive and fibrotic niche that supports tumor persistence and resistance to therapy [108].
1. The glutamate–glutamine axis is a central route for stromal nutrient exchange and can couple to immune regulation.
2. Aspartate, asparagine and proline pathways support ECM synthesis and can be reinforced by mechanical stiffness.
3. NAD+, adenosine, kynurenine and arginine metabolism connect stromal metabolism to T-cell suppression and fibrotic remodeling.
4. Metabolic circuits in CAFs are plastic and can trigger compensatory responses when a single node is inhibited.
5. Translational Implications: Collectively, AA/nucleotide remodeling highlights druggable nodes including GLS, CD73/A2A signaling and ARG1/2. Stratification may leverage stromal GS/GLS balance, CD73-high stroma or arginine-depleted/adenosine-rich niches, which could help prioritize rational combinations with immune checkpoint blockade.
Standard CAF classifications use surface markers and secreted factors to define myCAF, iCAF and apCAF. However, their metabolic features are still not well defined. With the study dominated by correlative analyses, we summarize the subtype-linked metabolic signatures as a practical framework.
The inflammatory iCAF phenotype shows broad metabolic activity. The iCAF state is defined by a prominent inflammatory program. These CAFs show increased glycolytic activity probably associated with cytokine production [109]. However, OXPHOS CAF states can also display cytokine and inflammatory readouts, indicating that a high-secretory phenotype is not unique to a single metabolic mode [15, 20]. iCAFs also use lipid and FAO pathways [109]. For example, lipid accumulation is linked to the release of factors like VEGFA and HGF [62]. Additionally, CPT1A- or CPT1C-associated FAO has been linked to an immunosuppressive environment [61, 69].
The myCAF phenotype focuses on contraction and matrix remodeling. These roles often rely on OXPHOS and the TCA cycle [109]. myCAFs also engage in amino-acid remodeling. For instance, proline synthesis via PYCR1 and glutamine flux from GS are linked to collagen production [90]. Within this group, the senescent TSPAN8+ myCAF subset shows an AA-remodeling program that contribute to nutrient support for tumor cells [86].
apCAFs show moderate metabolic activity compared to the iCAF and myCAF groups [110]. apCAFs focus on antigen presentation and immune regulation. Their specific energy requirements are currently less defined.
Metabolic rewiring is not only a feature of CAF biology but can also accompany switching between canonical CAF subtypes (e.g., myCAF and iCAF) [107]. This plasticity underscores that metabolic states and CAF identity are coupled to microenvironmental constraints.
Advances in understanding CAF–tumor metabolic crosstalk have revealed several stromal vulnerabilities with translational potential. Because CAFs and tumor cells rely on overlapping metabolic enzymes, interventions targeting CAF metabolism can also affect tumor-cell metabolism. This dual effect can be leveraged therapeutically: modulating CAF metabolism not only limits tumor growth but also reshapes the microenvironment [21, 48]. Below, we group strategies by targetable metabolic nodes.
The most clinically advanced CAF-targeted approach is the use of FAP inhibitors for imaging and potential therapy. FAP is selectively overexpressed in CAFs and radiolabeled FAP inhibitors (e.g., 68Ga-FAPI-46) have shown high tumor-to-background ratios in PET imaging [111]. However, the clinical translation of CAF-targeted metabolic therapies remains at an early stage, with most evidence derived from biomarker studies, imaging trials and retrospective analyses rather than large-scale interventional clinical trials [112, 113, 114, 115].
Many FAP-targeted therapeutics, including ADCs, radioligands and antibody-photosensitizer conjugates, aim to eliminate FAP-positive CAFs. Several agents have already entered phase I studies. For example, the anti-FAP ADC OMTX705 shows efficient uptake by FAP-positive CAFs, cytotoxic activity and immune-contexture changes in preclinical models [116]. FXX489 is a FAP-targeted radioligand with high tumor selectivity and retention, and it is now being evaluated across multiple solid tumors.
However, these depletion approaches overlook CAF heterogeneity: some CAF subtypes are linked to poor prognosis and therapy resistance, whereas others may be neutral or even protective. FAP-targeted nanomedicine offers a way to modulate CAF metabolism rather than eliminate all CAFs. The FAP-C NPs system uses a FAP single-chain antibody fragment for active targeting and its payloads (such as the vitamin D analog calcipotriol) activate the vitamin D receptor in CAFs, shifting activated CAFs toward a quiescent state and reducing their pro-tumorigenic signaling [117].
Since CAF-derived lactate supports tumor metabolism and weakens antitumor immunity, blocking the lactate shuttle is an attractive therapeutic strategy. In the CAF–tumor lactate shuttle, monocarboxylate transporters (MCTs) serve as key regulators. MCT1, mainly expressed in tumor cells, supports lactate uptake, while MCT4, enriched in glyCAFs, enables lactate export [118, 119]. Together, their complementary distribution forms a lactate-recycling loop whose disruption may weaken CAF–tumor metabolic coupling [120].
AZD3965, a selective MCT1 inhibitor, shows preclinical activity across multiple tumor models by limiting lactate uptake in cancer cells [121, 122]. In a phase I trial (NCT01791595), AZD3965 showed manageable safety and clear target engagement, but the small cohort (approximately 40 patients) prevented firm conclusions on efficacy [123].
However, growing evidence indicates that MCT4 expression may influence the response to MCT1 inhibition. Since MCT4 preserves lactate export and sustains the CAF side of the shuttle, increased MCT4 could lessen the effect of MCT1 blockade [124]. In line with this idea, blocking both MCT1 and MCT4 has been reported to produce a more durable metabolic disruption in preclinical models [124].
In addition, tumors with a high MCT1-to-MCT4 ratio have shown better responses in vitro , but transporter expression varies widely across tumor regions, possibly mirroring differences in CAF abundance, making biomarker development challenging and clear predictors of MCT1 inhibitor response are still lacking [123]. Integrating transporter expression with stromal composition in patient samples will be essential for identifying tumor that may respond to lactate-transport inhibition.
Beyond lactate transport, additional steps in glycolysis have been examined as therapeutic targets. CAFs rely on glucose uptake and inhibiting transporters such as GLUT1 can reduce their glycolytic activity and limit stromal support [48]. However, glucose transport is broadly shared across cell types and compensatory changes in other GLUT isoforms may restrict the feasibility of selective inhibition, keeping most efforts at the preclinical stage [125, 126].
Lactate dehydrogenase (LDH), particularly LDHA, has also been investigated as a means to curb glycolytic flux in both tumor cells and CAFs. LDHA blockade can lessen local immunosuppression and improve antitumor responses in experimental models, but clinical evidence remains limited [127, 128]. Although direct glycolytic targets remain limited, broader metabolic regulators that influence CAF activation and energy usage have shown translational potential.
Metformin, a mitochondrial complex I inhibitor originally used as an
antidiabetic drug, has also been shown to modulate tumor metabolism. In
preclinical models, metformin enhances the activity of immunotherapy and other
systemic treatments, and several clinical trials are evaluating its use in
combination therapy (Table 2, Ref. [123, 129, 130, 131, 132, 133, 134, 135, 136, 137, 138]) [139, 140]. In the stroma,
metformin disrupts CAF–tumor metabolic coupling by reducing CAF activation. For
example, metformin reduces the secretion of pro-tumor mediators, including
TGF-
| Metabolic pathway | Agent | Tumor type(s) | Phase | Trial identifier | Combination strategy | Status | Results | Ref | Primary CAF-related rationale |
| Glucose | AZD3965 (MCT1 inhibitor) | PRAD (Prostatic adenocarcinoma), BRCA (Breast carcinoma) and DLBCL (Diffuse large B cell lymphoma) | I | NCT01791595 | - | Completed | Among 30 evaluable patients, 9 had stable disease (SD), 18 had progressive disease (PD), and 3 experienced early progression. The median time to progression for the 24 assessable patients was 57 days. | [123] | CAF-driven nutrient shuttling: Targets the CAF–tumor lactate shuttle. |
| metformin | MSS CRC | II | NCT03800602 | Nivolumab (anti-PD-1 antibody) | Completed | Among 18 evaluable patients, 2 achieved RECIST v1.1 SD; the remaining patients met criteria for PD. The median OS and PFS were 5.2 months (95% CI, 3.2–11.7) and 2.3 months (95% CI, 1.7–2.3). In the intent-to-treat cohort (n = 24), OS and PFS were 5.2 months (95% CI, 3.2–8.4) and 2.2 months (95% CI, 1.7–2.3). | [129] | Dampening CAF activation: Lowers | |
| SCLC (Small cell lung cancer) | II | NCT03994744 | Pembrolizumab (anti-PD-1 antibody) | Recruiting | - | ||||
| SKCM (Skin cutaneous melanoma) | I | NCT03311308 | Pembrolizumab | Recruiting | - | ||||
| NSCLC (Non-small cell lung cancer) | II | NCT02115464 | Chemoradiotherapy | Terminated | The addition of metformin did not improve, and may even have reduced, the efficacy of chemoradiotherapy (CRT), with higher toxicity observed. | [130] | |||
| Lipid | TVB‑2640 (FASN inhibitor) | Advanced KRAS-mutant NSCLC, BRCA and OV (Ovarian carcinoma) | II | NCT03808558, NCT03179904 | Chemotherapy | Active, not recruiting | Among 76 patients receiving monotherapy, complete or partial responses (CR or PR) were observed. In combination with paclitaxel, the PR rate was 11%. | [131] | CAF lipid reprogramming: Limits lipogenesis-linked stromal support and lipid-rich CAF programs. |
| High Grade Astrocytoma | II | NCT03032484 | Bevacizumab | Active, not recruiting | Among 25 patients, the objective response rate (ORR) for the combination therapy was 56%, including CR 17% and PR 39%. The 6-month progression-free survival (PFS6) was 31.4%, significantly higher than historical bevacizumab monotherapy (16%). The 6-month overall survival (OS6) was 68%, with no significant difference versus historical control. | [132] | |||
| HER2 positive BRCA | II | NCT03179904 | Trastuzumab (anti-HER-2 antibody) + chemotherapy/endocrinotherapy | Active, not recruiting | - | ||||
| MTI-301 (SCD1 inhibitor) | Solid Cancers | I | NCT06911008 | - | Not yet recruiting | - | |||
| IOA-289 (ATX inhibitor) | PRCA | I/II | NCT05586516 | Chemotherapy | Active, not recruiting | - | Stromal remodeling: Blocks CAF-derived ATX/LPA production involved in fibrosis. | ||
| Glutamine | CB-839 (GLS1 inhibitor) | ccRCC (Clear cell renal cell carcinoma), SKCM and NSCLC | I/II | NCT02771626 | Nivolumab | Terminated | The ORR was 24% in 25 ICI-naïve patients with ccRCC, 5.9% in 17 patients with ccRCC after ICI, 0% in 9 patients with ccRCC after other prior ICI, 5.4% in 37 patients with melanoma after ICI, and 0% in 19 patients with NSCLC after ICI. | [133] | CAF metabolic invasion: Targets GLS1-linked CAF motility and coordinated tumor invasion. |
| Arginine | INCB001158 (ARG1 inhibitor) | Solid cancers | I/II | NCT02903914 | Pembrolizumab | Completed | The highest response rate was observed in the ICI–naive combination therapy group with HNSCC (n = 26), with an ORR of 19.2% (4 PR; 1 CR) | [134] | Stromal nutrient depletion: Counteracts stromal arginine depletion to support T-cell function. |
| OATD-02 (ARG1/ARG2 inhibitor) | advanced metastatic CRC, OV, RCC and PDAC | I | NCT05759923 | / | Recruiting | - | Stromal nutrient depletion: Targets ARG1/2-linked arginine depletion in CAF-rich niches. | ||
| Adenosine | Oleclumab (CD73 inhibitor) | CRC, PDAC and NSCLC | I | NCT02503774 | Durvalumab (anti-PD-L1 antibody) | Completed | In the CRC cohort, the ORR was approximately 2.4% (1 CR); PDAC, approximately 4.8% (1 CR and 1 PR); and in NSCLC, approximately 9.5% (4 PRs). The PFS6 rates were approximately 5.4%, 13.2%, and 16.0%, respectively. | [135] | CAF-enriched expression: CAFs are a major stromal source of CD73-driven adenosine signaling. |
| TNBC | I/II | NCT03616886 | Durvalumab + chemotherapy | Active, not recruiting | Among 127 evaluable patients, ORR was comparable between groups (63.5% vs 64.1%). Median PFS was 5.9 vs 7.0 months. Oleclumab combined with durvalumab + chemotherapy did not improve 24-week clinical benefit rate (CBR) or PFS. | [136] | |||
| Luminal B BRCA | II | NCT03875573 | Durvalumab + SBRT | Active, not recruiting | Pathologic response, based on residual cancer burden (RCB), was assessed in 6 patients: 2 RCB-0 (pathologic complete response [pCR]), 2 RCB-1, 1 RCB-2, and 1 RCB-3. | [137] | |||
| SARC (Sarcoma) | II | NCT04668300 | Durvalumab | Active, not recruiting | - | ||||
| Quemliclustat (CD73 inhibitor) | PDAC | I | NCT04104672 | Zimberelimab (anti-PD-1 antibody) + chemotherapy | Active, not recruiting | Over 120 patients, median OS was 15.7 months, compared with 9.8 months in a synthetic control arm, representing a 37% reduction in risk of death (HR |
[138] |
Given shared targets and compensatory metabolic shifts in CAFs, single-pathway inhibition may be insufficient to restrain their tumor-supportive functions [6, 42]. Accordingly, multi-pathway approaches will be necessary to suppress CAF-associated tumor progression.
CAF-focused metabolic interventions show potential, but their effectiveness may depend on overcoming shared metabolic targets and compensatory pathways within CAF subsets. Therapeutic approaches that combine lactate-transport inhibition, glycolytic modulation, and agents that dampen CAF activation may provide greater benefit, particularly when guided by stromal metabolic profiling.
The metabolic flexibility of CAFs allows them to shift toward lipid-based metabolism when glycolytic or oxidative pathways are inhibited. This adaptability has drawn attention to lipid metabolism as a shared metabolic vulnerability in both CAFs and tumor cells.
Inhibition of FASN (TVB 264) or SCD1 (MTI-301) suppresses de novo fatty-acid synthesis and membrane biogenesis, potentially limiting the emergence of both fatty-acid–oxidizing and lipid-rich CAF subsets [146, 147]. Early clinical studies of the FASN inhibitor TVB-2640 have shown signs of activity across several tumor types. In KRAS-mutant NSCLC, BRCA and OV, TVB-2640 monotherapy showed clinical activity in a subset of patients, while combination therapy with paclitaxel produced higher response rates (details in Table 2; NCT03808558, NCT03179904) [131]. In high-grade astrocytoma, the addition of bevacizumab similarly enhanced therapeutic outcomes compared with historical monotherapy controls (NCT03032484) [132]. These findings suggest that FASN inhibition may be more effective in combination settings.
Beyond lipogenesis, CAFs also export soluble lipid mediators. CAF-derived LPCs
are converted by ATX into LPA, forming a key stromal lipid–signaling pathway
that promotes fibrosis and tumor proliferation [67, 68]. Inhibiting this axis can
reduce CAF-derived LPA signaling and fibrosis. Its therapeutic effect is
strengthened when combined with agents that block fatty-acid uptake or transport,
such as CD36 or FABP antagonists, which further limit lipid fueling and suppress
tumor growth [65, 68]. In addition, in CAF-rich tumors such as PDAC, combining
lipid-targeted agents with anti-fibrotic therapy, TGF-
Lipid-targeted interventions show promise, but shared and compensatory lipid pathways in CAFs and tumor cells limit the effectiveness of single agents. Combination approaches may therefore be more likely to achieve meaningful therapeutic benefit.
Amino-acid exchange between CAFs and tumor cells is emerging as a promising therapeutic target. By modulating amino acid flux and influencing immune function, these pathways provide opportunities to reprogram both metabolism and immunity within the TME.
The reliance of both tumor cells and stromal components on glutamine has motivated clinical evaluation of GLS inhibitors. CAFs not only upregulate GS to supply glutamine to tumor cells under nutrient stress, but also increase GLS1 activity to sustain their own migration and invasion [79]. In preclinical models, glutamine deprivation or GLS1 inhibition reduces CAF motility and disrupts their coordinated invasion with cancer cells [149]. Clinically, the GLS1 inhibitor telaglenastat (CB-839) has been evaluated in patients with advanced cancers (Table 2). Although clinical activity has been modest, these studies suggest that glutamine metabolism is a tractable therapeutic target and support continued evaluation of GLS inhibition in rational combination strategies [133].
Arginine depletion within the TME, driven in part by stromal and myeloid ARG1/ARG2 activity, contributes to impaired T-cell function and immune suppression [150, 151]. This has led to the development of arginase inhibitors aimed at restoring intratumoral arginine availability. INCB001158, a selective ARG1 inhibitor, has been evaluated in combination with immune checkpoint blockade. In a phase I/II study of solid tumors (NCT02903914), combining INCB001158 with pembrolizumab showed the highest activity in PD-1/PD-L1–naïve head and neck squamous cell carcinoma (ORR 19.2%; including one complete response), whereas activity in previously treated cohorts was limited.
To more comprehensively counteract stromal arginine depletion, OATD-02, a dual ARG1/ARG2 inhibitor, is now under evaluation in a phase I study for solid malignancies (NCT05759923). Preclinical data indicate that dual arginase inhibition increases local arginine availability, reduces polyamine accumulation, and promotes a more immunostimulatory microenvironment [152].
Across multiple tumor types, CAFs constitute the predominant CD73-high stromal population [153, 154]. As CAFs are a major source of extracellular adenosine, CD73 blockade can diminish CAF-driven adenosine signaling and thereby relieve immune suppression [153].
Clinical development has centered on using CD73 inhibitors in combination with immunotherapy. Oleclumab has been evaluated with durvalumab across colorectal cancer, pancreatic cancer, NSCLC, TNBC and Luminal B breast cancer, reflecting a combination-based development path (Table 2). Across these studies, oleclumab showed limited activity as a single agent. When combined with PD-L1 blockade, with or without additional chemotherapy or radiotherapy, oleclumab produced variable immunomodulatory effects, although overall antitumor efficacy remained limited [135, 136, 137]. Quemliclustat (AB680) has shown similarly limited activity. By contrast, etrumadenant—a dual A2A/A2B antagonist acting downstream of CD73—has produced more encouraging results (NCT04660812), with improved outcomes when combined with immunotherapy and anti-angiogenic therapy in metastatic CRC.
Amino-acid and adenosine-pathway therapies show immunomodulatory potential but have demonstrated limited efficacy as monotherapies. As CAFs are key contributors to these metabolic circuits, their role in shaping therapeutic responses warrants greater attention.
A key question is whether CAF metabolic rewiring represents a universal stromal response or a cancer-type–specific adaptation. In desmoplastic and hypovascular tumors such as PDAC, glycolytic CAFs are frequently observed. These cells adapt to hypoxia and can provide lactate and alanine to tumor cells [78]. In contrast, in ovarian cancer, prostate cancer and melanoma, CAF subsets have been reported to rely more on oxidative phosphorylation or fatty acid oxidation [16, 17, 18]. However, these differences cannot be explained by tumor type alone. Spatial position, oxygen availability and nutrient distribution also shape CAF metabolic states. Within the same tumor, distinct CAF subsets may exhibit either glycolytic or oxidative phenotypes depending on local conditions [14, 155]. This spatial heterogeneity is increasingly revealed by single-cell and spatial transcriptomic studies. Therefore, CAF metabolism should be viewed as cancer-type biased but microenvironment regulated. It reflects an adaptive response to local ecological pressures rather than a fixed lineage program.
Targeting CAFs can produce opposite outcomes depending on context. In PDAC
models, depletion of
As a result, therapeutic efforts should shift from CAF depletion toward more selective approaches. The goal is to reprogram tumor-promoting stromal functions and minimize damage to tumor-restraining fibroblasts and systemic immunity.
Disagreement across studies often reflects how CAFs are defined. Early work used
single markers such as
Moreover, model systems influence outcomes. Standard 2D cultures provide abundant nutrients and oxygen and do not mimic the metabolic microenvironment of human tumors [161]. Findings from such systems should be validated in physiologically relevant models, including 3D cultures, organoids or in vivo settings.
Therefore, reconciling discordant findings require more precise CAF subclassification, spatially resolved analysis and validation across multiply model systems. These approaches can help distinguish biological heterogeneity from methodological artifacts.
This review highlights that CAFs act as metabolically active stromal components
that influence nutrient availability and modulate cancer cell behavior.
Glycolytic CAFs show increased aerobic glycolysis and lactate exchange with
neighboring cancer cells, which contribute to tumor metabolic adaptation.
Fatty-acid–oxidizing CAFs show increased fatty-acid uptake and
A primary challenge in targeting CAF metabolism is their intrinsic plasticity [162]. While our classification delineates dominant functional states, these identities are not static. For instance, glucose limitation potentially triggers compensatory transitions toward fatty acid oxidation or amino acid catabolism, sustaining CAF viability and their pro-tumorigenic roles [60, 163]. This context-dependent adaptability suggests that durable therapeutic efficacy likely requires combinatorial strategies targeting multiple nodes of this plastic network rather than isolated pathways [48, 164].
Despite the translational potential of stromal metabolic interventions, several challenges remain. Specificity is a core limitation: many metabolic enzymes are shared by fibroblasts, immune cells and epithelial cells, making on-target effects difficult to confine to CAFs. Metabolic redundancy further complicates targeting, as CAF programs can shift when one pathway is blocked. Finally, inter-patient and intra-tumor heterogeneity limits the generalizability of one metabolic vulnerability. Together, these findings support biomarker-guided stratification and monitoring, for example using FAP-targeted PET to assess CAF abundance and spatial distribution [111].
Future work should focus on state-informed intervention of CAFs. CAF-oriented imaging may support patient selection and on-treatment monitoring. Beyond 68Ga-FAPI-46 PET for CAF abundance, tracers linked to lactate transport or lipid uptake could help capture spatial metabolic activity in vivo. Besides, metabolic-state biomarkers derived from single-cell and spatial profiling may enable stratification, particularly when linked to outcomes and immune contexture. Spatially guided delivery strategies, including FAP-targeted nanoparticles and microenvironment-activated prodrugs, could improve local exposure while reducing systemic toxicity from shared metabolic targets.
CAFs are key components of the tumor metabolic landscape. Through rewiring of glucose, lipid and amino acid metabolism, CAFs support cancer cell growth and invasion while concurrently reshaping the ECM and suppressing antitumor immunity. These integrated metabolic adaptations enable tumors to persist under both nutrient limitation and immune pressure. Therefore, CAF-directed metabolic therapies, particularly when combined with immunotherapy, may hold promise for achieving more durable and selective reprogramming of the tumor microenvironment.
ZY and XB conceptualized and designed the study. ZY, XLu, RP, and HJ collected and analyzed the relevant literature. XZ, XLin, and WW provided critical input on data interpretation and manuscript structure. ZY drafted the initial manuscript. WW and XB critically revised the manuscript for important intellectual content. All authors contributed to manuscript editing, read and approved the final version, and agree to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflicts of interest.
During the preparation of this work the authors used ChatGpt-5.0 in order to check spell and grammar. After using this tool, the authors reviewed and edited the content as needed and takes full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.