



 , Haiyuan Li 2,*
, Haiyuan Li 2,*
1 Department of Gastroenterology, The Second Hospital and Clinical Medical School, Lanzhou University, 730030 Lanzhou, Gansu, China
2 Department of Tumor Surgery, Lanzhou University Second Hospital, 730030 Lanzhou, Gansu, China
Abstract
Nitric oxide (NO) is a key bioactive signaling molecule produced primarily by L-arginine by inducible NO synthase (iNOS), which is involved in signal transduction and performs a variety of biological functions. As the mainstay of the immune response, macrophage polarization is a dynamic and reversible process, allowing them to shift their functional states and immune-inflammatory properties in response to changing environmental cues. M1-type macrophages are polarized by stimuli such as lipopolysaccharide or interferon gamma. These pro-inflammatory cells secrete tumor necrosis factor alpha, interleukin-1β (IL-1β), IL-6, and other pro-inflammatory factors, which serve to initiate and amplify the inflammatory response. By contrast, M2-type macrophages are induced by IL-4 or IL-13 and express high levels of anti-inflammatory mediators such as IL-10 and transforming growth factor beta, which inhibit inflammation and promote tissue repair. Macrophages in the tumor immune microenvironment produce NO via arginine metabolism, with important impacts on tumorigenesis and immune escape. In turn, NO production regulates macrophage function by inducing a shift from the M2 type (which promotes tumor growth) to the M1 type (which suppresses tumor growth). In addition, NO affects the tumor immune response by regulating T-cell activity. Relevant therapeutic strategies for targeting NO include arginase (ARG) inhibitors, NO donors, and iNOS overexpression. This review summarizes the complex role of NO in tumor immunity and related therapeutic strategies. While the findings provide new directions for cancer immunotherapy, further research is needed to fully understand specific mechanisms, such as the dynamic regulation of the NO concentration threshold, the clinical translation of these findings, and the seemingly contradictory role of NO in immune cell function, ultimately leading to more effective treatment options.
Keywords
- tumor immune microenvironment
- macrophages
- nitric oxide
- arginine
- nitric oxide synthase
As a gaseous signaling molecule, nitric oxide (NO) plays diverse biological roles beyond modulating vascular tone and nerve conduction. Its dual role in the tumor immune microenvironment (TIME) has attracted increasing attention. Studies have shown that the concentration gradient of NO directly determines its pro-cancer or anti-cancer effects [1, 2]. This review systematically summarizes the metabolic regulatory network of NO within the TIME, elucidates its dual mechanisms of action and impact on immune cells (particularly macrophage polarization), and explores novel therapeutic strategies targeting the NO pathway.
The duality of NO stems from its spatiotemporal heterogeneity within the TIME, a phenomenon rooted in the competitive metabolism of the common substrate L-arginine by two key enzymes: inducible NO synthase (iNOS) and arginase (ARG) [3, 4, 5]. This competition creates discrete microenvironments where defined concentration thresholds dictate opposing functional outcomes: activating or inhibiting specific signaling pathways in both tumor and immune cells [6, 7].
Early studies on NO focused on its direct cytotoxic effects. For example,
activated macrophages produce high concentrations of NO from L-arginine using
iNOS, which kill tumor cells by inhibiting the mitochondrial respiratory chain
and DNA replication, thereby killing tumors. In addition, the synergistic effect
of NO with tumor necrosis factor alpha (TNF-
Recent studies have revealed the contradictory role of NO in immunosuppression. Although high concentrations of NO directly kill tumor cells, it simultaneously weakens the overall anti-tumor immune response. This weakening occurs because NO inhibits the activation and proliferation of T cells and can even induce the production of regulatory T cells, which suppress the immune system’s activity.
In addition, tumor cells competitively deplete L-arginine by upregulating ARG1. This depletion of L-arginine prevents macrophage iNOS from producing enough NO. To make matters more complicated, the remaining NO can hinder T cells from reaching the tumor core by nitrifying chemokine C-C motif ligand 2 (CCL2). These findings suggest that simply increasing the concentration of NO can worsen immune escape; therefore, it is necessary to explore spatiotemporal-specific regulatory strategies [10].
To address these abovementioned issues, researchers have significantly enhanced the therapeutic potential of NO through metabolic interventions. For example, L-arginine supplementation has been shown to not only effectively restore macrophage iNOS activity but also clearly improve survival rates in patients with gastric cancer in clinical applications [11]. However, the heterogeneity of arginine transporter expression in the TIME may limit efficient drug delivery. Furthermore, the short half-life and non-specific targeting of NO donors necessitate the development of intelligent delivery systems capable of adapting to the unique physicochemical properties of the TIME.
Therefore, this review first delineates the sources and regulation of NO in the TIME. It then provides a detailed, concentration-dependent analysis of its pro-tumor and anti-tumor mechanisms, with a particular focus on its role in orchestrating macrophage polarization and T-cell function. Finally, we critically evaluate the current landscape of NO-based therapeutic interventions and discuss future directions for harnessing NO in cancer immunotherapy.
Macrophages play a critical and undeniable role in tumor development and progression, a process significantly influenced by their uptake of NO. Additionally, immune cells within the TIME are integral components of the body’s anti-tumor defense mechanisms [12]. Arginine enters the cell via the cationic amino acid transporter (CAT) proteins CAT-1 and CAT-2B. Once inside, the intracellular metabolism of arginine is a pivotal control point in tumor immunity, primarily governed by the enzymatic competition between NOS and ARG, which drive divergent immunological outcomes (Fig. 1). Depending on the iNOS/ARG ratio, L-arginine can be catalytically metabolized to NO and citrulline by NOS or to ornithine and urea by ARG1 or ARG2. Neuronal NO synthase (nNOS), endothelial NOS (eNOS), and iNOS are isozymes of NOS [13]. The first two types of NOS, nNOS and eNOS, are continuously expressed in neuronal cells, endothelial cells, and platelets under normal physiological conditions. By contrast, iNOS is primarily produced by immune cells and is not expressed under physiological conditions. Instead, iNOS is highly expressed in tumors and during inflammation, suggesting a strong association with these pathological conditions [5]. The competition between iNOS and ARG for their shared resource, L-arginine, establishes a key regulatory axis within the TIME.
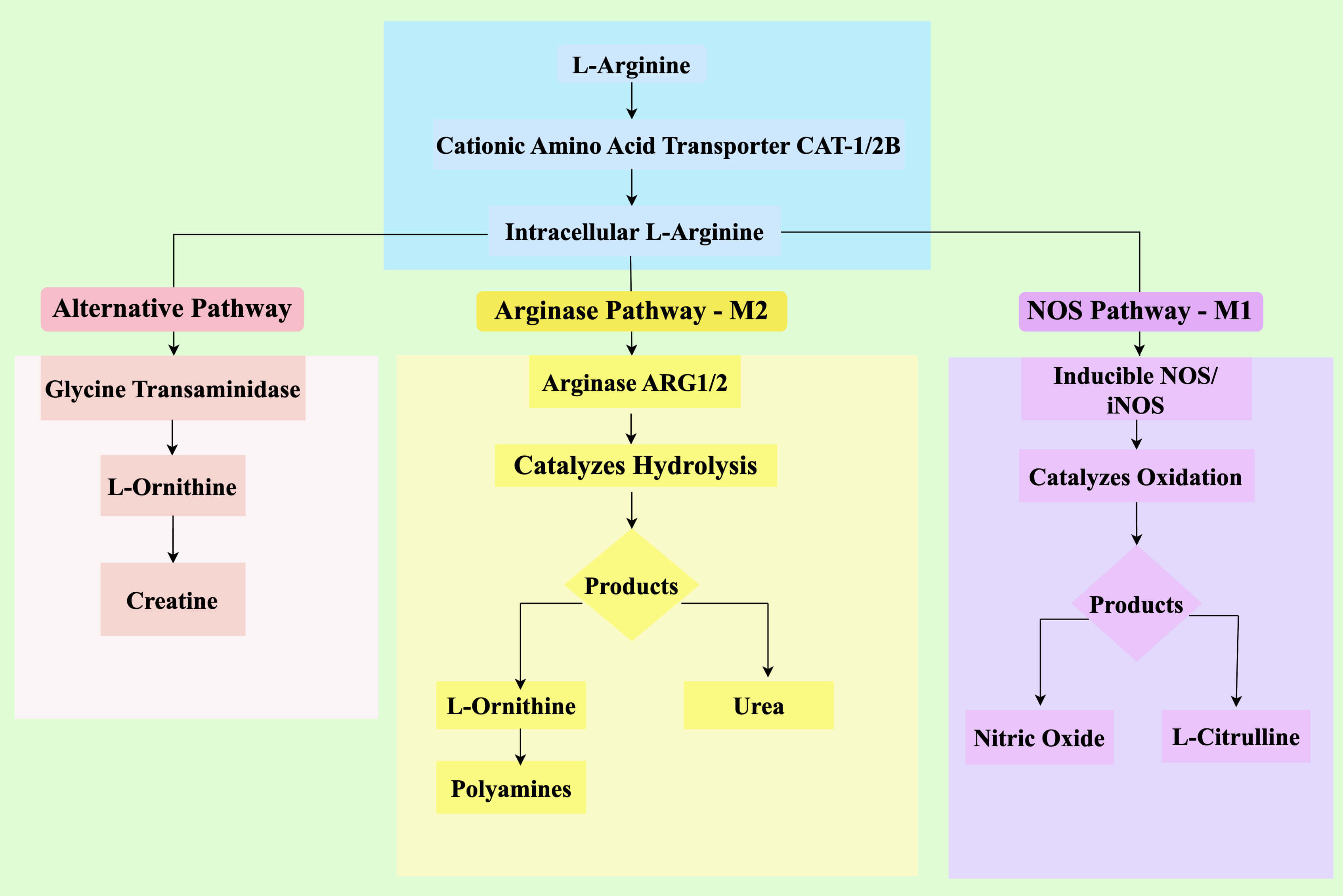 Fig. 1.
Fig. 1.
Metabolic pathways of L-arginine in the TIME and their
immunologic consequences. L-arginine is imported via CATs and serves as a
substrate for three major metabolic pathways. Left panel (Alternative pathway): A third pathway involving glycine transaminase converts arginine to creatine. Middle panel (Arginase pathway, M2): ARG1/2 catalyzes the hydrolysis of L-arginine to ornithine and urea. Ornithine fuels polyamine synthesis, supporting tumor proliferation. Right panel (NOS pathway, M1): iNOS catalyzes the oxidation of L-arginine to NO and L-citrulline. NO mediates direct tumor cytotoxicity and promotes M1-like macrophage polarization, driving anti-tumor immunity. The competition between iNOS and ARG for L-arginine is a critical determinant of the overall immune phenotype in the TIME. TIME, tumor immune microenvironment;
CAT, cationic amino acid transporter; NO, nitric oxide; iNOS, inducible NO
synthase; IFN-
iNOS is upregulated in classically activated (M1) macrophages by
inflammatory stimuli such as interferon gamma (IFN-
ARG (with the cytosolic ARG1 isoform being dominant in this context) is highly
expressed in alternatively activated (M2) macrophages and myeloid-derived
suppressor cells in response to cytokines such as interleukin 4 (IL-4) and IL-13.
ARG hydrolyzes L-arginine to L-ornithine, which serves as a precursor for
polyamines that fuel tumor cell proliferation and tissue remodeling [14, 15]. A
critical immunosuppressive mechanism of ARG is depletion of the local L-arginine
pool. This substrate starvation impairs T-cell function by downregulating
expression of the T-cell receptor CD3
Three known metabolic pathways are involved in the in vivo metabolism of arginine: one primarily mediated by ARG, another by NOS, and a third pathway involving glycine transaminase (Fig. 1).
ARG1 and ARG2 are two types of the enzyme ARG, which plays an essential role in the metabolic functions of organisms. In the liver, ARG1, sometimes referred to as “hepatic ARG”, is highly active and hydrolyzes L-arginine into urea and L-ornithine, a process known as the urea cycle. ARG2, also referred to as “extrahepatic ARG”, is found in many tissues such as the kidney, brain, small intestine, mammary gland, and macrophages. It primarily controls the amount of intracellular arginine, which in turn controls the production of NO, proline, polyamines, and other chemicals in living organisms [14, 15]. However, it does not participate in the urea cycle.
NO-mediated immunometabolic reprogramming extends beyond arginine metabolism; rather, NO directly modulates the tricarboxylic acid (TCA) cycle in immune cells by S-nitrosylating key enzymes (e.g., aconitase and succinate dehydrogenase), thereby impairing mitochondrial respiration in T cells while promoting glycolysis in M1 macrophages [8]. This metabolic switch reinforces the pro-inflammatory phenotype of M1 macrophages but concurrently suppresses effector T-cell function, illustrating NO’s paradoxical role in immune regulation. Furthermore, NO-derived peroxynitrite (ONOO–) enhances the immunosuppressive activity of programmed cell death protein 1 (PD-1) in the TIME by nitrating the tyrosine residues on PD-1 [9].
Due to its source, timing, and concentration, NO functions as a dual mediator in
cancer. It is also linked to the sensitivity of tumor cells to the NO they
produce themselves [16, 17, 18]. The pleiotropic effects of NO in the TIME are
governed by its local concentration, which can be categorized into distinct
functional windows: low-level (pM to low nM) NO primarily mediates signaling
functions that can promote tumor progression; intermediate-level NO can exert
context-dependent pro- or anti-tumor effects; and high-level (high nM to
µM) NO induces nitrosative stress and direct cytotoxicity [6]. The
transition between these functional states is dynamically regulated by the
competition for L-arginine between iNOS and ARG, the intrinsic stability of NO,
and the expression of downstream sensors such as soluble guanylyl cyclase (sGC)
and hypoxia inducible factor 1 alpha (HIF-1
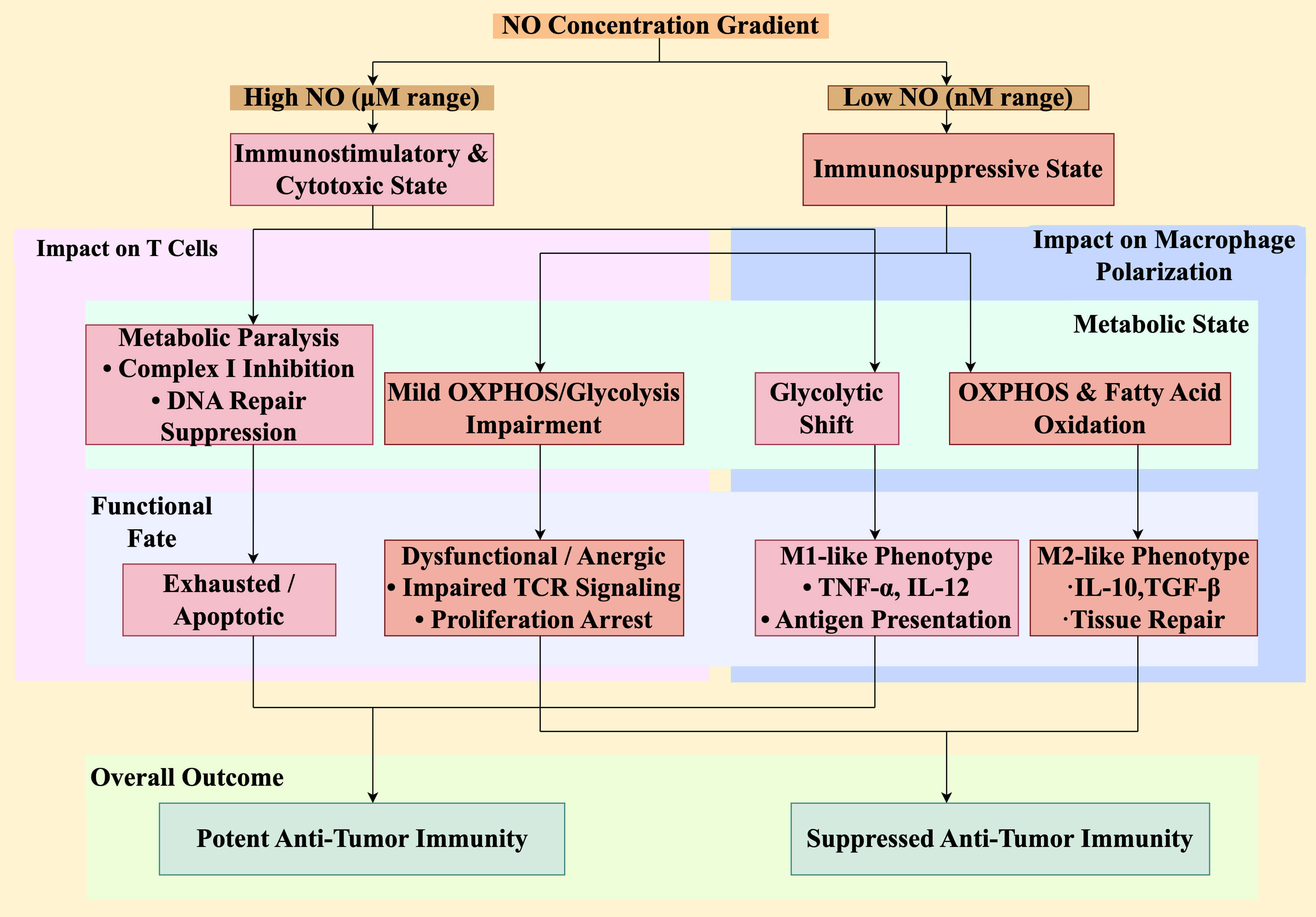 Fig. 2.
Fig. 2.
NO concentration determines immune cell metabolic fitness and
functional fate. The level of NO directly and differentially regulates the
metabolism and function of key immune cells, creating a concentration-dependent
immunological switch. (Right) In low NO environments (nM range), T cells
experience mild metabolic impairment leading to a dysfunctional or anergic state,
characterized by defective TCR signaling and proliferation arrest. Concurrently,
macrophages are skewed towards an M2-like phenotype, supported by oxidative
metabolism (oxidative phosphorylation [OXPHOS] and fatty acid oxidation), and
secrete immunosuppressive cytokines such as IL-10 and TGF-
The concept of concentration thresholds is central to understanding NO’s duality; thus, this subsection details the basis of the concentration-dependent effects of NO. The molecular basis for these thresholds lies in the differential affinity of NO for its molecular targets. For instance, low-level NO primarily activates the high-affinity sGC-cyclic guanosine monophosphate (cGMP) pathway, influencing cell proliferation and vascular tone [19, 20]. By contrast, high-level NO saturates these pathways and engages lower-affinity targets, leading to pervasive protein S-nitrosylation, mitochondrial dysfunction, and DNA damage. The dynamic interplay between iNOS (driving NO production) and ARG (limiting the L-arginine substrate) creates spatially and temporally restricted concentration gradients within the TIME, establishing discrete microenvironments where either pro- or anti-tumor programs dominate [7].
Low NO levels can encourage the growth and spread of tumors. These pro-tumor effects operate through two interconnected axes: direct stimulation of tumor cells and suppression of anti-tumor immunity.
First, low NO concentrations directly stimulate tumor growth through several mechanisms. They directly stimulate cancer cell proliferation and survival via the NO/sGC/cGMP-dependent pathway [3]. In addition, low NO levels induce tumor protein p53 (p53) mutations and other pathways to prevent apoptosis. The p53 gene normally expresses various microRNAs (miRNAs), and its encoded p53 protein is crucial for mediating cell cycle arrest and apoptosis. This process can cause tumor cells to undergo apoptosis. However, when the p53 gene is mutated, the ability of NO to cause tumor cell apoptosis is blocked. Finally, at low concentrations, NO participates in the activation and phosphorylation of extracellular signal-regulated kinase, phosphoinositide 3-kinase (PI3K)/Akt/mammalian target of rapamycin, signal transducer and activator of transcription (STAT), and rat sarcoma virus oncogene (Ras) pathways; dilates blood vessels in tumor tissues; and promotes tumor angiogenesis [5].
Second, and perhaps more critically, low concentrations of NO potently suppress
anti-tumor immunity, fostering an immunosuppressive microenvironment. This
multi-faceted suppression occurs on several fronts. On T cells, low-dose NO can
impair T-cell receptor (TCR) signaling by S-nitrosylating key proximal signaling
molecules such as lymphocyte-specific protein tyrosine kinase (Lck) and
zeta-chain-associated protein kinase 70 (ZAP-70), leading to defective T-cell
activation and effector function [21]. On macrophages, the low-level NO/sGC/cGMP
axis helps sustain the immunosuppressive M2 phenotype, promoting the secretion of
anti-inflammatory mediators such as IL-10 and transforming growth factor beta
(TGF-
The growth and spread of tumors are inhibited by high NO concentrations; the NO concentration that inhibits tumor growth is 10–100 times greater than the concentration promoting tumor growth. The anti-tumor mechanisms of high-level NO operate through two synergistic arms: direct cytotoxicity toward tumor cells and reprogramming of the immunosuppressive TIME into an anti-tumor state.
The direct cytotoxic effects are well-established and multifaceted. The
cytotoxic effector molecule NO, which is produced by activated macrophages in the
TIME, can inhibit the metabolic processes of tumor cells, including DNA
replication and aerobic respiration, which causes tumor cells to lose a
significant amount of iron and ultimately die [24]. Moreover, NO and
TNF-
NO influences macrophage polarization through metabolic rewiring. When the M1
inflammatory state occurs, iNOS produces NO, which helps stabilize
HIF-1
Macrophages constitute the most prominent phagocytic cell population within the
innate immune system and are found throughout the body’s tissues [35]. Upon tumor
development, contingent upon the specific microenvironmental context, monocytes
originating from the bone marrow are mobilized to the tumor site where they
undergo differentiation into either the classically activated M1-type
macrophages, which suppress tumor growth, or into the alternatively activated
M2-type macrophages, which facilitate tumor progression, expansion, and
metastasis [34]. NO regulates metabolic reprogramming of macrophages in the TIME,
mainly through the TCA cycle. Tumor-associated macrophages (TAMs) can be
reprogrammed into the tumor-killing M1-type through stimulation with
M1-associated signals such as IFN-
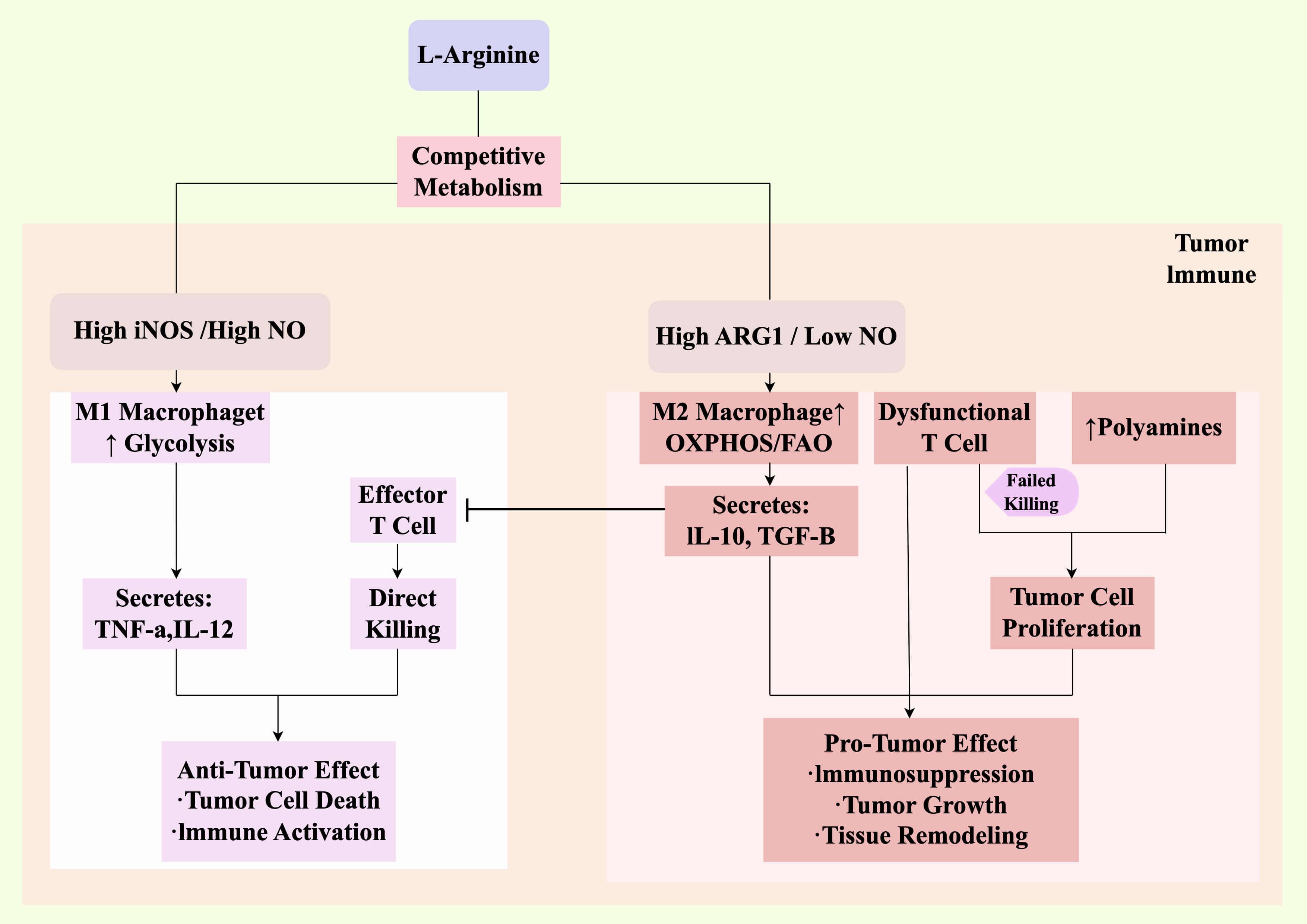 Fig. 3.
Fig. 3.
Integrated dual role of NO in shaping the TIME. This schematic
synthesizes the metabolic and cellular mechanisms to show how NO contributes to
spatial heterogeneity and functional outcomes within the TIME. The competition
for L-arginine (as detailed in Fig. 1) creates two distinct microenvironments.
(Left, anti-tumor niche) In regions with high iNOS activity and elevated NO,
macrophages are polarized to an M1 state (glycolytic metabolism), secreting
pro-inflammatory cytokines that activate and recruit effector T cells. High NO
also directly induces tumor cell death. This synergy creates a localized
anti-tumor niche. (Right, pro-tumor niche) In regions with high ARG1 activity and
low NO, macrophages are polarized to an M2 state (oxidative metabolism),
secreting immunosuppressive cytokines such as IL-10 and TGF-
Arginine utilization is the most researched metabolism in terms of distinguishing M1 from M2 macrophages. M1 and M2 macrophages in tumor microcircuits use arginine in various ways. M1 TAMs are regulated primarily by iNOS, which converts L-arginine to NO and citrulline [37, 38], as well as by reactive oxygen species (ROS) production and the expression of major histocompatibility complexes I and II (MHC-I/MHC-II), which activate the Th1 immune response and play a tumor-killing role. Type M2 TAMs are primarily controlled by ARG, which converts L-arginine to ornithine and urea, dictating the quantity of NO generated by TAMs [39]. NO generated by activated macrophages in response to iNOS plays a role in cytotoxicity and anti-tumor activity [40, 41]. However, macrophages themselves are also targets of self-initiated NO production, and NO has been shown to induce apoptosis in activated macrophages [42, 43, 44].
The genotoxic potential of NO metabolites was confirmed because NOS inhibitors prevented DNA damage in activated macrophages. NO produces localized reactive nitrogen species (RNS) at high fluxes of 1–5 µM. These RNS react with sulfhydryl-containing peptides to form nitrosothiols, which change the functions of ion channels [45], p21ras [45], protein tyrosine phosphates [46], and cyclooxygenases [47]. These changes can damage activated macrophage DNA, which can cause apoptosis and mitochondrial damage. The suppression of DNA repair proteins by protein S-nitrosylation, such as zinc-containing formamidopyrimidine DNA glycosylase (Fpg protein) [48] and O-6-methylguanine-DNA methyltransferase [49], also increases the DNA-damaging activity of NO and its metabolites. The natural vascular system causes immune cell attachment and extravasation into nearby tissues when exposed to pathogens or LPS. However, by downregulating the expression of adhesion molecules in the tumor vasculature, low levels of NO inhibit immune cell adhesion and homing, preventing immune cells from entering the tumor and thus potentially contributing to the growth of additional cancers [50].
Low-dose NO donor treatment modulates M2 to M1 polarization [46, 51], and NO donors (e.g., S-nitrosoglutathione [GSNO] and S-nitrosothiol) can be converted to NO by intracellular metabolic processes to modulate the macrophage polarization status; however, the exact mechanism remains unclear. Collectively, these findings highlight that fine-tuning NO levels in the TIME can reprogram TAMs into a tumor-suppressive state, enhancing both local and systemic anti-tumor immunity.
In addition to metabolic reprogramming, NO directly reshapes the expression of
co-stimulatory molecules on macrophages. Low-dose SNAP significantly upregulates
M1 phenotypic markers (cluster of differentiation 80 [CD80], CD86, and MHC-II)
while downregulating M2 markers (CD206 and programmed death-ligand 1 [PD-L1]),
thereby enhancing their antigen presentation and T-cell co-stimulation
capabilities [52, 53]. Mechanistically, NO promotes the nuclear translocation of
nuclear factor kappa B (NF-
At the cytokine level, NO fine tunes the M1/M2 cytokine balance. In M1
polarization, iNOS-derived NO stabilizes HIF-1
In the tumor microenvironment (TME), NO affects the polarization state of macrophages by regulating their metabolic reprogramming (such as the iNOS/ARG1 balance). Recent studies have indicated that NO can further strengthen the polarization of macrophages by altering their phenotype through epigenetic modifications. This form of epigenetic regulation provides a more durable molecular basis for NO-mediated immune regulation.
NO not only regulates macrophage polarization through classical metabolic pathways (such as the TCA cycle and arginine metabolism), but also affects macrophage polarization through dynamic modification of epigenetic regulatory factors, such as DNA methylation, histone modification and non-coding RNA, thereby influencing the long-term functional status of cells. NO influences all three major layers of epigenetic regulation as follows.
NO can modulate the activity of DNA methyltransferases (DNMTs),
which are enzymes that add methyl groups to cytosine-phosphate-guanine (CpG)
islands, typically leading to gene silencing. For example, under hypoxia
conditions, NO-derived RNS can inhibit DNMT1 activity, potentially through
S-nitrosylation or by altering the cell’s redox state. This inhibition reduces
the methylation level in the promoters of key genes (e.g., M1-associated genes
like TNF-
NO directly and indirectly alters “histone marks” (chemical tags on the proteins that package DNA), which control chromatin accessibility.
NO can modify and inactivate histone deacetylases (HDACs),
through S-nitrosylation. A specific example of this modification is inactivation
of the Cys274 site on the HDAC2 protein after it is nitrosylated. This
inactivation prevents HDAC2 from removing acetyl groups from histones, which
causes the acetyl groups to accumulate and results in increased levels of histone
H3 lysine 9 acetylation (H3K9ac) and H3K27ac. These active chromatin marks,
specifically H3K9 acetylation, occur at the promoters of pro-inflammatory genes,
leading to the increased production of cytokines such as TNF-
The NO-ARG balance influences metabolites that act as co-factors for histone-modifying enzymes. For instance, ARG1-driven production of polyamines can consume S-adenosylmethionine (SAM), a universal methyl donor. A reduction in SAM availability can lead to H3K4me3 (an active mark) at M1 gene promoters. Furthermore, M2-associated signals can promote the activity of histone methyltransferases like Enhancer of Zeste Homolog 2 (EZH2), which catalyzes the repressive mark H3K27me3 at the iNOS and IL-12 promoters, silencing them. NO, by inhibiting ARG1 and shifting metabolism, can reverse these repressive methylation patterns [60, 61].
NO can influence the expression of miRNAs that form feedback loops in polarization. For instance, high concentrations of NO have been shown to induce the expression of miR-21-5p. This miRNA targets and inhibits the tumor suppressor Phosphatase and Tensin Homolog (PTEN), leading to activation of the PI3K/Akt pathway, which can paradoxically promote cell survival and potentially impair the sustained tumor killing capacity of M1 macrophages, illustrating a negative feedback mechanism.
These findings suggest that NO plays a dual role in macrophage polarization through epigenetic mechanisms, potentially enhancing both anti-tumor immunity and promoting immune escape. More importantly, these epigenetic mechanisms demonstrate that NO does not merely trigger transient phenotypic changes but can “lock in” macrophage characteristics by altering the epigenetic landscape, offering novel targets for durable reprogramming of the TIME.
NO acts as a signaling molecule and plays an important regulatory role in multiple aspects of T-cell development, differentiation, and activation. NO synthesized by iNOS in T cells, as well as NO produced by dendritic cells (DCs) and macrophages in the TME, can act on T cells. High concentrations of NO have broad immunosuppressive effects, including impacts on the differentiation and effector functions of Th1, Th2, Th17, and Treg cells (Tregs) [65].
In the TME, the interaction between macrophages and T cells is a key aspect of
NO-mediated immune regulation. IFN-
NO not only affects tumor immunity by modulating T-cell function but also regulates the behavior and activity of CD8+ T cells by promoting vascular normalization and degradation of the extracellular matrix, thereby exerting anti-tumor effects. In NOD/SCID (severe combined immune deficiency) mouse models, low doses of exogenous NO donors fail to inhibit tumor growth. However, anti-tumor effects have been observed in immunocompetent mouse models, and these effects disappeared with the depletion of CD8+ T cells [2, 67]. These findings indicate that the anti-tumor effects of NO depend on the presence of CD8+ T cells. Moreover, low-dose treatment with SNAP can promote the depletion of Tregs, thereby enhancing the immune system’s response to tumors. NO, as an immunotherapeutic agent, can also synergize with anti-PD-1 therapy [68]. Therefore, the combination of low-dose SNAP and anti-PD-1 antibodies may have a synergistic effect on tumor regression.
NO exerts concentration-dependent effects on T-cell function through distinct metabolic and signaling mechanisms, creating a biphasic regulatory paradigm. The molecular basis for this duality lies in the differential engagement of specific pathways.
At
physiologically low concentrations, NO primarily activates the high-affinity
soluble sGC, leading to a sustained increase in intracellular cGMP. Elevated cGMP
levels activate protein kinase G, which phosphorylates downstream targets. This
pathway can promote T-cell migration by modulating cytoskeleton dynamics and
integrin affinity, and mildly enhance T-cell proliferation by regulating
transcription factors like nuclear factor of activated T cells. In the context of
anti-tumor immunity, this pathway contributes to vascular normalization,
improving perfusion and enhancing CD8+ T-cell infiltration into tumor cores.
Concurrently, low-level NO can mediate the nitrosative inactivation of
TGF-
As NO concentrations rise, it modifies lower-affinity targets primarily through S-nitrosylation, a redox-based post-translational modification that can irreversibly alter protein function, resulting in widespread suppression of cellular activities.
Key proximal signaling molecules in the TCR cascade, such as Lck and ZAP-70, are susceptible to S-nitrosylation, which inhibits their kinase activity. This results in defective TCR signal transduction, impaired immunological synapse formation, and ultimately, defective T-cell activation and effector function [4].
NO directly S-nitrosylates and inhibits key components of the mitochondrial respiratory chain, particularly complex I. This inhibition disrupts oxidative phosphorylation (OXPHOS), severely damaging the metabolic fitness of T cells which require robust mitochondrial function for sustained activity and memory formation [8].
High levels of NO can cause DNA damage directly or through RNS. Furthermore, S-nitrosylation of DNA repair enzymes, such as poly(ADP-ribose) polymerase-1, suppresses their activity, exacerbating genomic instability and potentially leading to T-cell apoptosis or functional exhaustion [8]. Thus, the impact of NO on T-cell fate is heavily concentration-dependent. At low levels, it can support anti-tumor immunity by enhancing T-cell trafficking and altering the cytokine milieu. However, as concentrations rise into the pathological range, a dramatic shift occurs: the cumulative inhibition of TCR signaling, mitochondrial metabolism, and DNA repair machinery leads to a state of metabolic paralysis and functional exhaustion in T cells, effectively crippling their anti-tumor capacity.
This biphasic regulation explains why strategies to boost NO in TIME must be precisely controlled, as excessive production can inadvertently suppress the very immune cells needed for tumor eradication. It also highlights the therapeutic potential of precisely tuned NO modulation, particularly when combined with immune checkpoint blockade to simultaneously overcome metabolic immunosuppression and enhance T-cell effector function in the TIME [68].
NO exerts a concentration-dependent dual modulation of NK cell functions. Low concentrations of NO enhance NK cell cytotoxicity by upregulating activating receptors (e.g., NK group 2D) and granzyme B expression, thereby potentiating direct tumor cell lysis. Conversely, high concentrations of NO may impair NK cell activity by inhibiting the mitochondrial respiratory chain or triggering apoptotic signaling cascades [69, 70]. Within the TIME, tumor-derived NO downregulates NK cell chemokine receptors such as C-X-C motif chemokine receptor 3, restricting NK cell infiltration into tumor sites. By contrast, inducible NO produced by macrophage or DC-derived iNOS can promote NK cell-mediated tumor lysis via STAT5 activation [71]. These mechanisms provide a rationale for combination therapies, and it is postulated that NO donors (e.g., GSNO) combined with IL-2 could significantly augment the anti-tumor efficacy of NK cells.
NO alleviates tumor-associated inflammation by inhibiting mast cell
degranulation through S-nitrosylation modification of key signaling proteins
(e.g., spleen tyrosine kinase), which in turn reduces the release of histamine
and pro-inflammatory factors (e.g., TNF-
NO affects the anti-tumor immune response by regulating the maturation state of
DCs. The physiological concentration of NO promotes the expression of MHC-II and
co-stimulatory molecules (CD80/CD86) on the surface of DCs by activating the
NF-
NO participates in tumor immune balance by regulating neutrophil function. On the one hand, NO inhibits NADPH oxidase activity and reduces neutrophil extracellular trap (NET) formation, thereby reducing the risk of NET-mediated tumor metastasis [79, 80]. On the other hand, low concentrations of NO promote neutrophil chemotaxis to tumor sites by upregulating CD11b expression, whereas high concentrations of NO induce their apoptosis through caspase-3 activation, limiting chronic inflammatory damage [81]. In addition, NO donors, such as nitroglycerin, attenuate chemotherapy-induced neutropenia, suggesting its potential value in protecting immune cells [82, 83].
ARG generated from TAMs is becoming a significant therapeutic target for a number of malignancies [84]. T-cell function is inhibited when ARG breaks down arginine. Certain inhibitors of arginine metabolic enzymes (e.g., NG-monomethyl-L-arginine [L-NMMA] and L-N5-(1-iminoethyl)ornithine [L-NIO]) not only compete to inhibit the CAT-1 transporter but also enable iNOS in arginine. Clinically, the upregulation of ARG activity and the expression of ARG are linked to a number of cancers, including gastric cancer, breast cancer [85], hepatocellular carcinoma [86], renal carcinoma [87, 88], and head and neck cancers [89]. There is an advantage in the competitive regulation of acid metabolic enzymes (iNOS and ARG), which further elevates the concentration of NO, inhibiting tumorigenesis [90].
Given the dual role of NO in the growth, proliferation, and metastasis of tumor cells [3], controlling parameters such as the site of NO delivery, NO concentration, and the rate of NO release is crucial for developing NO-related therapies. Inducing iNOS overexpression (via gene therapy) or administering NO donors are emerging strategies for NO-based tumor treatment [91, 92, 93]. Clinical studies have shown that NO donors and NO synthesis induced by iNOS can kill certain hematologic tumor cells, such as leukemia, lymphoma, and myeloma cells [94]. The following are the applications and mechanisms of different NO donors in tumor immunotherapy (Table 1, Ref. [3, 8, 22, 64, 66, 67]). These NO donors demonstrate the diversity and potential of NO-based therapies, providing new ideas and approaches for future cancer treatment.
| NO donor type | Mechanism of action in the TIME | Reference |
| Organic Nitrates | Release NO to inhibit tumor cell DNA replication and aerobic respiration, inducing apoptosis. | [3] |
| S-Nitrosothiols | Modulate immune cell function in the TME, enhancing anti-tumor immunity. | [8] |
| Nitrosoamines | Release NO locally at the tumor site to inhibit tumor cell proliferation, invasion, and metastasis. | [22] |
| Metal–NO Complexes | Release NO to enhance tumor cell sensitivity to radiotherapy and chemotherapy. | [8] |
| Nanoparticle-Delivered NO | Release NO locally at the tumor site to inhibit tumor cell growth, avoiding systemic toxicity. | [64] |
| Gene Therapy-Induced NO Production | Enhance tumor cell sensitivity to chemotherapeutic drugs through iNOS gene transfection. | [8] |
| Cationic Liposome-Mediated iNOS Transfection | Enhance the anti-tumor effects of cisplatin in lung cancer. | [66] |
| eNOS Activators | Target eNOS activation to prevent lung metastasis in breast cancer. | [67] |
TME, tumor microenvironment; eNOS, endothelial NOS.
Current NO donors may cause systemic toxicity, such as cytokine release syndrome, necessitating the development of novel NO donors with localized action to prevent systemic effects [91]. Recent approaches have focused on controlling the release and delivery of NO to inhibit cancer cell growth. For example, the use of nanoparticles for NO delivery has shown promising results. Another method involves targeting NO production via gene therapy to avoid systemic toxicity. A study found that NO deficiency due to reduced eNOS activity may be an early cause of lung metastasis in breast cancer, suggesting that targeting eNOS activation can prevent lung metastasis. Additionally, research has developed gene therapies that transfect cancer cells with the iNOS gene. For example, cationic liposome-mediated iNOS transfection can enhance the anti-tumor effects of cisplatin on lung cancer [95].
An appealing strategy for cancer immunotherapy is the re-culturing of TAMs from the M2 to M1 phenotype. By promoting the conversion of arginine to NO, NOS in M1 macrophages can increase the amount of NO in the TIME, which helps suppress tumor growth. One intriguing new treatment to enhance tumor immunotherapy is the reprogramming of TAM cells to restore their ability to attack tumors. Various TAM-targeting strategies are currently being investigated at the preclinical and clinical levels. These include reprogramming TAMs to become anti-tumor macrophages [26, 96], such as Toll-like receptor (TLR) ligands, anti-signal regulatory protein alpha, and anti-CD40 antibodies [34], interfering with TAM survival, blocking monocyte recruitment, and thus recruiting TAMs to tumors (e.g., colony-stimulating factor 1 receptor and C-C chemokine receptor type 2 inhibitors).
Future research should focus on developing spatiotemporally controllable NO delivery systems, such as photoresponsive NO-releasing metal–organic frameworks or TME enzyme-triggered (e.g., matrix metalloproteinase 2-activated) nanoparticles, to precisely regulate local NO concentration windows [97, 98]. This approach requires integration with real-time monitoring tools (e.g., implantable NO microsensors) to establish closed-loop feedback systems [99], thereby avoiding pro-tumor risks caused by concentration fluctuations. Concurrently, leveraging single-cell multi-omics technologies (e.g., single-cell RNA sequencing [scRNA-seq] coupled with spatial metabolomics) will enable the systematic dissection of NO’s heterogeneous regulatory effects on immune cell subsets, particularly the crosstalk between metabolic reprogramming and epigenetic modifications, providing novel targets for combination immunotherapy [100].
Future clinical translations should prioritize combinatorial strategies that synergize NO modulators with existing immunotherapies to overcome the immunosuppressive TME. For example, low-dose NO donors or iNOS inducers can enhance the efficacy of immune checkpoint inhibitors (ICIs) by reprogramming TAMs to the M1 phenotype and restoring CD8+ T-cell infiltration [101]. To guide these strategies, robust biomarkers are critical. Real-time imaging of iNOS activity (e.g., 18F-labeled positron emission tomography [PET] tracer) and multiplexed analysis of nitrosine stress markers (e.g., 3-nitrotyrosine) can enable the dynamic monitoring of treatment response and patient stratification [102, 103].
However, key challenges remain in translating these preclinical advances. First, the dichotomous effect of NO requires the precise control of its spatiotemporal transfer. For example, while NO-releasing nanoparticles have shown efficacy in repolarizing TAMs in murine models, their clinical applicability is limited by the lack of humanized models that recapitulate the complexity of NO gradients in the human TME. Addressing these barriers requires combining multi-omics profiling (e.g., metagenomics of the microbiota and scRNA-seq of immune cell states) with intelligent delivery systems (e.g., matrix metalloproteinase-2 [MMP-2]-responsive NO nanocarriers) for environment-dependent NO regulation.
In recent years, it has been found that the gut microbiome can affect tumor immunotherapy response by regulating host NO metabolism [104]. Specific probiotics (e.g., Lactobacillus and Bifidobacterium) can upregulate the expression of host iNOS, promote the production of NO in the TME, and in turn enhance the M1 polarization of macrophages and the infiltration of CD8+ T cells [105, 106]. In addition, microbial metabolites such as short‑chain fatty acids (SCFAs) have been shown to enhance the efficacy of ICIs, for example by promoting CD8+ T cell effector function through HDAC inhibition and metabolic reprogramming [107]. However, certain pathogenic bacteria such as Enterobacteriaceae may promote immunosuppressive microenvironment formation by limiting NO synthesis through the production of endogenous NOS inhibitors such as asymmetric dimethylarginine [108]. These findings suggest that targeting the microbe–NO axis (e.g., probiotics combined with NO donors) may become a new strategy to overcome tumor immunotherapy resistance; however, challenges remain in areas such as developing individualized flora intervention and precisely regulating NO concentrations. Future research should combine multi-omics technologies to resolve microbe–host–NO interaction networks to optimize combination therapy options.
Despite these advances, critical hurdles remain in translating NO-based therapies. A primary challenge lies in controlling the dichotomous effects of NO, where precise maintenance of its concentration within the therapeutic window (100–500 nM) is essential to avoid accidental pro-tumorigenic switching due to threshold fluctuations. This necessitates advanced delivery systems with real-time feedback control, as discussed in Section 6. Furthermore, translational bottlenecks persist in preclinical models to clinical application. Although both organoid and murine models provide valuable mechanistic insights, they fail to fully recapitulate the complexity of human TMEs, particularly regarding immune cell recruitment and accurately replicating the spatial heterogeneity of NO gradients. Addressing these gaps requires developing humanized models that integrate vascularized tumor–stroma interactions and microbiome components.
This review systematically elucidated the complex, concentration-dependent role of NO in tumor immunity, highlighting its context-specific functions as both a tumor suppressor and tumor promoter. At high concentrations, NO exerts direct cytotoxic effects by damaging tumor DNA and mitochondria while polarizing macrophages toward the anti-tumor M1 phenotype. Conversely, low concentrations of NO promote immunosuppression by inhibiting T-cell function, facilitating angiogenesis and sustaining M2 macrophage polarization. The spatiotemporal heterogeneity of NO production, governed by the competitive interplay between iNOS and ARG pathways, underscores the need for precise delivery strategies. Emerging evidence has further revealed NO’s epigenetic regulation of immune cells (e.g., via HDAC S-nitrosylation and DNA methylation), adding another layer to its immunomodulatory potential. These findings collectively position NO as a master regulator of the TIME, with implications for targeted therapy.
Current NO-based therapeutic strategies, including ARG inhibitors, NO donors, and macrophage reprogramming, demonstrate promise in preclinical models but face significant translational hurdles. For instance, nanoparticle-delivered NO donors enhance TAM repolarization and synergize with immune checkpoint inhibitors; however, their clinical efficacy is limited by poor pharmacokinetics and off-target effects. Similarly, while gut microbiota modulation (e.g., probiotics to boost iNOS) offers a novel avenue to optimize NO levels, interspecies differences in microbiome–immune crosstalk complicate human applications. Critical gaps remain in understanding the threshold dynamics of NO concentration, the impact of tumor-specific mutations (e.g., p53 status) on NO sensitivity, and the long-term consequences of epigenetic modifications induced by NO. To address these challenges, advanced tools such as real-time NO microsensors and humanized mouse models incorporating microbial ecosystems are urgently needed to bridge preclinical and clinical research.
Future research should prioritize the following concrete directions. First, developing smart NO-releasing nanocarriers responsive to specific TME cues (e.g., MMP-2, low pH) for spatiotemporally controlled delivery. Second, integrating single-cell multi-omics approaches (e.g., scRNA-seq coupled with spatial transcriptomics) to systematically dissect the heterogeneous responses of immune cell subsets to NO and its associated metabolic and epigenetic rewiring. Third, employing implantable NO microsensors for real-time monitoring of NO fluxes in preclinical models to establish concentration-efficacy correlations. Additionally, combining NO modulators with existing immunotherapies (e.g., anti-PD-1) or ferroptosis inducers could overcome resistance mechanisms in “cold” tumors. Clinically, validating NO-related biomarkers, such as iNOS PET imaging or plasma nitrosative stress markers, will be essential for monitoring therapeutic efficacy.
Ultimately, a deeper mechanistic understanding of NO’s interplay with metabolism, epigenetics, and the microbiome will unlock its full potential as a cornerstone of next-generation cancer immunotherapies, paving the way for personalized and combination treatment paradigms.
XK and HL made substantial contributions to the conception and design of the work. XK conducted the literature acquisition and analysis, drafted and wrote of the paper; HL contributed to the interpretation of the literature and critically revised the manuscript for important intellectual content. Both authors contributed to editorial changes in the manuscript. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We would like to give our heartfelt thanks to the Cuiying Biomedical Research Center, Lanzhou University Second Hospital.
This review was funded by Cuiying Scientific Training Program for Undergraduates of Lanzhou University Second Hospital (CYXZ2022-60), Cuiying Scientific and Technological Innovation Program of Lanzhou University Second Hospital (CY2024-MS-B09) and Gansu Province Health Industry Scientific Research Plan Project (GSWSKY2021-016).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.