







1 920th Hospital of Joint Logistics Support Force, PLA, 650032 Kunming, Yunnan, China
2 Yunnan Key Laboratory of Stomatology, Kunming Medical University, 650500 Kunming, Yunnan, China
3 Department of Oral and Maxillofacial Surgery, The First People’s Hospital of Yunnan Province, 650032 Kunming, Yunnan, China
4 Shiyan Key Laboratory of Comprehensive Prevention and Treatment of Oral Cancer, Department of Stomatology, Taihe Hospital, Hubei University of Medicine, 442000 Shiyan, Hubei, China
5 Institute of Oral Diseases, School of Dentistry, Hubei University of Medicine, 442000 Shiyan, Hubei, China
†These authors contributed equally.
Abstract
Sporadic venous malformations are a prevalent vascular anomaly in the oral and maxillofacial region. Current therapeutic strategies are associated with high recurrence rates and limited applicability in critical anatomical regions. Therefore, there is a pressing demand for more effective treatment modalities to address these challenges.
This study utilized transcriptome sequencing to analyze samples from venous malformation patients and combined lesion tissue analysis, cell culture, and xenograft mouse models to investigate the pathogenic mechanisms and potential therapeutic approaches for venous malformations.
Our findings indicated that the primary pathological features of venous malformations include abnormal angiogenesis and excessive activation of the phosphoinositide 3-kinase (PI3K) signaling pathway. In endothelial cells with the most common pathogenic mutation, TIE2-L914F, this mutation activates the PI3K pathway, promoting cell proliferation, inhibiting normal angiogenesis, and suppressing apoptosis. Treatment with PI3K inhibitors effectively reversed these pathological changes. More importantly, the combination of rapamycin and alpelisib exhibited superior therapeutic efficacy, not only significantly inhibiting the PI3K pathway but also activating P53 expression, thereby effectively preventing disease progression. Further validation through in vitro 3D angiogenesis assays and xenograft mouse models confirmed the therapeutic potential of this combination. The results demonstrated a marked reduction in vessel sprouting in the 3D model and inhibited both lesion size and angiogenesis in the xenograft mouse model.
This study is the first to demonstrate that the combination of rapamycin and alpelisib, through multi-target synergy, effectively inhibits the PI3K pathway and activates P53 expression, offering new insights and therapeutic options for the treatment of venous malformations.
Keywords
- venous malformation
- TIE2 protein
- phosphatidylinositol 3-kinases
- target of Rapamycin protein
Venous malformations (VMs) represent a common category of congenital vascular anomalies that affect the skin and mucosa of the oral and maxillofacial region, while they may also involve deeper anatomical structures including skeletal muscles and internal organs [1, 2]. Clinically, patients typically exhibit blue-purple, soft, and compressible vascular masses, and a considerable proportion of them may also present with symptoms such as pain, phleboliths or coagulopathy [3]. Histopathology shows characteristic features, including abnormal dilation of venous lumens, endothelial cell hyperplasia, reduced vascular smooth muscle, and adventitial fiber degeneration [4, 5]. Conventional treatments include surgical resection and sclerotherapy. However, sclerotherapy can induce local inflammation and fibrosis, often requiring multiple sessions to alleviate symptoms, while surgical intervention is limited by lesion size and location and is associated with a high risk of recurrence [6, 7, 8, 9]. Therefore, investigating the pathogenesis of VMs and identifying effective targeted therapies have significant clinical implications.
Receptor tyrosine kinase with immunoglobulin and epidermal growth factor homology domains-2(TIE2), a member of the angiopoietin (ANGPT) family, is primarily expressed in endothelial cells. Somatic mutations in TIE2 are responsible for over half of VMs, with the L914F mutation accounting for 77% of cases [10, 11]. These mutations cause receptor hyperphosphorylation, leading to activation of the Phosphoinositide 3-Kinase/Protein Kinase B/Mammalian Target of Rapamycin (PI3K/AKT/mTOR) signaling pathway and subsequent VM development [12]. Additionally, activating mutations in PIK3CA are observed in approximately 50% of TIE2 mutation-negative VMs, which also promote VM formation through chronic activation of the PI3K/AKT/mTOR pathway [13]. Some patients may exhibit dual mutations in both TIE2 and Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha (PIK3CA) [14].
Thus, dysregulation of the PI3K pathway plays a critical role in various VMs, forming the molecular pathological basis of these lesions. Although VMs associated with PIK3CA mutations have been widely investigated [12, 13, 15, 16, 17], the precise role of the PI3K pathway in TIE2 mutation-driven VMs remains incompletely elucidated [18, 19, 20]. This study seeks to explore the function of the PI3K pathway in TIE2-L914F mutation-related VMs and provide a theoretical foundation for developing targeted therapeutic strategies. We delivered the TIE2-L914F mutation into venous endothelial cells using a lentiviral vector and analyzed the morphological, proliferative, angiogenic, and downstream signaling alterations in these cells both in vitro and in vivo. Furthermore, we employed PI3K pathway inhibitors to determine their ability to inhibit the pathological phenotypes of VMs. Notably, we discovered that the combination of Rapamycin and Alpelisib demonstrated remarkable efficacy in treating VMs, which was further confirmed in an in vitro 3D angiogenesis model and a nude mouse xenograft model.
Human umbilical vein endothelial cells (HUVECs) (iCell, iCell-h110, Shanghai, China) were isolated from human umbilical cords. All cell lines were authenticated by short tandem repeat (STR) analysis and tested negative for mycoplasma. Optical microscopy showed that endothelial cells exhibited a spindle-shaped morphology (Supplementary Fig. 1A). These cells were characterized by flow cytometry for surface markers (Supplementary Fig. 1B) and stained positive for von Willebrand factor (vWF) (Supplementary Fig. 1C). The cells were cultured in endothelial cell medium (ECM, iCell, Shanghai, China) containing 5% fetal bovine serum under standard conditions of 37 °C, 5% CO2, and controlled humidity. The medium was refreshed every three days, and subsequent experiments were conducted when the cell density reached 80%–90%.
Approximately 1.5
The concentrations of experimental drugs were determined by referencing previous studies and conducting preliminary screening experiments [21, 22]. The experimental design for drug combinations is presented in Table 1.
| Grouping | Concentration | Source | Item number |
| Dimethyl Sulfoxide (DMSO) | 1‰ | Solarbio | D8371 |
| Rapamycin | 10 nM | Laboratories | R-5000 |
| Alpelisib | 2 µM | GLPbio | GC16462 |
| Rapamycin + Alpelisib | 5 nM +1 µM | Laboratories + GLPbio | R-5000 + GC16462 |
| Rapamycin + Ponatinib | 5 nM + 50 nM | Laboratories + GLPbio | R-5000 + GC14396 |
Images were captured for TIE2-NC, TIE2-WT, and TIE2-L914F cells during the logarithmic growth phase. To investigate the effects of drugs on cell morphology, images were captured after 48 hours of drug treatment. The circularity index of cells was quantified with ImageJ software (National Institutes of Health, Bethesda, MD, USA).
TIE2-NC, TIE2-WT, and TIE2-L914F cells were plated in a 96-well plate coated with fibronectin at a density of 2000 cells per well, with three replicate wells. Each well was filled with 100 µL of complete ECM medium. At 0 h, 24 h, 48 h, 72 h, and 96 h, 100 µL of CCK-8 solution (GLPbio, GK10001, Montclair, California, USA) was added per well, followed by incubation at 37 °C for 2 hours. The optical density (OD) at 450 nm was determined with a microplate reader, and growth curves for the three cell types were plotted. To investigate the effects of drugs on cells, drug-containing ECM medium was added post-attachment. At 0 h, 24 h, 48 h, 72 h, and 96 h, 100 µL of CCK-8 solution was added per well, followed by incubation at 37 °C for 2 hours. The OD at 450 nm was determined, and growth curves under different drug treatments were plotted.
TIE2-NC, TIE2-WT, and TIE2-L914F cells were plated in a 48-well plate at a
density of 3
TIE2-NC, TIE2-WT, and TIE2-L914F cells were plated in a six-well plate at a
density of 3
TIE2-NC, TIE2-WT, and TIE2-L914F cells were plated in a 96-well plate pre-coated
with 50 µL of Matrigel (Corning, CLS356234, Corning, New York ,USA) at a
density of 2
Following Phosphate Buffered Saline (PBS) washing, cells were lysed using
Rapamycin lysis buffer containing protease and phosphatase inhibitors for protein
extraction. The extracted cellular proteins were subjected to Western blot
analysis with the following antibodies:
HUVECs in the logarithmic growth phase were plated in a culture plate, and fixation and blocking were carried out when cell confluence reached 80%. The primary antibody, Von Willebrand Factor (ab6994, 1:250, Abcam, Cambridge , Cambridgeshire, UK), was applied overnight at 4 °C. Subsequently, the secondary antibody, Cy3-conjugated goat anti-rabbit IgG (GB21303, Servicebio, 1:250, Wuhan, HuBei, China), was added at 37 °C in the dark for 1 hour, followed by washing. Next, 4′,6-Diamidino-2-phenylindole (DAPI) staining solution (G1012, Servicebio, Wuhan, HuBei, China) was applied in the dark for 10 minutes, followed by another wash. For paraffin-embedded tissue sections, following standard blocking procedures, the primary antibodies CD31 (ab9498, 1:250, Abcam, Cambridge, Cambridgeshire, UK) and p-AKT (Thr308) (ab38449, 1:250, Abcam, Cambridge, Cambridgeshire, UK) were applied overnight at 4 °C. The secondary antibodies, Cy3-conjugated goat anti-mouse IgG (GB21301, 1:250, Servicebio, Wuhan, HuBei, China) and FITC-conjugated goat anti-rabbit IgG (GB22303, 1:250, Servicebio, Wuhan, HuBei, China), were used for CD31 and p-AKT (Thr308), respectively, and were applied at 37 °C in the dark for 1 hour, followed by thorough washing. Finally, DAPI staining solution (G1012, Servicebio, Wuhan, HuBei, China) was applied in the dark for 10 minutes, and after washing, the sections were mounted with resin. All images were acquired with a Nikon microscope system, and the relative protein expression levels were analyzed with ImageJ software (National Institutes of Health, Bethesda, MD, USA).
p-AKT (Thr308) Paraffin-embedded tissue sections were incubated with the following primary antibodies: p-TIE2 (Y992) (ab321883, 1:250, Abcam, Cambridge, Cambridgeshire, UK), p-AKT (Thr308) (ab38449, 1:250, Abcam, Cambridge, Cambridgeshire, UK), p-AKT (Ser473) (ab81283, 1:250, Abcam, Cambridge, Cambridgeshire, UK), p-mTOR (Ser2448) (ab109268, 1:250, Abcam, Cambridge, Cambridgeshire, UK), and p53 (ab32049, 1:250, Abcam, Cambridge , Cambridgeshire, UK). Subsequently, the secondary antibody, Goat Anti-Rabbit IgG H&L (HRP) (ab6721, 1:5000, Abcam, Cambridge, Cambridgeshire, UK), was applied to detect the primary antibodies, and color development was carried out with DAB reagent (DAB-0031, Maixin, Fuzhou, Fujian, China). The sections were stained with hematoxylin for counterstaining. All images were acquired with a Nikon microscope system, and the relative protein expression levels were analyzed with ImageJ software (National Institutes of Health, Bethesda, MD, USA).
The transcriptome sequencing dataset GSE130807, comprising venous malformation
tissues, was retrieved from the Gene Expression Omnibus (GEO) database, including
5 venous malformation samples and 6 normal venous samples. Differential
expression analysis was conducted using the DESeq2 package in RStudio, with the
criteria for identifying DEGs set as
The fibrin bead assay was previously described in detail [23]. In this study, Cytodex® 3 microcarrier beads (Sigma, Louis, Missouri, USA) were coated with TIE2-L914F cells at 1500 cells per bead and embedded in fibrin gel. Fibroblasts (Cells are identified by surface marker analysis by flow cytometry, Supplementary Fig. 1D) were seeded onto the gel surface, and the ECM medium was refreshed every 2 days. On day 0, the designated drugs were added to the wells. After 2 days of treatment, the drugs were removed, and fresh medium was added. On day 7, the fibroblast layer overlaying the fibrin gel was removed by trypsin treatment. Images were acquired using a Nikon microscope, and the number of vascular sprouts per bead was quantified by two independent blinded observers.
The nude mice (nu/nu, Kunming Medical University Animal Research Institute,
China) used in this study were purchased from the Kunming Medical University
Animal Research Institute. First, TIE2-L914F cells were pretreated with
drugs or DMSO for 48 hours. Then, 2.5
Data are presented as mean
The heatmap revealed 310 significantly dysregulated genes in VMs tissues compared to normal tissues (Fig. 1A). KEGG pathway analysis demonstrated that the PI3K-AKT signaling pathway was significantly enriched in the disease group and ranked as the top enriched pathway (Fig. 1B). Additionally, pathways associated with cytoskeleton regulation and extracellular matrix interactions were also significantly enriched in the disease group (Fig. 1B). Further GSEA analysis indicated significant enrichment of the angiogenesis biological process (HALLMARK_ANGIOGENESIS, Fig. 1C), with a nominal p-value of 0.02088. However, the adjusted p-value (p.adjust = 0.1533) did not reach statistical significance, potentially due to the limited sample size or the gene ranking approach.
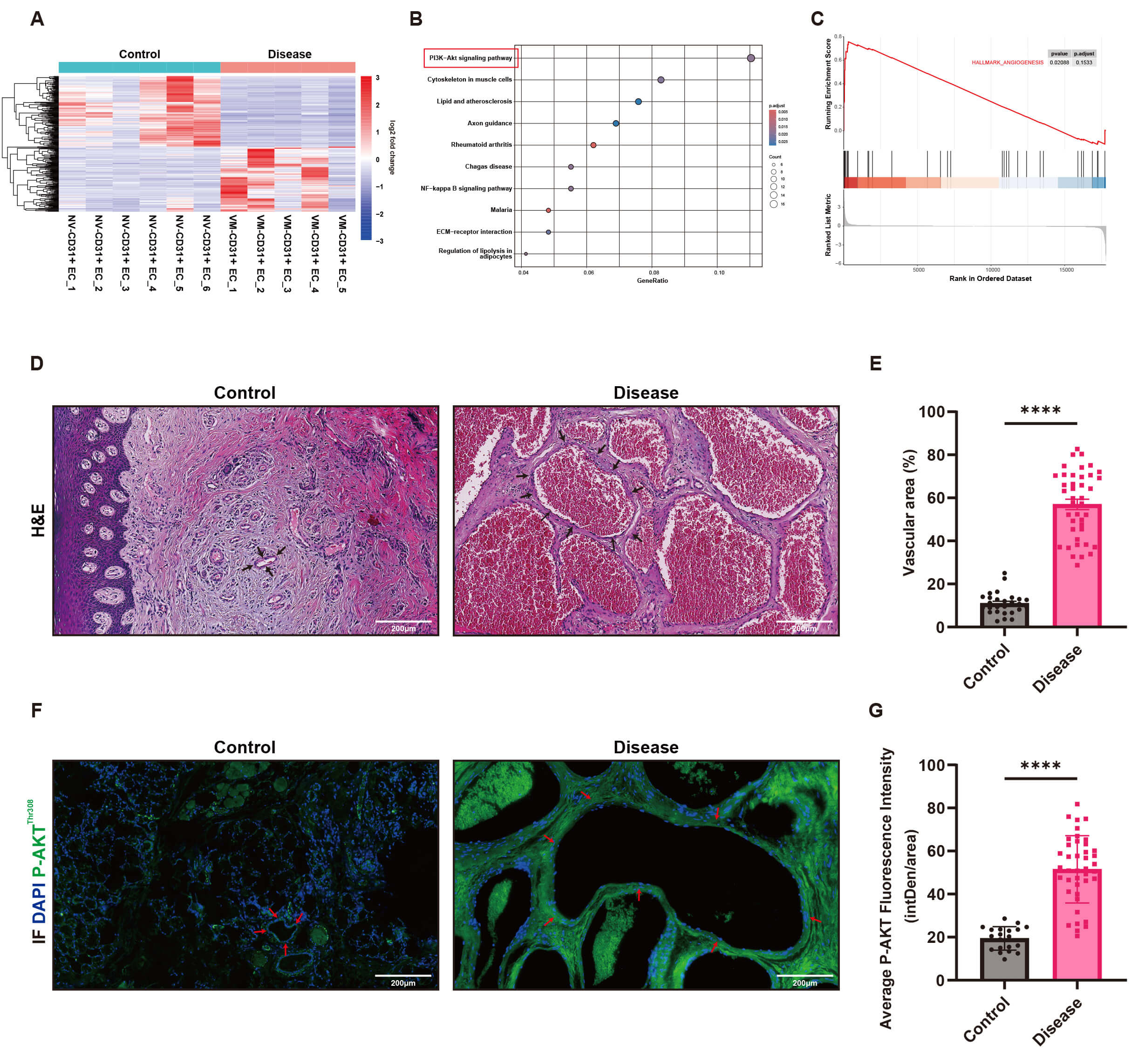 Fig. 1.
Fig. 1.
Overactivation of the Phosphoinositide 3-Kinase/Protein Kinase B
(PI3K-AKT) pathway and abnormal angiogenesis in venous malformation (VM) tissues.
(A) Heatmap depicting the expression profiles of 310 differentially expressed
genes in VM tissues (Disease, n = 5) and normal tissues (Control, n = 6). Red
represents high expression, and blue represents low expression. (B) Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathway analysis reveals that the
PI3K-AKT signaling pathway is significantly enriched and ranked first in the
disease group. (C) Gene Set Enrichment Analysis (GSEA) analysis demonstrates that
the biological process of angiogenesis (HALLMARK_ANGIOGENESIS) is exhibit marked biological enrichment tendency in the disease group (p = 0.02088, p.adjust = 0.1533).
(D) H&E staining reveals significant dilation of vascular lumens in VM tissues,
with clear differences compared to normal skin and gingival tissues (VM tissues n
= 41, normal tissues n = 21, Scale bar = 200 µm). (E) Quantitative analysis
reveals that the vascular area in the disease group is significantly higher than
in the normal group (mean
HE and immunofluorescence staining of VMs tissue sections demonstrated a significantly increased vascular area in VMs tissues compared to normal skin or gingival tissues (Fig. 1D,E), suggesting marked abnormal angiogenesis in the disease group. Furthermore, the expression of p-AKT protein was significantly elevated in VMs tissues compared to normal tissues (Fig. 1F,G), further supporting the hyperactivation of the PI3K pathway in VMs.
HUVECs were transduced via lentiviral-mediated gene delivery with a TIE2-L914F mutant construct, successfully establishing a TIE2-L914F mutant VM cell model and providing a solid experimental foundation for subsequent mechanistic studies. Initially, Western blot analysis was performed to assess the impact of the TIE2-L914F mutation on the PI3K signaling pathway (Fig. 2A). Analysis revealed that the expression of p-TIE2 was significantly elevated in the TIE2-L914F mutation group (L914F) compared to the control (NC) and wild-type TIE2 (WT) groups (Fig. 2C). Additionally, downstream signaling molecules, including p-AKT (p-AKT Thr308 and Ser473, Fig. 2D,E) and p-mTOR (Fig. 2F), were also significantly upregulated, indicating hyperactivation of the PI3K pathway in cells with the TIE2-L914F mutation. Analysis of apoptosis-related markers (Fig. 2B) demonstrated that p53 expression was significantly downregulated in the L914F group (Fig. 2G), along with reduced levels of Cleaved Caspase-3 (Fig. 2H) and the pro-apoptotic protein BAX (Fig. 2I), suggesting that the TIE2-L914F mutation inhibits apoptosis through p53 downregulation. Additionally, the L914F group exhibited significant morphological changes, with cells displaying elongated or spindle-shaped forms compared to the NC and WT groups (Fig. 2J), accompanied by a reduction in cellular roundness (Fig. 2O). EdU staining revealed that the TIE2-L914F mutation promoted cell proliferation, with the L914F group showing a significantly higher proliferation rate than the NC and WT groups (Fig. 2M,P). The cell proliferation curve further demonstrated accelerated proliferation in the L914F group (Fig. 2N). In the tubule formation assay, the tubule formation capacity in the L914F group was significantly impaired (Fig. 2K), with fewer vascular nodes (Fig. 2R) and ring-like structures (Fig. 2S) compared to controls. Flow cytometry revealed a significantly lower apoptosis rate in the L914F group than in the NC and WT groups (Fig. 2L,Q).
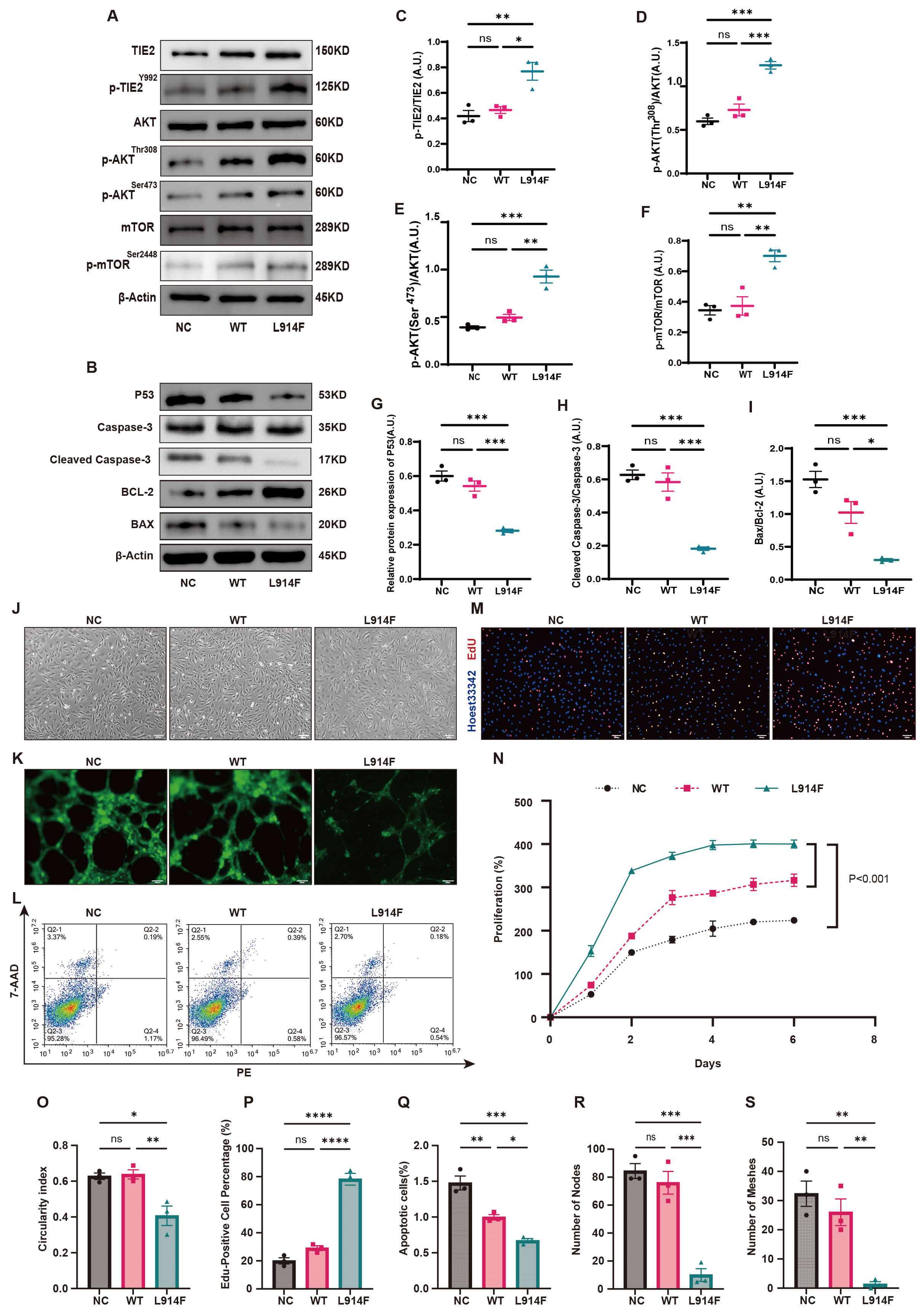 Fig. 2.
Fig. 2.
The TIE2 p.Leu914Phe (TIE2-L914F) mutation alters the
phenotype of normal cells and related signaling pathways. (A) Western blot
reveals that the expression of p-TIE2, p-AKT (Thr308 and Ser473), and p-mTOR in
the TIE2-L914F mutation group (L914F) is significantly higher than in the control
group (NC) and wild-type group (WT) (n = 3). (B) Western blot reveals that the
expression of P53, Cleaved Caspase-3, and Bcl-2-associated X protein (BAX) is
significantly reduced in the L914F mutation group (n = 3). (C–F) Quantitative
analysis reveals that p-TIE2/TIE2 (C), p-AKT (Thr308)/AKT (D), p-AKT (Ser473)/AKT
(E), and p-mTOR/mTOR (F) in the L914F mutation group are significantly higher
than in the NC and WT groups (mean
Collectively, the TIE2-L914F mutation upregulates p-TIE2 expression, hyperactivates the PI3K signaling pathway, and suppresses p53 in venous endothelial cells. These findings suggest a pathological mechanism underlying abnormal cell morphology, enhanced proliferation, impaired angiogenesis, and inhibited apoptosis.
The cell growth curve (Supplementary Fig. 1E) revealed that cell proliferation was significantly inhibited after 48 hours of drug treatment compared to controls, prompting the standardization of a 48-hour treatment period for subsequent experiments. Western blot analysis demonstrated that PI3K inhibitors, either alone or in combination, effectively suppressed the PI3K pathway (Fig. 3A). Notably, the combination of Rapamycin and Alpelisib exhibited the strongest inhibitory effect, characterized by significantly reduced expression of p-TIE2 (Fig. 3C), p-AKT (Thr308 and Ser473, Fig. 3D,E), and p-mTOR (Fig. 3F). Compared to single-agent treatments or the recommended combination (Rapamycin + Ponatinib), Rapamycin and Alpelisib showed enhanced synergistic effects, suggesting promising therapeutic potential against PI3K pathway hyperactivation. Analysis of apoptosis-related markers (Fig. 3B) revealed that the Rapamycin and Alpelisib combination significantly upregulated p53 expression (Fig. 3G) and increased levels of pro-apoptotic proteins Cleaved Caspase-3 (Fig. 3H) and BAX (Fig. 3I), further supporting its potent pro-apoptotic effects compared to other treatments. Functional assays confirmed the reversal of TIE2-L914F pathological phenotypes upon PI3K pathway inhibition. The Rapamycin and Alpelisib combination significantly improved cell morphology (Fig. 3J), increased cellular roundness (Fig. 3N), suppressed abnormal proliferation (Fig. 3K,O), and enhanced apoptosis (Fig. 3L,P). Tubule formation assays demonstrated that this combination restored normal angiogenesis (Fig. 3M), characterized by increased vascular nodes (Fig. 3Q) and ring-like structures (Fig. 3R), indicating superior therapeutic effects compared to single agents or the recommended combination.
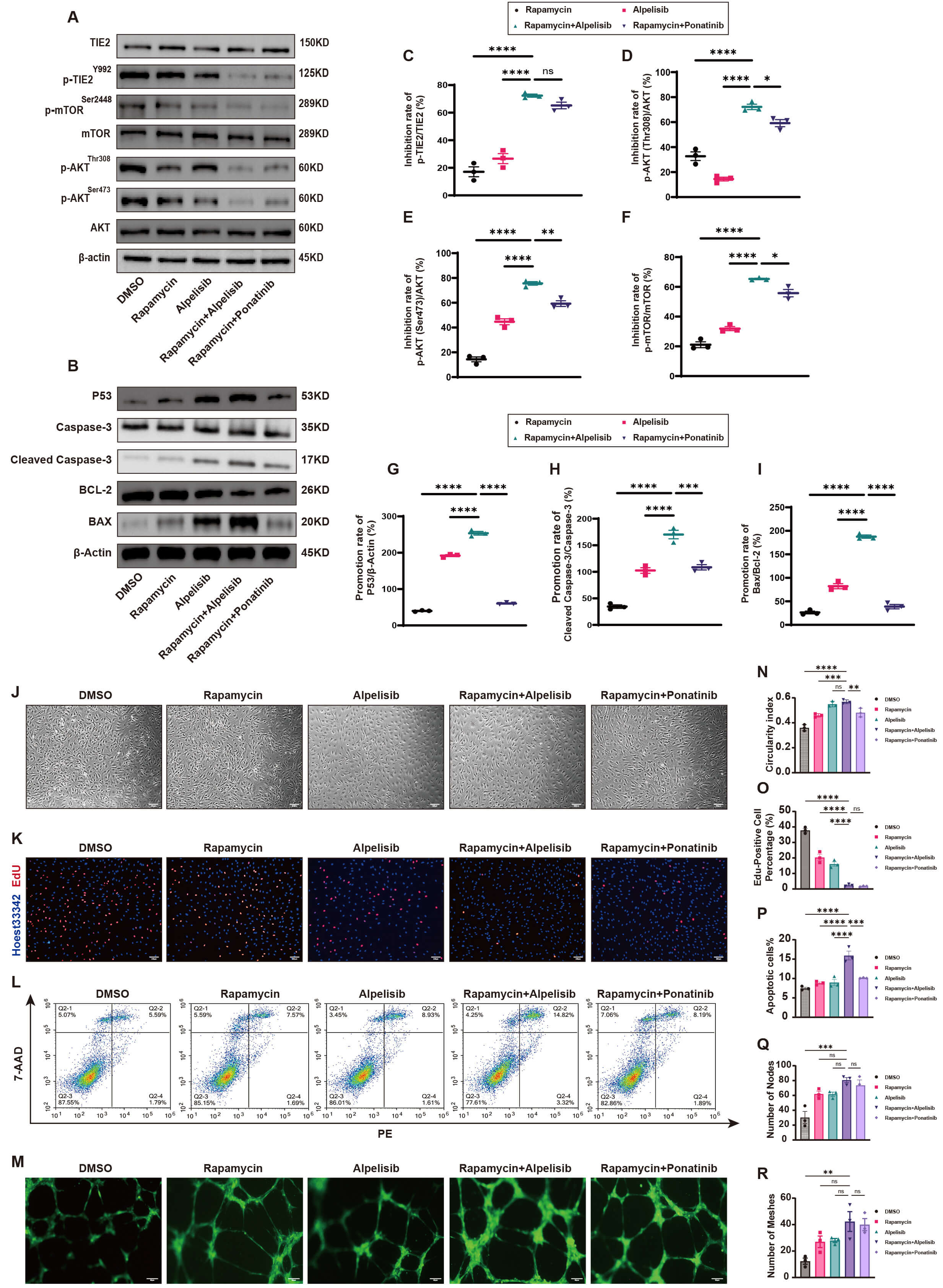 Fig. 3.
Fig. 3.
Rapamycin and Alpelisib combination ameliorates TIE2-L914F
pathological features via PI3K/AKT/mTOR inhibition and p53 restoration. (A)
Western blot reveals that PI3K inhibitors significantly suppress the activation
of the PI3K/AKT/mTOR pathway (n = 3). (B) Western blot reveals that PI3K
inhibitors significantly upregulate the expression of P53, Cleaved Caspase-3, and
BAX proteins (n = 3). (C–F) Quantitative analysis reveals that the Rapamycin and
Alpelisib combination significantly inhibits the expression of p-TIE2/TIE2 (C),
p-AKT (Thr308)/AKT (D), p-AKT (Ser473)/AKT (E), and p-mTOR/mTOR (F) (mean
Collectively, the Rapamycin and Alpelisib combination synergistically inhibits the PI3K pathway, effectively reversing pathological phenotypes in TIE2-L914F mutant endothelial cells and demonstrating significant therapeutic potential.
Regarding cell morphology, the Rapamycin and Alpelisib combination significantly reversed morphological abnormalities in TIE2-L914F mutant cells while exerting minimal effects on control (NC) and wild-type (WT) groups (Fig. 4A). Notably, cellular roundness was significantly increased in TIE2-L914F mutant cells (Fig. 4F), with no significant changes in NC and WT groups. Regarding cell proliferation, the combination significantly inhibited abnormal proliferation in TIE2-L914F mutant cells (Fig. 4B,C), showing significantly greater proliferation inhibition compared to NC and WT groups (Fig. 4G). Flow cytometry revealed that the combination significantly promoted apoptosis in TIE2-L914F mutant cells (Fig. 4D), exhibiting a significantly increased apoptosis rate compared to NC and WT groups (Fig. 4H). Regarding angiogenesis, the combination restored normal angiogenesis in TIE2-L914F mutant cells (Fig. 4E), significantly enhancing vascular node (Fig. 4J) and network structure formation (Fig. 4I), while exerting no significant effects on NC and WT groups. These results demonstrate that the Rapamycin and Alpelisib combination exhibits high selectivity and safety, exerting minimal effects on normal cells but significant therapeutic effects on TIE2-L914F mutant cells. Subsequently, transcriptome sequencing was performed on TIE2-L914F mutant cells treated with the Rapamycin and Alpelisib combination to further explore the underlying mechanisms. KEGG pathway analysis revealed that the PI3K-AKT signaling pathway was significantly enriched and ranked first (Fig. 4K), indicating that the combination primarily targets this pathway. GSEA further confirmed that the combination significantly inhibited the MTORC1 signaling pathway (HALLMARK_MTORC1_SIGNALING, Fig. 4L) and angiogenesis (HALLMARK_ANGIOGENESIS, Fig. 4M), validating the target specificity of this combination therapy against disease mechanisms and phenotypes.
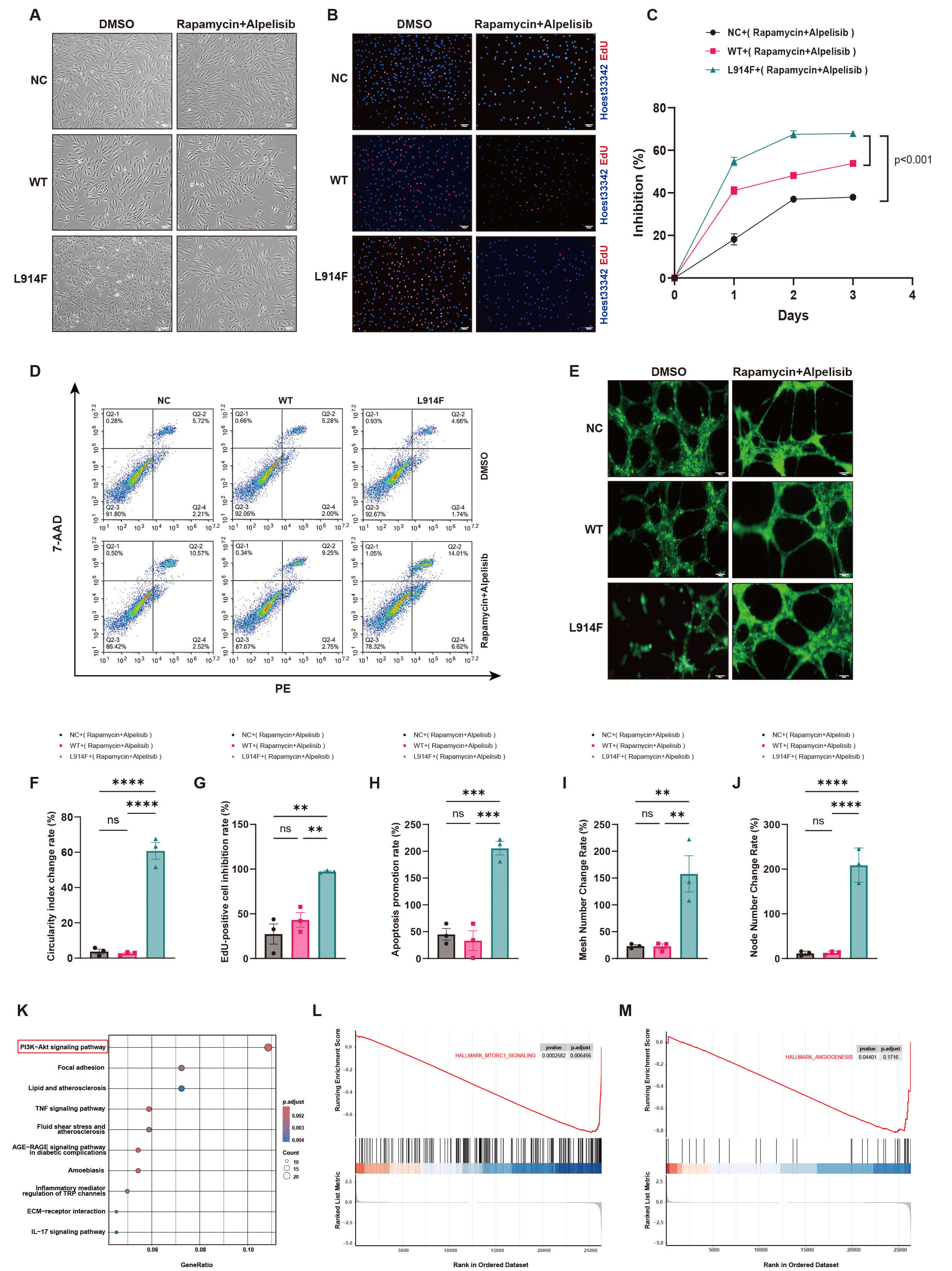 Fig. 4.
Fig. 4.
Rapamycin and Alpelisib combination exhibits high safety and
target specificity. (A) Inverted microscopy reveals that the combination
significantly improves the abnormal morphology of TIE2-L914F mutant cells but has
no significant effect on the morphology of normal control (NC) and wild-type (WT)
cells (n = 3), Scale bar = 200 µm. (B) EdU assay reveals that the
combination significantly inhibits the proliferation of TIE2-L914F mutant cells
but has minimal effects on NC and WT cells (red: EdU-positive cells, blue:
Hoechst-stained nuclei, n = 3), Scale bar = 200 µm. (C) Cell proliferation
curve reveals that the combination significantly inhibits the proliferation of
mutant cells but has minimal effects on NC and WT groups (mean
Collectively, within 48 hours, the low-concentration combination of Rapamycin and Alpelisib exhibits high selectivity and target specificity against the TIE2-mutated venous malformation cell model, while exerting minimal effects on normal endothelial cells. By targeting the PI3K pathway, it effectively inhibits proliferation, promotes apoptosis, and restores normal angiogenesis in TIE2-L914F mutant cells.
The efficacy of the Rapamycin and Alpelisib combination was further assessed using a 3D angiogenesis assay, as illustrated in Fig. 5A. In the 3D tubule formation assay, the combination significantly suppressed angiogenesis in TIE2-L914F mutant venous endothelial cells. Compared with the control (DMSO) and other treatment groups (Rapamycin, Alpelisib, and Rapamycin + Ponatinib), the Rapamycin and Alpelisib combination significantly decreased vascular branching (Fig. 5B), with quantitative analysis revealing a significant reduction in vascular sprout numbers (Fig. 5C). These results demonstrate that the combination exerts significant anti-angiogenic effects. Next, the efficacy of the combination was evaluated in nude mice, as illustrated in Fig. 5D. Lesions formed by TIE2-L914F mutant venous endothelial cells mixed with Matrigel were significantly suppressed in the Rapamycin and Alpelisib group (Fig. 5E), with lesion weight significantly reduced compared to controls (Fig. 5F). Immunofluorescence and immunohistochemical analysis further demonstrated that the combination significantly reduced the vascular area in lesion tissues (Fig. 5G,M), indicating effective suppression of lesion formation. Additionally, the combination significantly decreased the expression of p-TIE2 (Fig. 5H,N), p-AKT (Thr308 and Ser473, Fig. 5I,J,O,P), and p-mTOR (Fig. 5K,Q) in lesion tissues, while significantly increasing p53 expression (Fig. 5L,R). These findings indicate that the combination effectively suppresses lesion progression by targeting the PI3K/AKT/mTOR pathway and regulating p53 signaling.
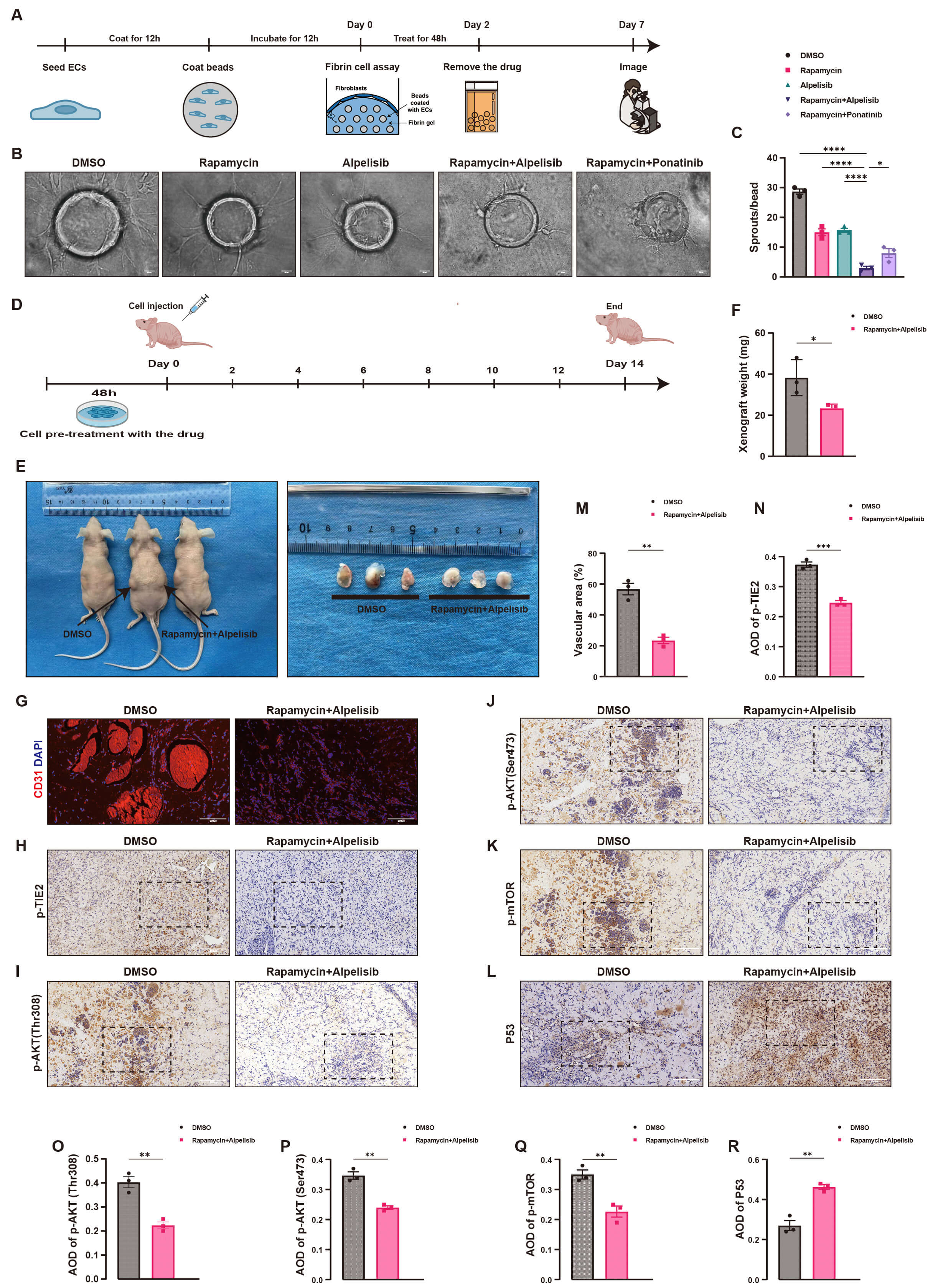 Fig. 5.
Fig. 5.
Rapamycin and Alpelisib combination suppresses angiogenesis in
3D tubule formation and lesion development in nude mice. (A) Flowchart of the 3D
tubule formation assay, illustrating the timeline of fibrin gel embedding, drug
treatment, and tubule formation analysis. (B) 3D tubule formation assay images
reveal that the Rapamycin and Alpelisib combination significantly inhibits
vascular sprout formation compared to the Dimethyl Sulfoxide (DMSO) control and
other drug groups (n = 3), Scale bar = 50 µm. (C) Quantitative analysis
reveals that the number of vascular sprouts in the Rapamycin and Alpelisib group
is significantly lower than in the DMSO control and other drug groups (mean
Collectively, the Rapamycin and Alpelisib combination significantly inhibits angiogenesis in 3D assays and suppresses lesion formation in nude mice. Through precise targeting of the PI3K/AKT/mTOR pathway and regulation of p53 expression, this combination therapy provides critical experimental evidence for treating TIE2-L914F mutation-related VMs.
Dysfunction of the PI3K pathway is prevalent in various VMs and represents one of the key molecular pathological mechanisms [12]. Although the PI3K pathway has been extensively studied in VMs associated with PIK3CA activating mutations [13, 15, 16], its role in VMs caused by TIE2 activating mutations remains poorly understood [24]. This study provides important experimental data on the role of the PI3K pathway in VMs with TIE2 activating mutations and provides a theoretical foundation for further targeted therapeutic strategies.
Initially, we compared the gene expression profiles of VM tissues and normal venous tissues through transcriptome sequencing analysis. The significant enrichment of the PI3K-AKT signaling pathway further supported the critical role of this pathway in the pathogenesis of VMs. The significant enrichment of pathways related to cytoskeleton regulation and extracellular matrix interactions further suggested that VMs may be associated with abnormal cell morphology and structure. Additionally, the significant activation of angiogenesis highlighted its critical role in the pathological process of VMs, consistent with the clinically observed characteristics of abnormal vascular proliferation [25]. To validate these findings, we performed HE and immunofluorescence staining on VM tissue sections, further supporting that the main pathological features of the disease are abnormal angiogenesis and overactivation of the PI3K pathway, providing important clues for further research into its pathogenic mechanisms.
The TIE2-L914F mutation represents the most frequent pathogenic mutation in sporadic VMs [18]. Although the PI3K pathway has been extensively studied in VMs associated with PIK3CA activating mutations, its specific role in VMs caused by TIE2-L914F mutations remains elusive. Therefore, we constructed a VM cell model with the TIE2-L914F mutation using lentiviral technology and demonstrated that the TIE2-L914F mutation hyperactivates the PI3K pathway by activating p-TIE2. This led to elongated or spindle-shaped changes in endothelial cell morphology, promoted cell proliferation, downregulated p53 to inhibit apoptosis, and impaired normal angiogenesis, driving lesion development. This cellular phenotype further supports the fundamental nature of venous malformations as congenital developmental vascular anomalies, which are non-neoplastic lesions, and in which abnormal endothelial cell proliferation constitutes one of their core histopathological features [26, 27]. The genetic basis of VMs involves acquired pathogenic mutations that lead to overactivation of the PI3K pathway, thereby triggering the disease. Therefore, therapeutic strategies targeting the PI3K pathway hold significant promise in the treatment of VMs [28, 29, 30].
Current research has shown that Rapamycin has been used in clinical trials, but
as a monotherapy, it has certain limitations in therapeutic efficacy [31, 32].
Alpelisib is a selective PI3K
Unexpectedly, we observed that the combination of Rapamycin and Alpelisib significantly inhibited the abnormal activation of the PI3K/AKT/mTOR pathway. This combination restored p53 signaling, significantly inhibited the proliferation of mutant cells, promoted apoptosis, and restored angiogenesis. Previous studies have shown that Rapamycin in combination with ponatinib, an ABL kinase inhibitor, can induce regression of venous malformations in mice and suppress AKT phosphorylation [22]. Compared with monotherapy or the previously reported combination regimen (Rapamycin + ponatinib), the combined treatment of Rapamycin and Alpelisib exerted significantly stronger synergistic inhibitory effects on cell apoptosis, the PI3K pathway, and the p53 pathway. These findings suggest that this dual-drug combination holds promising therapeutic potential for targeting hyperactivation of the PI3K pathway and inhibition of the p53 pathway, providing an important experimental basis for the clinical translation of this strategy. In our preliminary experiments, we also tested the triple combination of Rapamycin, Alpelisib, and ponatinib in HUVEC-TIE2-L914F cells. However, even at lower drug concentrations, extensive cell death occurred within 24 h of treatment. We hypothesize that this outcome may result from drug–drug interactions among the three agents, leading to enhanced cytotoxicity. Although traditional tubule formation assays provide some insights into angiogenesis, they are limited in simulating the real disease environment in vivo. Therefore, we further assessed the efficacy of the Rapamycin and Alpelisib combination using 3D tubule formation assays and a nude mouse xenograft model. The results demonstrated that the combination significantly inhibited 3D angiogenesis and effectively slowed the formation and progression of lesions in nude mice. Mechanistic studies demonstrated that the combination exhibited multi-target synergistic potential by efficiently inhibiting the PI3K/AKT/mTOR pathway and activating p53 signaling.
The PI3K-AKT signaling pathway and p53 signaling pathway are key intracellular signal transduction pathways that play a central role in regulating fundamental cellular processes including cell growth, proliferation, apoptosis, and metabolism. In recent years, studies have confirmed that aberrant activation or dysfunction of these two pathways is closely associated with the initiation and progression of various diseases, and there exists intricate subcellular crosstalk between them. Genotoxic stress can induce nuclear AKT activation, a process dependent on p53-mediated regulatory mechanisms, which is significantly distinct from the canonical membrane-localized activation mode of the PI3K-AKT pathway. Specifically, upon genotoxic stress, nuclear PI3K can bind to p53 in the non-membranous nucleoplasm to form a complex of p53 and phosphatidylinositol 3,4,5-trisphosphate (PtdIns(3,4,5)P3). This complex recruits AKT, Pyruvate Dehydrogenase Kinase 1 (PDK1), and mechanistic target of Rapamycin complex 2 (mTORC2), thereby activating AKT and phosphorylating Forkhead box O1 (FOXO) proteins, ultimately inhibiting DNA damage-induced cell apoptosis [35] (A p53-phosphoinositide signalosome regulates nuclear AKT activation). In the context of venous malformations, researchers have demonstrated that Rapamycin ameliorates the physiological functions of TIE2-mutant endothelial cells by inhibiting the AKT-mTOR pathway; meanwhile, it promotes the nuclear translocation of FOXO1 and the expression of PDGFB in cells, further improving the paracrine interaction between endothelial cells and smooth muscle cells (SMCs). Based on these findings, the AKT-FOXO1 axis has been proposed as a potential therapeutic target for TIE2-mutated venous malformations [36].
In the present study, we found that hyperactivation of the AKT-mTOR signaling pathway and suppression of p53 signaling occurred in TIE2-mutant HUVECs. Notably, combined treatment with Rapamycin and Alpelisib significantly inhibited the aberrant activation of the PI3K/AKT/mTOR pathway and restored p53 signaling expression in these mutant cells. Therefore, we hypothesize that the intricate subcellular crosstalk between the PI3K-AKT and p53 pathways may ameliorate the phenotypic abnormalities of venous malformation endothelial cells and exert potential therapeutic effects via the mechanism of nuclear PI3K binding to p53 to form a complex, which in turn recruits AKT/PDK1/mTORC2, activates AKT, and phosphorylates FOXO proteins. This feedback regulatory mechanism reveals the complex interactive pattern of the two pathways at the level of gene transcription in TIE2-mutated venous malformations. In fact, the PI3K-AKT and p53 signaling pathways exhibit multi-dimensional and tight mutual regulatory relationships: they not only achieve direct molecular crosstalk through mechanisms such as p53-dependent nuclear AKT activation and PI3K-AKT-mediated p53 degradation, but also display widespread co-enrichment characteristics and synergistic regulatory effects in various disease states. We thus speculate that the close association between these two pathways in venous malformations may lead to compensatory activation of one pathway when the other is targeted alone, thereby compromising the therapeutic efficacy. Future studies are warranted to clarify the specific interactive mechanisms of these two pathways in venous malformations through additional experiments, which will provide a theoretical basis for the development of novel targeted therapeutic agents and combination treatment strategies.
However, this study is subject to several specific limitations that may affect the interpretation of the results, which are described objectively as follows. First, the in vivo experiments were restricted to two groups: a negative control group (DMSO group) and a combination therapy group (Rapamycin + Alpelisib group), with the absence of a positive control group (e.g., Rapamycin + Ponatinib combination). This experimental design inherently limits the ability to directly compare the therapeutic efficacy of different combination regimens, as there is no reference positive treatment to benchmark the performance of the Rapamycin + Alpelisib combination. Nevertheless, the results clearly demonstrated that, compared with the negative control group, the Rapamycin + Alpelisib combination significantly inhibited lesion progression, which was mediated by the regulation of the PI3K/AKT/mTOR signaling pathway and p53 expression. To address this limitation, future studies should refine the experimental design by incorporating a positive control group, thereby enabling a more comprehensive evaluation of the therapeutic advantages of the Rapamycin + Alpelisib combination relative to other potential treatment strategies.
Additionally, to initially explore the synergistic effects of Rapamycin and Alpelisib and evaluate the therapeutic efficacy of their combination, the dose of the combination therapy was set to half of the experimental concentration used for each single drug. This approach was based on the hypothesis that the two drugs might exert synergistic effects at low doses, allowing for significant therapeutic efficacy while reducing potential toxic side effects. However, this dose selection lacks precise optimization, which may affect the accuracy of assessing the true synergistic potential and optimal therapeutic effect of the combination. Although the current results confirmed the presence of synergistic effects between the two drugs, future studies should further optimize the combination dose by calculating the combination index (CI), which will help to accurately determine the optimal ratio of the two drugs and further improve the therapeutic efficacy and safety of the combination therapy. Furthermore, future studies should conduct more systematic and large-scale in vivo animal experiments to further validate the long-term therapeutic efficacy, biosafety, and in vivo pharmacodynamic mechanisms of this combination regimen, so as to provide more solid experimental evidence for its potential clinical transformation.
The overactivation of the PI3K pathway drives venous malformations. This study reveals that Rapamycin and Alpelisib synergistically inhibit PI3K and activate P53, offering significant therapeutic potential for venous malformations.
All data generated or analyzed during this study are included in this article. For further inquiries, please contact the corresponding author directly.
YL and YH conceived and designed the experiments, performed the experiments, collected the data, and drafted the manuscript; JL, TW, and WW performed part of the experimental operations, collected and analyzed some of the data; ZCT conceived and designed the experiments, provided experimental guidance, analyzed the experimental data, translated the manuscript, and revised it critically. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work. All authors contributed to editorial changes in the manuscript.
All pathological tissue sections of venous malformations (VMs) were provided by the Department of Pathology at the First Affiliated Hospital of Kunming Medical University. We have signed a scientific research cooperation agreement with the Department of Pathology at the First Affiliated Hospital of Kunming Medical University and submitted the approval document (Approval No.: KHLL2023-KY198) from the Medical Ethics Committee of the First People’s Hospital of Yunnan Province. All gingival/skin tissues were obtained as discarded surgical materials from oral and maxillofacial surgeries at the First People’s Hospital of Yunnan Province, and Human umbilical vein endothelial cells (HUVECs) were isolated from umbilical cords provided by the Obstetrics Department of the First People’s Hospital of Yunnan Province, with approval from the Medical Ethics Committee of the First People’s Hospital of Yunnan Province (Approval No.: KHLL2023-KY198). Human Oral Mucosal Fibroblasts (HOMFs) were kindly provided by Professor Jiemei Zhai’s research group at the Affiliated Stomatology Hospital of Kunming Medical University. All experiments were conducted in strict accordance with the Declaration of Helsinki and relevant ethical guidelines and regulations, ensuring compliance with ethical requirements for human experimentation and the use of human tissue samples. Informed consent was obtained from all patients or their families, agreeing to the use of their pathological tissue sections or tissue samples for this study. The nude mice (nu/nu, Kunming Medical University Animal Research Institute, China) used in this study were purchased from the Kunming Medical University Animal Research Institute. All animal experiments were approved by the Animal Welfare Ethics Committee of Kunming Medical University (Approval No.: Kmmu20230003) and were conducted in strict compliance with principles of animal protection, animal welfare, and ethical guidelines. The study strictly adhered to the principles of Replacement, Reduction, and Refinement (3R).
We would like to express our sincere gratitude to all the individuals, institutions and fundings who contributed to this study. We sincerely acknowledge Professor Hefeng Yang, Professor Zheyi Sun, Professor Rongqiang Yang, Professor Zichao Dai, and Professor Weihong Wang from Kunming Medical University for their critical technical expertise provided during the experimental phase of this study.
This work was supported by the Yunnan Provincial Science and Technology Talent and Platform Planning Project (Grant No. 202305AF150120), and the Xingdian Talents Program Famous Medical Specialty Project (Grant No. XDYC-MY-2022-0049).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL48694.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.